

Abstract
AIMS
The aim of this study was to determine the inhibitory effect of itraconazole, a P-glycoprotein (P-gp) inhibitor, on the stereoselective pharmacokinetics of fexofenadine.
METHODS
A two-way double-blind, placebo-controlled crossover study was performed with a 2-week washout period. Twelve healthy volunteers received either itraconazole 200 mg or matched placebo in a randomized fashion with a single oral dose of fexofenadine 60 mg simultaneously. The plasma concentrations and the amount of urinary excretion (Ae) of fexofenadine enantiomers were measured up to 24 h after dosing.
RESULTS
After placebo administration, mean AUC(0,24 h) of S- and R-fexofenadine was 474 ng ml−1 h (95% CI 311, 638) and 798 ng ml−1 h (95% CI 497, 1101), respectively. Itraconazole affected the pharmacokinetic parameters of S-fexofenadine more, and increased AUC(0,24 h) of S-fexofenadine and R-fexofenadine by 4.0-fold (95% CI of differences 2.8, 5.3; P < 0.001) and by 3.1-fold (95% CI of differences 2.2, 4.0; P = 0.014), respectively, and Ae(0,24 h) of S-fexofenadine and R-fexofenadine by 3.6-fold (95% CI of differences 2.6, 4.5; P < 0.001) and by 2.9-fold (95% CI of differences 2.1, 3.8; P < 0.001), respectively. Additionally, the R : S ratio for AUC(0,24 h) and Ae(0,24 h) were significantly reduced in the itraconazole phase, while tmax, t1/2 and renal clearance were constant during the study.
CONCLUSIONS
This study indicates that the stereoselective pharmacokinetics of fexofenadine are due to P-gp-mediated transport and its stereoselectivity is altered by itraconazole, a P-gp inhibitor. However, further study will be needed because the different affinities of the two enantiomers for P-gp have not been supported by in vitro studies.
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Recently, we have shown that the plasma concentration of R-fexofenadine is greater than that of the S-enantiomer.
Although itraconazole co-administration is known to increase the bioavailability of a racemic mixture of fexofenadine, little is known about the stereoselective inhibition of P-gp activity by itraconazole.
WHAT THIS STUDY ADDS
This study indicates that the stereoselective pharmacokinetics of fexofenadine are due to P-gp-mediated transport and its stereoselectivity is altered by itraconazole, a an inhibitor of P-gp.
Keywords: enantiomer, fexofenadine, itraconazole, P-glycoprotein
Introduction
There is growing evidence that various membrane transporters play an important role in the absorption and disposition of many drugs [1–3]. Among these transporters, the efflux transporter P-glycoprotein (P-gp), the product of the human MDR1 gene, has been particularly investigated. In in vitro models, P-gp has been reported to transport fexofenadine [4–6], a selective histamine H1-receptor antagonist [7]. Previous clinical studies including ours reported that co-administration of itraconazole, an inhibitor of P-gp function, increased fexofenadine AUC without change in fexofenadine renal clearance or elimination half-life [8–11]. Since an increase in plasma itraconazole concentration did not affect the increase of fexofenadine AUC after repeated administration of itraconazole [10] and a single co-administration of itraconazole with fexofenadine led to maximal effect on fexofenadine pharmacokinetics [11], the increase in fexofenadine AUC by itraconazole is probably due to the reduced first-pass effect by inhibiting P-gp activity in the gastrointestinal tract.
In previous in vitro studies, stereoselective permeability was reported in P-gp-mediated transport [12, 13]. For some P-gp substrates such as verapamil [14, 15] and talinolol [16], their pharmacokinetics are known to be stereoselective. However, for both verapamil and talinolol, it has been previously reported that P-gp is unlikely to discriminate between the stereoselectivity of these compounds [14, 17–19]. Fexofenadine is also a racemic mixture of R- and S-enantiomers, possessing equal potency in their clinical effects [20], There are only a few studies on the pharmacokinetics of fexofenadine enantiomers [20, 21]. Recently, we have shown that the plasma concentration of R-fexofenadine is greater than that of the S-enantiomer and P-gp is likely to have the ability for chiral discrimination [22]. Thus, although itraconazole co-administration is known to increase the bioavailability of a racemic mixture of fexofenadine, little is known about the stereoselective inhibition of P-gp activity by itraconazole.
R/S-fexofenadine is a substrate of the drug uptake transporter, such as human organic anion-transporting polypeptide (OATP) 1A2 and OATP2B1 [4, 23–25], while there is no information that itraconazole is an inhibitor of OATPs to date. By comparing the stereoselective pharmacokinetics of R/S-fexofenadine with and without itraconazole co-administration, the stereoselective contribution of P-gp function to fexofenadine pharmacokinetics could be estimated. We therefore investigated the potential stereoselective pharmacokinetics of R/S-fexofenadine in healthy volunteers, and evaluated how itraconazole co-administration affected its stereoselective pharmacokinetics.
Methods
Subjects
Twelve healthy Japanese volunteers (seven males and five females) were enrolled in this study after giving written informed consent. Each subject was physically normal by clinical examination and routine laboratory testing and had no history of significant medical illness or hypersensitivity to any drugs. The mean (±SD) values of age and body weight of the volunteers were 21.6 (± 1.1) years (range 21–24 years) and 56.6 (± 7.0) kg (range 46–67 kg), respectively. This study was approved by the Ethics Committee of Hirosaki University School of Medicine.
Study design
A randomized, double-blind placebo-controlled crossover study design was conducted at interval of at least 2 weeks. Twelve healthy volunteers received either itraconazole 200 mg in capsule form containing four 50 mg itraconazole capsules (Janssen Pharmaceutical K.K., Tokyo, Japan) or matched placebo (in capsule form with the appearance and size the same as itraconazole) with a tablet of 60 mg fexofenadine hydrochloride (Aventis Pharma Ltd, Tokyo, Japan) at 08.00 h after an overnight fast. Compliance with taking the test drug was confirmed by pill count. The volunteers did not take any medication or fruit juices for at least 7 days before the placebo or the treatment phases, and no meal or beverages were allowed until 4 h after the administration of fexofenadine.
Plasma and urine collections and determination of fexofenadine enantiomers, itraconazole and hydroxyitraconazole
Blood samples (10 ml each) were drawn into heparinized tubes before and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12 and 24 h after administration of fexofenadine, and plasma was separated immediately. Just before fexofenadine administration, a spot of urine was collected as a blank sample. After fexofenadine administration urine was collected from 0 to 24 h. The plasma and urine samples were stored at −20°C until assayed.
The plasma concentration of fexofenadine enantiomers was determined according to the HPLC method of Miura et al.[21]. In brief, following the addition of diphenhydramine (50 ng) as an internal standard in methanol (10 μl) to 400 μl of plasma, the plasma sample was diluted with 600 μl water and vortexed for 30 s. For urine, a sample was diluted with 900 μl water after diphenhydramine (50 ng) in methanol (10 μl) was added to a 100 μl urine sample. These mixtures were applied to an Oasis HLB extraction cartridge that had been activated previously with methanol and water (1.0 ml each). The cartridge was then washed with 1.0 ml water and 1.0 ml 40% methanol in water, and eluted with 1.0 ml 100% methanol. Eluates were evaporated to dryness in a vacuum at 40°C using a rotary evaporator (Iwaki, Tokyo, Japan). The residues were then dissolved in 50 μl methanol and vortex-mixed for 30 s; 50 μl mobile phase was added to each sample and samples were vortex-mixed for another 30 s. An aliquot of 50 μl of each sample was then processed on the HPLC apparatus. The HPLC column was a Chiral CD-Ph (250 mm × 4.6 mm i.d., Shiseido, Tokyo, Japan) and the mobile phase was 0.5% KH2PO4 (pH 3.5)-acetonitrile (65 : 35, v : v). The flow-rate was 0.5 ml min−1 at ambient temperature and sample detection was carried out at 220 nm. The lower limit of quantification was 25 ng ml−1 for (R)- and (S)-fexofenadine. The validated concentration ranges of this assay from plasma and urine were 25–625 ng ml−1 for both enantiomers. The within- and between-day coefficients of variation were less than 13.6% and accuracies were within 8.8% over the linear range for both analytes.
Plasma concentrations of itraconazole and hydroxyitraconazole were determined by the HPLC method developed in our laboratory [26]. In brief, after 30 μl of an internal standard (R051012, 10 μg ml−1) and 0.1 ml of 0.5 M disodium hydrogen phosphate were added to 1 ml of plasma, the mixture was extracted with n-heptane-chloroform (60 : 40, v : v). The organic phase was evaporated, and the samples were dissolved and injected into a column I (TSK precolumn BSA-ODS/S, 5 μm, 10 mm × 4.6 mm i.d.) for clean-up and column II (Develosil C8-5 column, 5 μm, 150 mm × 4.6 mm i.d.) for separation. The mobile phase consisted of phosphate buffer-acetonitrile (68 : 32 v : v, pH 6.0) and phosphate buffer-acetonitrile (35 : 65 v : v, pH 6.0) for clean-up and separation, respectively. The peak was detected with an ultraviolet detector set at a wavelength of 263 nm. The validated concentration ranges of this method were 3–500 ng ml−1 and 3–1000 ng ml−1 for itraconazole and hydroxyitraconazole, respectively. Intra- and interday coefficients of variation (CV) at 4, 80, 250 and 400 ng ml−1 were less than 4.6 and 5.0% for itraconazole, 4.6 and 4.9% for hydroxyitraconazole, respectively. The limit of quantification was 2 ng ml−1 for both compounds.
Pharmacokinetic data analysis
The maximum plasma concentration (Cmax) and the time to reach Cmax (tmax) of fexofenadine enantiomers, itraconazole, and hydroxyitraconazole were determined directly from the observed data. The elimination rate constant (λz) of fexofenadine was obtained by linear regression analysis by use of at least three sampling points of the terminal log-linear declining phase to the last measurable concentration. The elimination half-life (t1/2) was calculated as 0.693 divided by λz. The area under the plasma concentration–time curve from time zero to the last sampling time (AUC(0,24 h)) was calculated by the trapezoidal rule. The apparent oral clearance (CL/F) was obtained from the equation CL/F = Dose/AUC(0,24 h), where Dose is 30 mg for each fexofenadine enantiomer. The apparent volume of distribution (Vd/F) was calculated from the equation Vd/F = CL/F/λz. The renal clearance (CLrenal) was obtained from the following equation: CLrenal = Ae/AUC(0,24 h), in which Ae is the amount of fexofenadine excreted into the urine within 24 h.
Statistical analysis
The results are expressed as mean and 95% confidence interval in the tables, and mean ± SD in the figures. After the Kolmogorov-Smirnov goodness of fit test was used to assess normal distribution, a paired t-test was used for the comparison of the pharmacokinetic parameters between the two enantiomers, and two phases, i.e. placebo and itraconazole. The comparison of tmax was performed using the Wilcoxon signed-sample test. A P value of 0.05 or less was regarded as significant. Geometric mean ratios to corresponding values in the placebo phase with 95% confidence intervals were used for detection of significant difference. When the 95% confidence interval did not cross 1.0, the result was also regarded as significant. All data were analyzed with the statistical program SPSS for Windows, version 11.5 J (SPSS Inc. Chicago, III).
Results
None of the enrolled subjects reported any adverse events in the study and they completed all phases according to the study protocol.
Pharmacokinetics of fexofenadine enantiomers
Plasma concentration−time profiles of fexofenadine enantiomers after placebo and itraconazole treatments are shown in Figure 1, and the pharmacokinetic parameters are summarized in Table 1.
Figure 1.
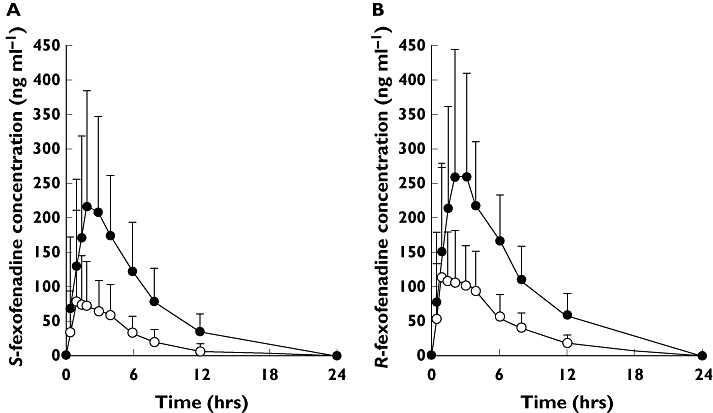
(A) Mean (+SD) plasma concentration–time curves of S-fexofenadine following a single oral administration of 60 mg fexofenadine hydrochloride in 12 healthy volunteers treated with placebo (open circles) or itraconazole (closed circles). (B) Mean (+SD) plasma concentration–time curves of R-fexofenadine following a single oral administration of 60 mg fexofenadine hydrochloride in 12 healthy volunteers treated with placebo (open circles) or itraconazole (closed circles)
Table 1.
Effects of itraconazole on pharmacokinetic parameters of fexofenadine enantiomers after a single oral 60 mg dose of fexofenadine hydrochloride. Data are shown as mean and 95% confidence interval; tmax data are shown as a median with a range
| S-fexofenadine | R-fexofenadine | |||||
|---|---|---|---|---|---|---|
| Parameters | Placebo | Itraconazole | P value | Placebo | Itraconazole | P value |
| tmax (h) (range) | 2.0 (1–4) | 3.0 (1–4) | 0.195 | 1.5 (1–4) | 3.0 (1.5–4) | 0.129 |
| Cmax (ng ml−1) | 111 (39, 182) | 236 (147, 326) | <0.001 | 160 (75, 245)*** | 290 (195, 384)††† | 0.005 |
| Mean difference (%) | 388 (184, 592) | 273 (148, 398) | 0.031 | |||
| t1/2 (h) | 3.4 (2.7, 4.2) | 3.4 (3.0, 3.9) | 0.594 | 3.9 (3.3, 4.5)* | 4.2 (3.7, 4.8)† | 0.147 |
| Mean difference (%) | 111 (86, 137) | 113 (98, 128) | 0.914 | |||
| AUC(0,24 h) (ng ml−1 h) | 474 (311, 638) | 1559 (990, 2129) | <0.001 | 798 (497, 1101)*** | 2119 (1500, 2738)††† | 0.002 |
| Mean difference (%) | 401 (277, 526) | 307 (220, 395) | 0.014 | |||
| Vd/F (l) | 445 (276, 613) | 120 (89, 150) | 0.019 | 268 (185, 350)* | 100 (77, 123)† | 0.016 |
| Mean difference (%) | 39 (25, 53) | 49 (32, 67) | 0.013 | |||
| CL/F (l h−1) | 95 (55, 135) | 25 (19, 31) | 0.017 | 50 (32, 68)** | 17 (13, 21)††† | 0.016 |
| Mean difference (%) | 33 (23, 43) | 44 (28, 60) | 0.005 | |||
| R : S ratios of AUC | 1.84 (1.69, 1.99) (Placebo phase) | 1.43 (1.30, 1.55) (Itraconazole phase) | 0.009 | |||
| Ae (0,24 h) (mg) | 3.5 (2.4, 4.6) | 10.5 (8.5, 12.4) | <0.001 | 3.3 (2.3, 4.3) | 8.1 (6.4, 9.9)††† | <0.001 |
| Mean difference (%) | 356 (260, 451) | 294 (210, 377) | 0.006 | |||
| CLrenal (l h−1) | 9.0 (6.4, 11.7) | 8.4 (6.1,10.8) | 0.669 | 4.6 (3.4, 5.7)*** | 4.4 (3.3, 5.5)††† | 0.688 |
| Mean difference (%) | 106 (82, 130) | 105 (83, 127) | 0.654 | |||
| R : S ratios of Ae | 0.95 (0.91, 0.98) (Placebo phase) | 0.77 (0.72, 0.82) (Itraconazole phase) | <0.001 | |||
tmax, observed time to reach the maximum plasma concentration; Cmax, observed maximum plasma concentration; t1/2, elimination half-life; AUC(0,24 h), area under plasma drug concentration–time curve from 0 to 24 h; Vd/F, apparent volume of distribution; CL/F, apparent oral clearance; Ae, the amount of fexofenadine excreted into urine from 0 to 24 h; CLrenal, the renal clearance; Mean difference (95% confidence interval), the within-subject ratio (itraconazole phase/placebo phase). P values, compared with the placebo phase, and compared with mean difference percentages of S-fexofenadine.
P < 0.05, between R-fexofenadine and S-fexofenadine in the placebo phase.
P < 0.01, between R-fexofenadine and S-fexofenadine in the placebo phase.
P < 0.001, between R-fexofenadine and S-fexofenadine in the placebo phase.
P < 0.05, between R-fexofenadine and S-fexofenadine in the itraconazole phase.
P < 0.001, between R-fexofenadine and S-fexofenadine in the itraconazole phase.
After placebo administration, mean plasma concentrations of R-fexofenadine were higher than those of S-fexofenadine, and mean AUC(0,24 h) (95% CI of differences −422, −202; P < 0.001) and Cmax (95% CI of differences −60, −28; P < 0.001) of R-fexofenadine were significantly greater than those of the S-enantiomer. Mean CL/F (95% CI of differences 17, 84; P = 0.009) and Vd/F (95% CI of differences 48, 288; P = 0.012) of S-fexofenadine were significantly greater than those of R-fexofenadine, and mean t1/2 of S-fexofenadine was significantly shorter than that of the R-enantiomer (95% CI of differences −0.9, −0.3; P = 0.033).
Itraconazole co-administration increased plasma concentrations of both enantiomers of fexofenadine, compared with those during the placebo phase (Figure 1), and itraconazole treatment significantly altered the pharmacokinetic parameters except the tmax and t1/2 of each enantiomer (Table 1).
The R : S ratios of plasma concentration at each sampling point ranged from 1.58 to 2.06 in the placebo phase, but itraconazole co-administration decreased the ratios from 1.20 to 1.56 (Figure 2A). The mean R : S ratio of AUC(0,24 h) was 1.84 (95% CI 1.69, 1.99) in the placebo phase, and significantly decreased to 1.43 (95% CI 1.30, 1.55) in the itraconazole phase (95% CI of differences 0.15, 0.62; P = 0.009). For the pharmacokinetic parameters except t1/2, the difference between the placebo and itraconazole phase for the parameters of S-fexofenadine was greater than those of R-fexofenadine. Itraconazole co-administration did not affect the mean t1/2 of each enantiomer, but the t1/2 of S-fexofenadine was significantly shorter than that of the R-enantiomer (95% CI of differences −1.8, −0.1; P = 0.034).
Figure 2.
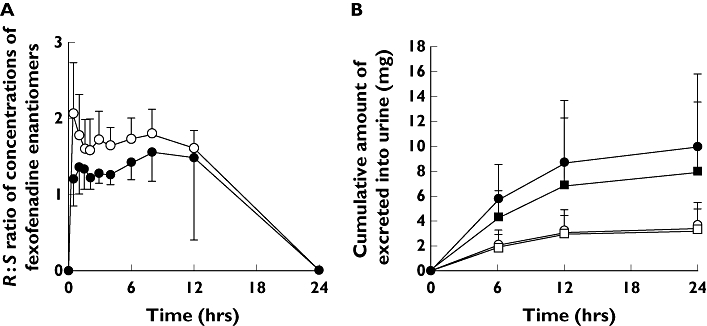
(A) Mean (±SD) R : S ratios of plasma concentrations following a single oral administration of 60 mg fexofenadine hydrochloride in 12 healthy volunteers after placebo administration (open circles) or itraconazole administration (closed circles). (B) Mean (+SD) cumulative amount of fexofenadine enantiomers excreted into urine during the placebo phase (S-fexofenadine; open circles, R-fexofenadine; open squares) or itraconazole phase (S-fexofenadine; closed circles, R-fexofenadine; closed squares) following a single oral administration of 60 mg fexofenadine hydrochloride in 12 healthy volunteers (S-placebo, (○); S-itraconazole, (•); R-placebo, (□); R-itraconazole, (▪))
Urinary excretion of fexofenadine enantiomers
The mean Ae (Figure 2B) of S-fexofenadine was significantly greater than that of R-fexofenadine in the itraconazole phase (95% CI of differences 1.5, 3.1; P < 0.001) but not after placebo (95% CI of differences −0.1, 0.5; P = 0.101), and the CLrenal of S-fexofenadine was greater than that of R-fexofenadine (95% CI of differences 2.8, 6.5; P < 0.001) (Table 1). Although itraconazole co-administration affected neither of the enantiomers with respect to CLrenal (95% CI of differences −3.1, 4.5; P = 0.669 for S-fexofenadine and 95% CI of differences −1.3, 1.8; P = 0.688 for R-fexofenadine), the co-administration significantly increased the Ae of S-fexofenadine more than that of R-fexofenadine, and the difference between the placebo and the itraconazole phase for the Ae of S-fexofenadine was greater than that of R-fexofenadine (95% CI of differences 29, 94; P = 0.006, Table 1). The R : S ratio of Ae was 0.95 (95% CI 0.91, 0.98) in the placebo phase and significantly decreased to 0.77 (95% CI 0.72, 0.82) in the itraconazole phase (95% CI of differences 0.11, 0.31; P < 0.001).
Pharmacokinetics of itraconazole and hydoxyitraconazole
The pharmacokinetic parameters of itraconazole and hydroxyitraconazole following 200 mg itraconazole are summarized in Table 2. Similar to the results in previous reports [8, 10, 11], the Cmax and AUC of hydroxyitraconazole were greater than the corresponding values of itraconazole.
Table 2.
Pharmacokinetic parameters of itraconazole and hydroxyitraconazole following 200 mg itraconazole. Data are shown as mean and 95% confidence interval; tmax data are shown as a median with a range
| Parameters | |
|---|---|
| Itraconazole | |
| tmax (h) (range) | 3 (2–6) |
| t1/2 (h) | 16 (9, 23) |
| Cmax (ng ml−1) | 96 (77, 115) |
| AUC(0,24 h) (ng ml−1 h) | 817 (630, 1003) |
| CL/F (ml h−1 kg−1) | 4563 (3560, 5566) |
| Hydroxyitraconazole | |
| tmax (h) (range) | 4 (2–6) |
| t1/2 (h) | 11 (8, 13) |
| Cmax (ng ml−1) | 195 (152, 239) |
| AUC(0,24 h) (ng ml−1 h) | 2457 (1747, 3168) |
tmax, observed time to reach the maximum plasma concentration; t1/2, elimination half-life; Cmax, observed maximum plasma concentration; AUC(0,24 h), area under plasma drug concentration-time curve from 0 to 24 h after administration; CL/F, apparent oral clearance.
Discussion
We have demonstrated the stereoselective pharmacokinetics of S- and R-fexofenadine enantiomers in healthy volunteers, and the Cmax and AUC of the R-enantiomer were significantly greater than those of S-enantiomer, resulting in a mean R : S ratio of AUC(0,24 h) of 1.84 ± 0.26. In addition, the Ae(0,24 h) of R- and S-fexofenadine were similar (3.3 ± 1.8 mg for R-fexofenadine and 3.5 ± 2.0 mg for S-fexofenadine, respectively) and the R : S ratio of Ae(0,24 h) was 0.95 ± 0.06. These results are in line with previous papers [20–22], and suggest that the effects of transporters in relation to the absorption and the excretion of S-fexofenadine were greater than those of R-fexofenadine.
Fexofenadine is a substrate of P-gp [4–6] and OATPs [4, 23–25]; the former is an efflux transporter and the latter is an uptake transporter, and both are expressed in organs including the small intestine, the liver and the kidney [27, 28]. On the other hand, known P-gp substrates, S- and R-verapamil have been previously reported to be transported equally by P-gp [14, 17, 18, 29], and the stereoselective disposition of talinolol, as a substrate of P-gp [16] and OATPs [30], is unlikely to be due to P-gp-mediated transport [19]. However, in the present fexofenadine study, the pharmacokinetics of R/S-fexofenadine show stereoselectivity, and the stereoselectivity of R/S-fexofenadine, as indicated by the R : S ratio of AUC(0,24 h), was reduced by the co-administration of itraconazole, an inhibitor of P-gp and CYP3A. Furthermore, in some in vitro studies, the stereoselective permeability was observed in P-gp-mediated transport [12, 13], and it was reported that the inhibitory effects of the P-gp efflux-pump were significantly different between the two enantiomers of butaclamol, a CNS (central nervous system) active compound [31]. Fexofenadine is poorly metabolized [4, 32] and to date there are no data available indicating that itraconazole affects the activity of OATPs. Additionally, in previous in vitro and in vivo studies, the transporters in the small intestine were reported to be important determinants of the bioavailability and disposition of fexofenadine [4–6, 23, 32, 33]. Therefore, the stereoselective pharmacokinetics of fexofenadine appear to be due to the efflux activity of P-gp in the small intestine, suggesting that the P-gp activity may be stereoselective.
In addition to these results, itraconazole administration decreased the AUC R : S ratio for fexofenadine enantiomers from 1.84 to 1.43. This implies that the inhibitory effect of P-gp-mediated transport by itraconazole is insufficient to eliminate the stereoselectivity of fexofenadine enantiomers. In our previous paper [11], we showed that itraconazole exposure at a much lower dose (50 mg) compared with the clinical dose (200 mg once or twice daily) had the maximal effect on fexofenadine pharmacokinetics. Hence, it was unlikely that the present plasma concentration of itraconazole was insufficient to inhibit the P-gp-mediated transport of fexofenadine enantiomers. This result probably suggests that OATP transporters in the small intestine play some role in the stereoselective pharmacokinetics of fexofenadine. Recently, Glaeser et al. has reported that among OATP transporters, OATP2B1 and OATP1A2 are expressed in the small intestine and only OATP1A2 is likely to be the key intestinal uptake transporter for fexofenadine absorption, whose inhibition results in the grapefruit juice effect [34]. Therefore, these findings suggest that the stereoselectivity of fexofenadine enantiomers may be related to a combination of P-gp efflux transporter and OATP1A2 uptake transporter. However, since it has not yet been determined by an in vivo/in vitro study whether OATP1A2 has stereoselectivity, further studies will be required to establish the relationships of fexofenadine enantiomers.
In our present study, itraconazole co-administration increased Cmax, AUC(0,24 h), and Ae of both S- and R-enantiomers of fexofenadine, but the parameters of the S-enantiomer were affected more than the parameters of the R-enantiomer. These results suggest that P-gp has a key role in the pharmacokinetics of both R- and S-enantiomers and this different effect may be due to the affinity of both enantiomers for P-gp. In addition, the limited effect of itraconazole on t1/2 and CLrenal of the enantiomers, which was in line with the findings of previous studies [8–11], would support the assumption that the stereoselective activity of P-gp in the small intestine led to the stereoselective pharmacokinetics of fexofenadine, and then the inhibition of the P-gp efflux activity by itraconazole increased fexofenadine absorption and reduced the stereoselectivity in fexofenadine enantiomers.
Although the AUC of S-fexofenadine was less than that of R-fexofenadine, the Ae of S-fexofenadine was greater than for R-fexofenadine in the present study, resulting in the CLrenal of S-fexofenadine being greater than that for R-fexofenadine. Since itraconazole co-administration increased the bioavailability of S-fexofenadine more, the increase in the Ae of S-fexofenadine was greater than for R-fexofenadine. To date, it remains unknown which transporter(s) play(s) a main role in the renal excretion of R- and S-fexofenadine, but stereoselectivity in renal excretion of fexofenadine was also noted because the CLrenal of S-fexofenadine was greater than for R-fexofenadine. A recent study has reported that the increase in fexofenadine AUC caused by itraconazole pretreatment was greater in subjects with the 2677TT/3435TT haplotype than in those with the 2677GG/3435CC haplotype [8], and the effect of itraconazole on P-gp activity could be MDR1 genotype dependent. Although the MDR1 haplotype was not determined in this study, there is a potential possibility that the stereoselectivity of fexofenadine enantiomers might be affected by MDR1 genotype.
In conclusion, this study indicates that the stereoselective pharmacokinetics of fexofenadine are due to the P-gp-mediated transport and its stereoselectivity is altered by itraconazole, as an inhibitor of P-gp. In addition, itraconazole appears to affect this P-gp-mediated transport of S-fexofenadine to a greater extent compared with that of R-fexofenadine. However, further study will be needed because the different affinities of the two enantiomers for P-gp have not been supported by in vitro studies.
Acknowledgments
Competing interests: None declared.
This work was supported by a Grant for priority Research Designated by the President of Hirosaki University.
REFERENCES
- 1.Kim RB. Transporters and xenobiotic disposition. Toxicology. 2002;181–182:291–7. doi: 10.1016/s0300-483x(02)00296-2. [DOI] [PubMed] [Google Scholar]
- 2.Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet. 2003;42:59–98. doi: 10.2165/00003088-200342010-00003. [DOI] [PubMed] [Google Scholar]
- 3.Fromm MF. Importance of P-glycoprotein for drug disposition in humans. Eur J Clin Invest. 2003;33:6–9. doi: 10.1046/j.1365-2362.33.s2.4.x. [DOI] [PubMed] [Google Scholar]
- 4.Cvetkovic M, Leake B, Fromm MF, Wilkinson GR, Kim RB. OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab Dispos. 1999;27:866–71. [PubMed] [Google Scholar]
- 5.Putnam WS, Ramanathan S, Pan L, Takahashi LH, Benet LZ. Functional characterization of monocarboxylic acid, large neutral amino acid, bile acid and peptide transporters, and P-glycoprotein in MDCK and Caco-2 cells. J Pharm Sci. 2002;91:2622–35. doi: 10.1002/jps.10264. [DOI] [PubMed] [Google Scholar]
- 6.Perloff MD, von Moltke LL, Greenblatt DJ. Fexofenadine transport in Caco-2 cells: inhibition with verapamil and ritonavir. J Clin Pharmacol. 2002;42:1269–74. doi: 10.1177/009127002762491370. [DOI] [PubMed] [Google Scholar]
- 7.Simpson K, Jarvis B. Fexofenadine: a review of its use in the management of seasonal allergic rhinitis and chronic idiopathic urticaria. Drugs. 2000;59:301–21. doi: 10.2165/00003495-200059020-00020. [DOI] [PubMed] [Google Scholar]
- 8.Shon JH, Yoon YR, Hong WS, Nguyen PM, Lee SS, Choi YG, Cha IJ, Shin JG. Effect of itraconazole on the pharmacokinetics and pharmacodynamics of fexofenadine in relation to the MDR1 genetic polymorphism. Clin Pharmacol Ther. 2005;78:191–201. doi: 10.1016/j.clpt.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu M, Uno T, Sugawara K, Tateishi T. Effects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br J Clin Pharmacol. 2006;61:538–44. doi: 10.1111/j.1365-2125.2006.02613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimizu M, Uno T, Sugawara K, Tateishi T. Effects of single and multiple doses of itraconazole on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br J Clin Pharmacol. 2006;62:372–6. doi: 10.1111/j.1365-2125.2006.02689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uno T, Shimizu M, Sugawara K, Tateishi T. Lack of dose-dependent effects of itraconazole on the pharmacokinetic interaction with fexofenadine. Drug Metab Dispos. 2006;34:1875–9. doi: 10.1124/dmd.106.011023. [DOI] [PubMed] [Google Scholar]
- 12.Hooiveld GJ, Heegsma J, van Montfoort JE, Jansen PL, Meijer DK, Muller M. Stereoselective transport of hydrophilic quaternary drugs by human MDR1 and rat Mdr1b P-glycoproteins. Br J Pharmacol. 2002;135:1685–94. doi: 10.1038/sj.bjp.0704620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siccardi D, Kandalaft LE, Gumbleton M, McGuigan C. Stereoselective and concentration-dependent polarized epithelial permeability of a series of phosphoramidate triester prodrugs of d4T: an in vitro study in Caco-2 and Madin-Darby canine kidney cell monolayers. J Pharmacol Exp Ther. 2003;307:1112–9. doi: 10.1124/jpet.103.056135. [DOI] [PubMed] [Google Scholar]
- 14.Luurtsema G, Molthoff CF, Windhorst AD, Smit JW, Keizer H, Boellaard R, Lammertsma AA, Franssen EJ. (R)- and (S)- [11C]verapamil as PET-tracers for measuring P-glycoprotein function: in vitro and in vivo evaluation. Nucl Med Biol. 2003;30:747–51. doi: 10.1016/s0969-8051(03)00078-7. [DOI] [PubMed] [Google Scholar]
- 15.Toornvliet R, van Berckel BN, Luurtsema G, Lubberink M, Geldof AA, Bosch TM, Oerlemans R, Lammertsma AA, Franssen EJ. Effect of age on functional P-glycoprotein in the blood–brain barrier measured by use of (R) -[(11) C] verapamil and positron emission tomography. Clin Pharmacol Ther. 2006;79:540–8. doi: 10.1016/j.clpt.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Siegmund W, Ludwig K, Giessmann T, Dazert P, Schroeder E, Sperker B, Warzok R, Kroemer HK, Cascorbi I. The effects of the human MDR1 genotype on the expression of duodenal P-glycoprotein and disposition of the probe drug talinolol. Clin Pharmacol Ther. 2002;72:572–83. doi: 10.1067/mcp.2002.127739. [DOI] [PubMed] [Google Scholar]
- 17.Hollt V, Kouba M, Dietel M, Vogt G. Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem Pharmacol. 1992;43:2601–8. doi: 10.1016/0006-2952(92)90149-d. [DOI] [PubMed] [Google Scholar]
- 18.Sandström R, Karlsson A, Lennernas H. The absence of stereo selective P-glycoprotein-mediated transport of R/S-verapamil across the rat jejunum. J Pharm Pharmacol. 1998;50:729–35. doi: 10.1111/j.2042-7158.1998.tb07133.x. [DOI] [PubMed] [Google Scholar]
- 19.Zschiesche M, Lemma GL, Klebingat KJ, Franke G, Terhaag B, Hoffmann A, Gramatte T, Kroemer HK, Siegmund W. Stereoselective disposition of talinolol in man. J Pharm Sci. 2002;91:303–11. doi: 10.1002/jps.10054. [DOI] [PubMed] [Google Scholar]
- 20.Robbins DK, Castles MA, Pack DJ, Bhargava VO, Weir SJ. Dose proportionality and comparison of single and multiple dose pharmacokinetics of fexofenadine (MDL 16455) and its enantiomers in healthy male volunteers. Biopharm Drug Dispos. 1998;19:455–63. doi: 10.1002/(sici)1099-081x(199810)19:7<455::aid-bdd130>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 21.Miura M, Uno T, Tateishi T, Suzuki T. Determination of fexofenadine enantiomers in human plasma with high-performance liquid chromatography. J Pharm Biomed Anal. 2007;43:741–5. doi: 10.1016/j.jpba.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 22.Miura M, Uno T, Tateishi T, Suzuki T. Pharmacokinetics of fexofenadine enantiomers in healthy subjects. Chirality. 2007;19:223–7. doi: 10.1002/chir.20370. [DOI] [PubMed] [Google Scholar]
- 23.Dresser GK, Bailey DG, Leake BF, Schwarz UI, Dawson PA, Freeman DJ, Kim RB. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin Pharmacol Ther. 2002;71:11–20. doi: 10.1067/mcp.2002.121152. [DOI] [PubMed] [Google Scholar]
- 24.Nozawa T, Imai K, Nezu J, Tsuji A, Tamai I. Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J Pharmacol Exp Ther. 2004;308:438–45. doi: 10.1124/jpet.103.060194. [DOI] [PubMed] [Google Scholar]
- 25.Shimizu M, Fuse K, Okudaira K, Nishigaki R, Maeda K, Kusuhara H, Sugiyama Y. Contribution of OATP (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metab Dispos. 2005;33:1477–81. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- 26.Uno T, Shimizu M, Sugawara K, Tateishi T. Sensitive determination of itraconazole and its active metabolite in human plasma by column-switching hgh-performance liquid chromatography with ultraviolet detection. Ther Drug Monit. 2006;28:526–31. doi: 10.1097/00007691-200608000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA. 1987;84:7735–8. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest. 2003;33:1–5. doi: 10.1046/j.1365-2362.33.s2.5.x. [DOI] [PubMed] [Google Scholar]
- 29.Neuhoff S, Langguth P, Dressler C, Andersson TB, Regardh CG, Spahn-Langguth H. Affinities at the verapamil binding site of MDR1-encoded P-glycoprotein: drugs and analogs, stereoisomers and metabolites. Int J Clin Pharmacol Ther. 2000;38:168–79. doi: 10.5414/cpp38168. [DOI] [PubMed] [Google Scholar]
- 30.Bernsdorf A, Giessmann T, Modess C, Wegner D, Igelbrink S, Hecker U, Haenisch S, Cascorbi I, Terhaag B, Siegmund W. Simvastatin does not influence the intestinal P-glycoprotein and MPR2, and the disposition of talinolol after chronic medication in healthy subjects genotyped for the ABCB1, ABCC2 and SLCO1B1 polymorphisms. Br J Clin Pharmacol. 2006;61:440–50. doi: 10.1111/j.1365-2125.2006.02599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szabo D, Molnar J. The role of stereoselectivity of chemosensitizers in the reversal of multidrug resistance of mouse lymphoma cells. Anticancer Res. 1998;18:3039–44. [PubMed] [Google Scholar]
- 32.Hamman MA, Bruce MA, Haehner-Daniels BD, Hall SD. The effect of rifampin administration on the disposition of fexofenadine. Clin Pharmacol Ther. 2001;69:114–21. doi: 10.1067/mcp.2001.113697. [DOI] [PubMed] [Google Scholar]
- 33.Lemma GL, Wang Z, Hamman MA, Zaheer NA, Gorski JC, Hall SD. The effect of short- and long-term administration of verapamil on the disposition of cytochrome P450 3A and P-glycoprotein substrates. Clin Pharmacol Ther. 2006;79:218–30. doi: 10.1016/j.clpt.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Glaeser H, Bailey DG, Dresser GK, Gregor JC, Schwarz UI, McGrath JS, Jolicoeur E, Lee W, Leake BF, Tirona RG, Kim RB. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81:362–70. doi: 10.1038/sj.clpt.6100056. [DOI] [PubMed] [Google Scholar]