

Abstract
AIMS
To determine the time-lag between the EU authorization of new medicines and the publications of the main randomized active control trials (RaCTs) used in the authorization process and to compare unpublished with published RaCTs of the same medicine.
METHODS
All RaCTs for new medicines with a new active substance, authorized between 1999 and 2003, were extracted from the European Public Assessment Reports (EPAR). Information about the publication status of RaCTs was obtained from the MEDLINE and EMBASE databases.
RESULTS
We identified 116 RaCTs for 42 new medicines; 28% of the RaCTs had been published at the moment of market authorization, 59% after 1 year, 78% after 2 and 83% after 3 years. Most of the rest of the studies remained unpublished after 3 years of follow-up. Unpublished RaCTs differed from published trials of the same medicine especially regarding therapeutic use and/or comparator. In some cases unpublished trials have influenced the risk : benefit asssessment of the registration authorities.
CONCLUSIONS
Most of the main RaCTs, relevant for assessing the added value of a new medicine, are published subsequent to market entry; some of these trials remain unpublished. We argue for a standardized public registration of the results of the main premarketing clinical trials as a condition for market authorization.
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Randomized active control trials are used by health care professionals and reimbursement authorities for the assessment of the added value of a new medicine.
Failing to publish the results of clinical trials limits making an evidence based assessment and conducting systematic reviews.
WHAT THIS STUDY ADDS
About one-third of the comparative trials used in the authorization process are published at the moment of market authorization and about four out of five within 2 or 3 years. Most of the rest remain unpublished.
Unpublished trials contain information regarding a different therapeutic use or a different comparator of the same medicine and, in some cases, have influenced the risk : benefit assessment of the registration authorities.
A standardized public registration of results of the main premarketing trials is advocated to fill the publication gap.
Keywords: clinical trial results database, comparative information, drug development, publication time-lag, randomized active-control trial
Introduction
When a new medicine is marketed, it is important to know how it compares with existing medicines for the same indication [1, 2]. Prescribers, pharmacists, formulary committees and regulators all require this information soon after market authorization in order to make a therapy decisions on individual patients, to develop prescribing guidelines and to set reimbursement levels. Evaluating how a new medicine compares with an existing medicine for the same indication on certain outcomes under the same conditions, can best be studied in a randomized controlled trial with the existing medicine as the active control group (RaCT).
In the premarketing period clinical trials are conducted with the objective to show efficacy and safety in order to obtain a marketing authorization. For the EU market these studies are evaluated through the European Medicines Agency (EMEA) and for the US market by the Food and Drug Administration (FDA). Placebo-controlled trials are commonly used; active control trials are not compulsory, only desirable, and sometimes necessary when a placebo-controlled trial would be unethical [3–6]. Trials designed to confirm the preliminary evidence on safety and efficacy, are called the main or pivotal trials. Efficacy can be demonstrated by detecting a difference with an placebo or an active control group (superiority trial), by confirming the absence of a difference with an active control group (equivalence trial) or by showing that the new medicine is no worse than the active control group (noninferiority trial) [6].
At the moment of market entry, the main clinical trials with an active control group are the primary source of information for learning more about the comparative efficacy and safety of the new medicine. To assess the usefulness of these studies for prescribing and reimbursement decisions, the full data of these trials should be publicly available, preferably in the form of peer-reviewed publications. Failing to publish the results of clinical trials substantially limits the possibility of making an evidence-based assessment of a new medicine and conducting systematic reviews [7]. For this reason, there should be a scientific and moral obligation upon conductors of the studies to publish the results [8, 9]. This view is echoed in the ‘Good Publication Practice’ guideline for pharmaceutical companies, which points out the responsibility of companies to make an effort to publish the results of all studies [10].
Currently little is known about the publication rates of RaCTs that are used in the authorization process or about the time-lag between market authorization and publication as an article in a journal. Furthermore, it is unknown which comparative information used in the market authorization process remains unpublished.
The aim of this study was to determine the time-lag between the authorization of a new medicine in the European Union (EU) and the publication of RaCTs used in the authorization process in the period 1999–2003 and to compare unpublished with published main RaCTs in terms of relevant therapeutic characteristics.
Methods
Source of information
We selected products with a new active substance that were authorized through the European Commission's centralized procedure in the period 1999–2003. Diagnostics and vaccines were excluded. For information about the premarketing RaCTs we used the European Public Assessment Reports (EPAR). These reports give an overview of the clinical trials that applicants have submitted to the EMEA for market approval and summarize the scientific discussion in the Committee for Medicinal Products for Human Use (CHMP) [11]. The initial version of the EPAR, which was retrieved from the EMEA website, was used. All studies were included that were labelled as main/pivotal studies in the EPAR and in which the medicine under investigation was compared directly with a known active medicine [12]. We extracted information about the date of marketing authorization and characteristics (indication, study design, number of patients) of each RaCT. In order to compare, the EU authorization date with the authorization date in the USA, we retrieved the latter from the website of the FDA [13].
Literature search
To determine which of the RaCTs that were reported in the EPAR had been published as an article in a journal, we searched the MEDLINE and EMBASE databases using the new medicine's international nonproprietary name and the keywords ‘randomized controlled trial’. The search date was January 1, 2007.
One investigator (JvL) assessed the publication status of all RaCTs by comparing the study design, number of patients and study-results of the published RaCTs with those reported in the EPAR. A second investigator (PS) assessed whether a study was correctly identified as not published. Meta-analyses were regarded as publications of a RaCT when not separately published. The date of publication, both on-line or in print, was extracted and the latter date was used for the analysis.
Therapeutic analysis
To analyze possible useful comparative information on efficacy and safety in unpublished studies, relevant therapeutic characteristics of unpublished main RaCTs were compared with the published trials of the same medicine. We made this comparison at two different moments, the moment of market authorization and 3 years later. We classified these therapeutic characteristics into six categories: 1) unpublished RaCT is the only source of information on comparative efficacy and safety used in the authorization process; 2) different therapeutic use (studied in another indication or patient population than in published RaCTs); 3) different comparator (different substance or dose than in published RaCTs); 4) longer duration of treatment than in published RaCTs; 5) shorter duration of treatment than in published RaCTs; 6) same therapeutic use, comparator and duration as in published RaCTs. If a medicine was compared with another comparator in case of different indication than in a published RaCT, it was classified as ‘different therapeutic use’ (category 2). Categories 1, 2, 3 and 4 were regarded as relevant additional information, categories 5 and 6 as less relevant.
Statistical analysis
We constructed Kaplan-Meyer curves depicting publication probability. Trials published before the moment of EU-authorization were analyzed with a time to publication of 0.01 months.
Results
Between 1999 and 2003 we identified 116 randomized active control trials for 42 medicines with a new active substance. Table 1 gives an overview of relevant characteristics of these RaCTs and their publication status. At the moment of market authorization 33 (28%) RaCTs had been published, 1 year after market authorization 68 (59%), after 2 years 90 (78%) and after 3 years 96 (83%). The annual number of publications 2 and 3 years after authorization, is reasonably constant. In view of the small number of new medicines with a RaCT in 2003, it is not possible to assess whether a trend towards prompter publication of trials at the moment of authorization exists.
Table 1.
Publication status of premarketing randomized active-control trials of new medicines
| Number of published trials (%) | ||||||
|---|---|---|---|---|---|---|
| Characteristics | Med | RaCT | At MA | After 1 year | After 2 years | After 3 years |
| Year of authorization | ||||||
| 1999 | 10* | 18 | 2 (11%) | 9 (50%) | 15 (83%) | 15 (83%) |
| 2000 | 9† | 25 | 5 (20%) | 12 (48%) | 17 (68%) | 18 (72%) |
| 2001 | 11‡ | 31 | 10 (32%) | 18 (58%) | 27 (87%) | 29 (94%) |
| 2002 | 10§ | 31 | 12 (39%) | 22 (71%) | 23 (74%) | 25 (81%) |
| 2003 | 2¶ | 11 | 4 (36%) | 7 (64%) | 8 (73%) | 9 (82%) |
| All years | 42 | 116 | 33 (28%) | 68 (59%) | 90 (78%) | 96 (83%) |
| Therapeutic indication | ||||||
| Bacterial infections | 2 | 13 | 1 (8%) | 5 (39%) | 11 (85%) | 13 (100%) |
| Diabetes mellitus | 5 | 18 | 4 (22%) | 8 (44%) | 10 (56%) | 11 (61%) |
| Glaucoma | 3 | 10 | 7 (70%) | 8 (80%) | 9 (90%) | 9 (90%) |
| HIV-1 infections | 5 | 8 | 0 | 3 (38%) | 5 (63%) | 5 (63%) |
| Other (n = 25) | 27 | 67 | 21 (31%) | 44 (66%) | 55 (82%) | 58 (87%) |
| Study design RaCT | ||||||
| Noninferiority | 17 | 32 | 7 (22%) | 19 (59%) | 26 (81%) | 27 (85%) |
| Equivalence | 10 | 23 | 6 (26%) | 11 (48%) | 19 (83%) | 20 (87%) |
| Superiority | 5 | 10 | 4 (40%) | 8 (80%) | 8 (80%) | 9 (90%) |
| No information | 20 | 51 | 16 (31%) | 30 (59%) | 37 (73%) | 40 (79%) |
| Market authorization | ||||||
| EU first | 16 | 41 | 7 (17%) | 19 (46%) | 32 (78%) | 35 (85%) |
| FDA first | 26 | 75 | 26 (35%) | 49 (65%) | 58 (77%) | 61 (81%) |
Medicines with new active substance (number of trials):
abacavir (1), cetrorelix (2), deferiprone (1), efavirenz (1), emedastine (2), insulin aspart (3), interferon alfacon-1(1), leflunomide (3), temozolomide (1), zaleplon (3);
amprenavir (1), atosiban (3), brinzolamide (4), ganirelix (3), insulin glargine (10), peginterferon alfa−2b (1), pioglitazone (1), rosiglitazone (1), sevelamer (1);
capecitabine (2), choriogonadotrophin alfa (4), darbepoetin alfa (4), lopinavir (2), nateglinide (3), rasburicase (1), sirolimus (1), telithromycin (8), tenecteplase (1), travoprost (3), zoledronic acid (2);
bimatoprost (3), epoetin delta (1), ertapenem (5), fondaparinux (4), norelgestromin (2), olopatadine (2), parecoxib (6), pegfilgrastim (2), peginterferon alfa-2a (4), voriconazole (2);
emtricitabine (3), valdecoxib (8); valdecoxib: suspension in marketing authorization October 13, 2005. MA, market authorization; Med, medicine; RaCT, randomized active control trial.
The new medicines were intended to treat a number of different indications. Indications with eight or more studies are bacterial infections, HIV-1 infections, diabetes mellitus and glaucoma. For new antiretroviral and antidiabetic medicines the publication rates after 3 years were lower than for antibacterial and antiglaucoma agents.
Looking at the design of the RaCTs, superiority trials have a comparatively higher publication rate than noninferiority and equivalence trials during the first year after market authorization. For 51 (44%) trials there was a lack of information in the EPAR on the design of the trial; on the basis of the CHMP's opinion on the results of the studies, we assumed that almost all studies were noninferiority or equivalence trials. This lack of clarity restricts a sound conclusion on differences in publication rate in relation to the study design. However, the results show that most of the studies were not designed to demonstrate superiority. We found 10 superiority trials for five new medicines: bimatoprost (2), fondaparinux (4), olopatadine (1), peginterferon alfa-2a (2) and voriconazole (1).
Figure 1 shows two Kaplan-Meyer curves depicting the publication probability of a RaCT after market authorization for medicines that were first authorized by the FDA and for those first authorized by the EMEA. We found that 26 (62%) medicines were authorized by the FDA before they were authorized by the EMEA. In these cases, the US authorization date was, on average, 13 months earlier than the EU authorization date. At the moment of EU market authorization, there were more publications for medicines with a prior FDA authorization. Both curves show that after 2 or 3 years few additional RaCTs are published; at the end of the follow-up 18 (16%) of the premarketing RaCTs remained unpublished.
Figure 1.
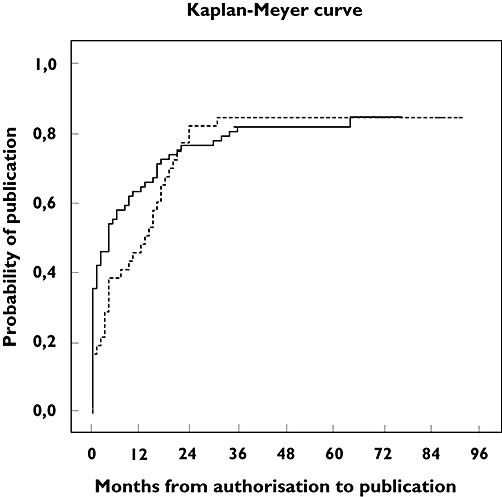
Kaplan-Meyer curves depicting the publication probability of a RaCT after market authorization for medicines that were first authorized by the FDA (—) and for those first authorized by the EU (- - -)
To evaluate the importance of the unpublished main clinical trials, we compared relevant therapeutic characteristics of these RaCTs with the characteristics of the trials published at that moment for the same medicine. Table 2 shows the results of this analysis at two different moments in the time. For 75 of the 83 (90%) RaCTs unpublished at the moment of market authorization it would be interesting to know the full data from the trial especially for a different indication, patient population or comparator. After 3 years, 17 of the 20 (85%) unpublished RaCTs possibly contained relevant therapeutic information. For amprenavir, deferiprone, epoetin delta, pioglitazone and rosiglitazone the unpublished RaCT was the only RaCT used in the authorization process. Also not published were: (i) trials with a different comparator, namely for: darbepoetin alfa (comparator with another dose), emtricitabine, insulin glargine, lopinavir, olopatadine, parecoxib and valdecoxib; (ii) trials in which medicines were studied in a different therapeutic indication, this was the case for bimatoprost in adjunctive therapy of glaucoma and insulin aspart in diabetes type II; (iii) trials in which medicines were studied in a special population, this was found for zaleplon in elderly patients and parecoxib in patients requiring postorthopaedic surgery analgesia. Less interesting are the results of three studies on insulin glargine as they had the same design or a shorter duration as the published trials.
Table 2.
Comparison of therapeutic characteristics of unpublished to published randomised active control trials (RaCT) of the same new medicine
| Number of unpublished trials | ||
|---|---|---|
| Category therapeutic characteristics of unpublished vs. published RaCTs, used in marketing authorization process | At moment of authorization n = 83 (number of medicines) | After 3 years n = 20 (number of medicines) |
| 1 Only comparative information | ||
| One RaCT/new medicine | 11 (11) | 5 (5) |
| More RaCTs/new medicine | 37 (12) | 0 |
| 2 Different therapeutic use | ||
| Other therapeutic indication | 12 (6) | 2 (2) |
| Other patient population | 5 (2) | 3 (2) |
| 3 Different comparator (same therapeutic use) | ||
| Other active substance | 9 (6) | 6 (6) |
| Other dose | 1 (1) | 1 (1) |
| 4 Longer duration of treatment | 0 | 0 |
| 5 Shorter duration of treatment | 3 (3) | 2 (1) |
| 6 Same therapeutic use, comparator and duration | 5 (3) | 1 (1) |
Discussion
Once market authorization for a new medicine has been obtained, there is considerable pressure on health care professionals and regulators to make the new medicine available so that it can be applied in clinical practice. At the same time there is a great need for information to make an evidence-based assessment of the therapeutic position of the new medicine in relation to products already on the market. In this study we found that less than one third of the main RaCTs used in the EU market authorization process, had been published at the moment of market authorization, and 78% after 2 years. About one in five RaCTs remained unpublished even after 3 years of follow-up.
Overall this is good news. It reflects a strong commitment by pharmaceutical companies to publish the main RaCTs. However, we found that for most of the unpublished RaCTs it was still relevant to take note of the results as a peer-reviewed publication. This conclusion is based on a comparison between published and unpublished RaCTs in terms of a different therapeutic use, comparator and duration. The usefulness of the results of these studies for prescribing and reimbursement decisions depends on the quality of the data. Therefore, the full data of the trial should be publicly available, to make a critical evaluation possible. For example, relating to the study design, assay sensitivity, the ability to distinguish active from inactive medicines, is a very critical issue in noninferiority and equivalence trials for a correct interpretation of the efficacy results [6, 14].
The finding that 3 years after market authorization less than one-fifth of the RaCTs remained unpublished, raises this question of whether trials with positive results are more likely to be published than trials with a negative result [15–17]. We could not study this problem as detailed information is needed on the statistical significance between the trial arms. For most studies the EPAR did not provide the basic details of trial design and results in a uniform fashion, as was mentioned earlier [18]. Therefore, we analyzed the results of some of the unpublished RaCTs qualitatively. We found that the superiority trial with olopatadine had failed to show a difference in efficacy to levocabastine. Moreover, some unpublished studies have influenced the risk : benefit assessment of the registration authorities, resulting in a restrictive therapeutic indication. For amprenavir the results of the comparative study were reason to mention in the approved indication that in protease inhibitor naive HIV-patients, amprenavir is less effective than indinavir. The comparative studies of rosiglitazone and pioglitazone, compared with suboptimal doses of glibenclamide, gave insufficient evidence of efficacy in monotherapy in diabetes mellitus; only 4 years later, based on new comparative studies, this indication was accepted by the EMEA. It is interesting to note that, based on the same comparative study on rosiglitazone and placebo-controlled studies, in an earlier authorization the FDA, unlike the EMEA, accepted the use of both glitazones for the treatment of diabetes mellitus type II in monotherapy [13, 19]. These examples show that publication of all available data from RaCTs that were reviewed in the context of the authorization process, is important for an evidence-based assessment of the position of a new medicine in therapy.
When interpreting the results of this study, there are four limitations that have to be taken into account. Firstly, this analysis was restricted to new medicines studied during the premarketing period in randomized active control trials. In an earlier study we found that only 48% of new medicines have been studied in the premarketing period in comparison with an existing medicine [20]. When the results of the present study were considered against these data, it means that for most new medicines very little comparative information is available at the moment of market authorization. Obviously, conducting RaCTs and making the results public through the scientific literature are two different systems. Secondly, we only included medicines that were centrally authorized in the EU. This choice was made because the assessment reports of these products are available in the public domain. Until October 2005, there was no obligation to provide a public assessment report under the decentralized procedure. Thirdly, we were not able to evaluate whether the publicly available RaCT information was complete as the trials submitted for market approval are chosen by the applicant and all commercially confidential information is deleted from the EPAR [11]. However, pharmaceutical companies are likely to be the only sponsors for premarketing studies and they must incorporate all relevant studies into a marketing authorization application. Finally, as this analysis was restricted to peer reviewed publications we excluded alternative methods for dissemination of trial results such as abstracts/posters at scientific meetings and databases sponsored by pharmaceutical companies.
The question is what needs to happen to improve, soon after market entry, the public availability of the information contained in main RaCTs. Firstly, improvements in the process of journal publication are an option, for example by reducing the duration of the peer-review process [21]. Secondly, time could also be gained by publishing articles electronically in advance. In our study, we found that only 5% of the articles were published on-line prior to printed publication. Thirdly, some suggest that registration authorities should be enabled to require publication of every clinical trial submitted [9]. However, although these possible solutions would improve the current situation, they still mean a delay in publication with the consequence of fewer data being available to make a decision on prescribing, guideline review or reimbursement at the moment of market entry. Therefore, in addition to peer reviewed publications and separated from the public clinical trial registry, we strongly support initiatives to make public the results of the main premarketing clinical trials in public trial results databases [22, 23]. However, to ensure the usefulness as reference for evidence-based decision-making and conducting systematic reviews, we feel that trial results databases should have to meet the same requirements as stated by the International Committee of Medical Journal Editors for an acceptable clinical trial registry [24, 25]. The database must be electronically searchable and accessible to the public at no charge; it should be open to all those who wish to register, it should be nonprofit-making, and it should have a mechanism for ensuring the validity of the registration data. Moreover, the results should be reported in a comprehensive and uniform format. To give health care professionals and regulators the opportunity to use the same information as registration authorities, we argue for a standardized public registration of the results as a condition for market authorization.
Is the urge of acquiring comparative treatment information exclusively a European feature? Surely this is not the case. Within the US, Canada and other health care systems, demands are also being made for more comparative evidence when more than one treatment option is available in order to support prescribing guidelines and reimbursement decisions [26]. It is difficult to control health care costs and guarantee access to necessary treatment possibilities in daily practice, particularly when they are expensive, without comparing medicines in terms of added value for patients and society.
Although the added value of a new medicine may not be part of the formal market authorization process, either in Europe or in the US, there is no doubt about the great need to address both comparative safety and efficacy between medicinal products subsequent to market authorization [27–29]. Therefore, it is not only necessary to invest in studies that provide comparative evidence, but also to identify and develop incentives for building comparative information as soon as possible in the drug development process and to make the results of such comparisons available as soon as possible to the public domain. This study showed that about four out of five of such main RaCTs were published within 2 or 3 years after market authorization. However, we need to evaluate the impact of the information gap due to the nonpublished RaCTs. Moreover, we need also to study the quality of the comparative information, both with regard to the choice of comparator and study design.
Placebo-controlled trials are commonly used in clinical drug development because they have important advantages; if patients are not harmed, such trials can ethically be carried out [3, 5]. But that is not the end of the story. There is ample need for innovative and comparative learning on drug effects when the confirming route has already been paved in a significant way [30].
Acknowledgments
The authors are grateful to Ms Marli van Amsterdam for the literature search.
REFERENCES
- 1.International Society of Drug Bulletins. ISDB Declaration on Therapeutic Advance in the Use of Medicines. Paris: International Society of Drug Bulletins; 2001. [Google Scholar]
- 2.National Institute of Clinical Excellence. Guide to the Methods of Technology Appraisal. London: NICE; 2004. [PubMed] [Google Scholar]
- 3.Ellenberg SS, Temple R. Placebo-controlled trials and active-control trials in the evaluation of new treatments. Part 2: practical issues and specific cases. Ann Intern Med. 2000;133:464–70. doi: 10.7326/0003-4819-133-6-200009190-00015. [DOI] [PubMed] [Google Scholar]
- 4.Koopmans PP, de Graeff PA, Zwieten-Boot BJ, Lekkerkerker JF, Broekmans AW. Clinical evaluation of efficacy and adverse effects in the (European) registration of drugs: what does it mean for the doctor and the patient? Ned Tijdschr Geneeskd. 2000;144:756–61. (in Dutch). [PubMed] [Google Scholar]
- 5.Temple R, Ellenberg SS. Placebo-controlled trials and active-control trials in the evaluation of new treatments. Part 1: ethical and scientific issues. Ann Intern Med. 2000;133:455–63. doi: 10.7326/0003-4819-133-6-200009190-00014. [DOI] [PubMed] [Google Scholar]
- 6.The European Agency for the Evaluation of Medicinal Products. Note for Guidance on Choice of Control Group in Clinical Trials (CPMP/ICH/364/96) London: EMEA; 2000. [Google Scholar]
- 7.Reidenberg MM. Conflict of interest and medical publication. Sci Eng Ethics. 2002;8:455–7. doi: 10.1007/s11948-002-0067-5. [DOI] [PubMed] [Google Scholar]
- 8.Chalmers I. Underreporting research is scientific misconduct. JAMA. 1990;263:1405–8. [PubMed] [Google Scholar]
- 9.Reidenberg MM. Improving how we evaluate the toxicity of approved drugs. Clin Pharmacol Ther. 2006;80:1–6. doi: 10.1016/j.clpt.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Wager E, Field EA, Grossman L. Good publication practice for pharmaceutical companies. Curr Med Res Opin. 2003;19:149–54. doi: 10.1185/030079903125001767. [DOI] [PubMed] [Google Scholar]
- 11.The European Agency for the Evaluation of Medicinal Products. European Public Assessment Reports. 2007. [1 January 2007]. Available at http://www.emea.eu.International/index/index1.htm.
- 12.The European Agency for the Evaluation of Medicinal Products. Notice to Applicants. Volume 2A: Procedures for Marketing Authorisation Appendix III. London: EMEA; 2004. [Google Scholar]
- 13.U.S. Food and Drug Administration. Approved Drug Products. 2007. [1 January 2007]. Available at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/
- 14.Temple R. Policy developments in regulatory approval. Stat Med. 2002;21:2939–48. doi: 10.1002/sim.1298. [DOI] [PubMed] [Google Scholar]
- 15.Bardy AH. Bias in reporting clinical trials. Br J Clin Pharmacol. 1998;46:147–50. doi: 10.1046/j.1365-2125.1998.00759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hopewell S, Clark M, Stewart L, Tierney J. Time to publication for results of clinical trials. Cochrane Database Methodol Rev. 2007 doi: 10.1002/14651858.MR000011.pub2. (4): MR000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ioannidis JP. Effect of the statistical significance of results on the time to completion and publication of randomized efficacy trials. JAMA. 1998;279:281–6. doi: 10.1001/jama.279.4.281. [DOI] [PubMed] [Google Scholar]
- 18.Wieringa NF, Vos R, de Graeff PA. Comparative trials in registration files of cardiovascular drugs: comparator drugs and dosing schemes. Pharm World Sci. 2001;23:28–30. doi: 10.1023/a:1011228828969. [DOI] [PubMed] [Google Scholar]
- 19.Gale EA. Lessons from the glitazones: a story of drug development. Lancet. 2001;357:1870–5. doi: 10.1016/S0140-6736(00)04960-6. [DOI] [PubMed] [Google Scholar]
- 20.Luijn J, Cv Gribnau FW, Leufkens HG. Availability of comparative trials for the assessment of new medicines in the European Union at the moment of market authorization. Br J Clin Pharmacol. 2007;63:159–62. doi: 10.1111/j.1365-2125.2006.02812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark A, Singleton-Jackson J, Newsom R. Journal editing: managing the peer review process for timely publication of articles. Publish Res Q. 2000;16:62–72. [Google Scholar]
- 22.Pharmaceutical Research and Manufactures of America. Clinical Study Results Database. 2007. [1 January 2007]. Available at http://www.clinicalstudyresults.org.
- 23.Sim I, Detmer DE. Beyond trial registration: a global trial bank for clinical trial reporting. PLoS Med. 2005;2:e365. doi: 10.1371/journal.pmed.0020365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Angelis C, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, Kotzin S, Laine C, Marusic A, Overbeke AJ, Schroeder TV, Sox HC, Van Der Weyden MB. Clinical trial registration: a statement from the International Committee of Medical Journal Editors. N Engl J Med. 2004;351:1250–1. doi: 10.1056/NEJMe048225. [DOI] [PubMed] [Google Scholar]
- 25.De Angelis CD, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, Kotzin S, Laine C, Marusic A, Overbeke AJ, Schroeder TV, Sox HC, Van Der Weyden MB. Is this clinical trial fully registered? – a statement from the International Committee of Medical Journal Editors. N Engl J Med. 2005;352:2436–8. doi: 10.1056/NEJMe058127. [DOI] [PubMed] [Google Scholar]
- 26.Morgan SG, McMahon M, Mitton C, Roughead E, Kirk R, Kanavos P, Menon D. Centralized drug review processes in Australia, Canada, New Zealand, and the United kingdom. Health Aff (Millwood) 2006;25:337–47. doi: 10.1377/hlthaff.25.2.337. [DOI] [PubMed] [Google Scholar]
- 27.Buckman S, Huang SM, Murphy S. Medical product development and regulatory science for the 21st century: the critical path vision and its impact on health care. Clin Pharmacol Ther. 2007;81:141–4. doi: 10.1038/sj.clpt.6100085. [DOI] [PubMed] [Google Scholar]
- 28.Ray WA, Stein CM. Reform of drug regulation – beyond an independent drug-safety board. N Engl J Med. 2006;354:194–201. doi: 10.1056/NEJMsb053432. [DOI] [PubMed] [Google Scholar]
- 29.Wood AJ. A proposal for radical changes in the drug-approval process. N Engl J Med. 2006;355:618–23. doi: 10.1056/NEJMsb055203. [DOI] [PubMed] [Google Scholar]
- 30.Sheiner LB. Learning versus confirming in clinical drug development. Clin Pharmacol Ther. 1997;61:275–91. doi: 10.1016/S0009-9236(97)90160-0. [DOI] [PubMed] [Google Scholar]