

Abstract
The important anticancer drug Taxol® (paclitaxel, PTX) owes its unique activity to its ability to bind to tubulin in a stoichiometric ratio and promote its assembly into microtubules. The conformation of the microtubule-bound drug has been the focus of numerous research efforts, since the inability of polymerized tubulin to form crystals precludes structure proof by X-ray crystallography. Likewise, although the αβ-tubulin dimer structure has been solved by electron crystallography, the 3.7 Å resolution is too low to permit direct determination of either ligand conformation or binding pose. In this study we present experimental results from 2H{19F} REDOR NMR that provide direct confirmation that paclitaxel adopts a T-shaped conformation when it is bound to tubulin.
Introduction
The natural product paclitaxel (PTX) (Taxol®, 1) is a major cancer chemotherapeutic agent which is in clinical use for the treatment of breast, lung, and ovarian carcinomas.1 Its importance is underscored by the fact that a number of analogs with improved activities have been prepared, some of which are in clinical trials.2 PTX acts as a microtubule stabilizer, binding to microtubules and blocking microtubule dynamics, ultimately leading to damage to the mitotic spindle and apoptosis.3 Although PTX has other biological effects, its clinical activity is believed to be directly related to its microtubule-binding activity.3e,4
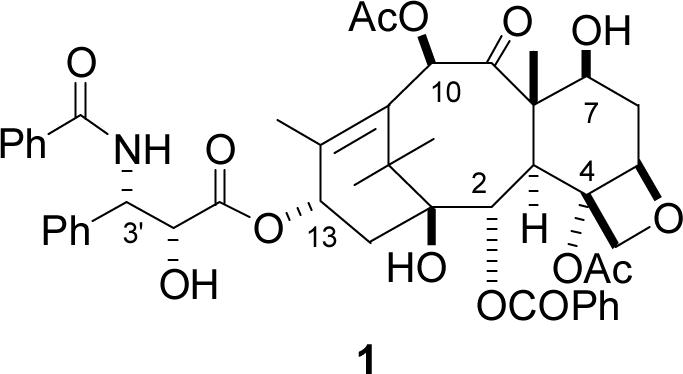
One aspect of the binding of PTX to microtubules that is still being subjected to test is the conformation of the various side chains in the bound molecule. Although PTX has a relatively rigid tetracyclic ring system, it has four flexible side chains at C2, C4, C10, and C13, and the presence of these freely rotatable units gives rise to numerous possible conformations. A detailed understanding of the microtubule-binding conformation of PTX could in principle lead to the development of improved and perhaps greatly simplified analogs. The elucidation of this conformation is thus a matter of potential practical as well as theoretical significance.
The direct approach of determining the binding conformation of the microtubule-PTX complex by X-ray crystallography is not available because of the polymeric and non-crystalline nature of the complex. Although it has proved possible to determine the structure of the tubulin dimer at 3.7 Å by electron crystallography of PTX-stabilized zinc-induced tubulin sheets,5 the structure lacks the resolution to define the detailed conformation of PTX on the tubulin polymer. It is thus necessary to use indirect methods to determine the bound conformation of PTX. Standard solution state NMR methods cannot be used for microtubule-bound PTX due to the aqueous insolubility of the polymeric receptor. Nonetheless, recent advances in NMR spectroscopy carry the promise of exploiting spin-diffusion to determine relative binding modes of microtubule-promoting ligands, including baccatin III, competing for the same binding site.6 In an alternative approach, solution state NMR analysis of PTX conformations in the absence of the protein coupled with molecular dynamics simulations and the electron crystallographic density of the tubulin-PTX complex led to the proposal of T-Taxol as the bound conformation.7
Most previous studies of the bioactive conformation have used evidence from solution NMR analyses and molecular modeling, and have led to the proposal of three different models for the solution conformation of PTX. NMR studies in nonpolar solvents suggested a “nonpolar” conformation with hydrophobic interactions between the C2 benzoate and the C3'-benzamido group.8 A “polar” or hydrophobic collapsed conformation was then proposed on the basis of NMR studies in polar solvents; this conformation features hydrophobic interactions between the C2 benzoate, the C3' phenyl group, and the C4 acetate.9-12 More recent studies using the NAMFIS deconvolution approach have shown that PTX adopts 9−10 conformations in CDCl3,13 none of which achieves a population above 30%.
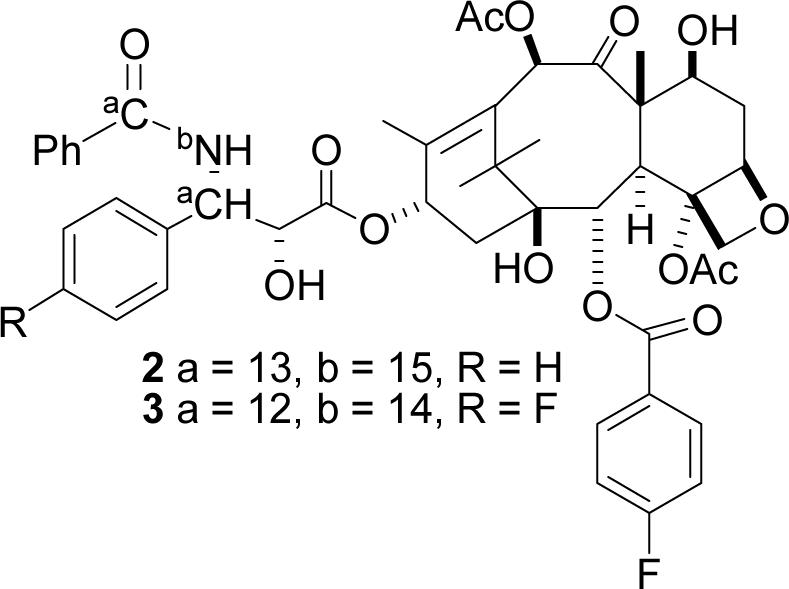
Although studies of the solution-phase conformations of PTX have been instructive, they cannot furnish direct information on the microtubule-bound conformation. The determination of the latter requires direct observation of microtubule-bound PTX, which necessitates the use of a technique that will permit measurement of internuclear distances on the ligand of a PTX-microtubule complex. Fortunately, rotational-echo double resonance (REDOR)14 allows just this measurement. One study using this technique has appeared to date. This study used the 2-(p-fluorobenzoyl)PTX 2 with 13C labels at the C3' amide carbonyl and C3' methine carbons, and yielded F-13C distances of 9.8 and 10.3 Å between the fluorine and the C3' amide carbonyl and C3' methine carbons, respectively.15,16
Analysis of the existing NMR data, both solution phase and REDOR, in conjunction with knowledge of the electron crystallographic density of β-tubulin, led to the postulation of the T-Taxol conformation as the microtubule-binding conformation.7,17 Subsequently, a slightly different structure consistent with the REDOR data was proposed as the bioactive tubulin-binding conformation of PTX.18 However, the same structure is inconsistent with the electron crystallographic density for PTX at the tubulin-taxane binding site.17,19 The T-Taxol conformation has been strongly supported by indirect evidence showing that the synthesis of constrained analogs based on this conformation yields compounds with improved activity.20 In this paper, we provide data on the synthesis and REDOR NMR analysis of additional PTX analogs which provide further experimental evidence for the T-Taxol conformation.
Results and Discussion
Design and synthesis of PTX analogs for REDOR analysis
An analysis of the proposed bioactive T-Taxol conformation (Figure 1)7,17 indicated that the C3' phenyl group on the side chain was positioned quite close to the methyl group of the C4 acetate unit. This observation, in fact, was the inspiration for the synthesis of highly active bridged taxanes matching and, in some cases, surpassing the activity of parent PTX.20 A critical test of the T-Taxol conformation would be determination of the distance between these two groups at the tubulin-taxane binding site. Unfortunately, the previous REDOR studies, with PTX analogs 2 and 3 labeled on the C2 benzoate and on the C13 side chain,15 provided no information on the spatial separation of the C4 acetate methyl group and atoms or groups located in other side chains.
Figure 1.
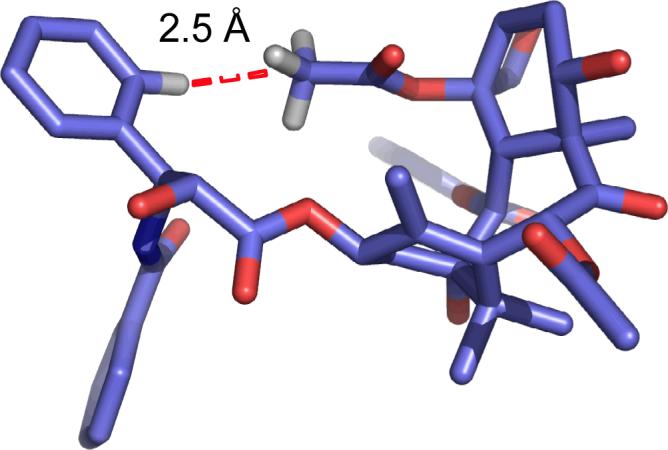
The T-Taxol conformation illustrating the short C-H distance between the centroid of the C4 acetate group and the meta-hydrogen of the C3' phenyl moiety.
These considerations led to the design of the two PTX analogs 4 − 5 as ligands to determine key distances in microtubule-bound PTX. Ligand 4 was particularly attractive, since in principle it allows the determination of two key distances; namely, between the C4 acetate and the C2 benzoate and between the C2 benzoate and the C3' phenyl. In the event, only a lower boundary on the latter distance could be determined for technical reasons, as discussed below. Ligand 5 was designed to allow a direct measurement of the distance between the C4 acetate and the C3' phenyl.

The labeled PTX derivatives were prepared from the available 10-deacetylbaccatin III. The retrosynthetic analysis of 4 is shown in Scheme 1 below.
Scheme 1.

Retrosynthesis of Ligand 4
The forward synthesis sequence is shown in Scheme 2. 10-deacetylbaccatin III was converted to the C2 debenzoylated compound 6 by the literature procedure,21 which was rebenzoylated to give 4–fluorobenzoate 7 in 75% yield. Deacetylation of the 4–acetate group by the published procedure22 gave the C4 alcohol 8, and this was reacetylated with deuterated acetyl chloride to give the acetate 9 in 66% yield. The low yield from the acetylation reaction is attributed to partial loss of the dimethylsilyl protecting group and to incomplete conversion of the starting material, even after a prolonged reaction period (72 hrs). Global deprotection was followed by selective acetylation23 of the 10–OH mediated by CeCl3 to give compound 10. Selective reprotection of the 7–OH group completed the synthesis of the labeled baccatin core 11.
Scheme 2.

Synthesis of Labeled Baccatin 11
The syntheses of the required β-lactam derivative was achieved by literature procedures (Scheme 3)24 The selected starting material was 2-bromo benzaldehyde (12), which was converted to the deuterated acetal 13 and thence to the deuterated β-lactam 14 through the N-(p-methoxy phenyl) (PMP) protected imines. Resolution of the (±)-β-lactam 14 was carried out with lipase PS (Amano) to yield the acetate of the desired (+)-enantiomer (15), along with the (-)-alcohols (not shown) in more than 95% combined yield. Functional group manipulations on 15 generated the triisopropylsilyl ether intermediate 16, which was benzoylated to give the deuterated (+)-(3R,4S)-1-benzoyl-3-triisopropylsilyloxy-4-(p-deuterophenyl)azetidin-2-one 17.
Scheme 3.

Synthesis of β-lactam 17
The final steps of the synthesis of the target taxol analog (4) consisted of a coupling reaction between the baccatin core (11) and β–lactam (17) followed by deprotection (Scheme 4).
Scheme 4.

Synthesis of Labeled PTX Derivative 4
Compound 5 was prepared by a similar route; the final steps are shown in Scheme 5, and full details of all synthetic procedures are included as Supporting Data.
Scheme 5.

Synthesis of Labeled PTX Derivative 5
Biological evaluation of REDOR analogs
The key activity criterion for the REDOR studies was the ability of the two compounds to promote tubulin assembly. In this respect both analogs proved to be effective agents; compound 4 was about twofold less active than paclitaxel while compound 5 was almost as active as paclitaxel (Table 1). Both compounds were also evaluated for cytotoxicity towards the A2780 ovarian cancer line. In this assay the differences were more marked. The analog 4 was more than two orders of magnitude less cytotoxic than paclitaxel against the A2780 ovarian cancer cell line, consistent with data on other similarly substituted analogs,25> while analog 5 was comparable in cytotoxicity to paclitaxel.26
Table 1.
Bioactivity of paclitaxel and analogs
| Compound | Tubulin Assembly activity, ED50, μM | A2780 Cytotoxicity IC50, μM |
|---|---|---|
| 1 (PTX) | 1.7 ± 0.4 | 0.023 |
| 4 | 3.5 ± 0.7 | 7.6 |
| 5 | 2.6 ± 0.5 | 0.047 |
REDOR Spectra and 2H-19F Distances
A symmetrical, full quadrupolar spectrum (broken up into spinning sidebands) is observed for the D-[2-2H,3-19F]alanine reference compound (Figure 2, bottom). This result indicates distortion-free 2H detection over a bandwidth of more than 150 kHz. Although strong n= ±8 spinning sidebands are observed for the alanine reference, they are weak in the spectrum of tubulin-bound 4 (Figure 2, top); the strong central part of the spectrum of tubulin-bound 4 is due to its CD3 group. We attribute this result to small-amplitude, low-frequency motions within the microtubule complex that interfere with echo formation for an aromatic 2H with a large quadrupolar interaction. We believe that the motion is small amplitude because the CPMAS 19F spectrum of tubulin-bound 5 is full strength, and the 19F of 5 is on the same ring as the 2H of 4. Such motions are not present in the rigid lattice of the alanine crystal. The 8% dephasing of the methyl-2H peak (Figure 3) translates into an R1-R2 distance of 7.8 ± 0.5 Å (Table 2). The R1-R3 distance in tubulin-bound 5 is 6.3 ± 0.5 Å (Figure 4, inset, and Table 2).
Figure 2.

Cross-polarization magic-angle spinning 2H NMR 2-Tr Hahn-echo spectra of tubulin-bound paclitaxel analog 4 (top) and D-[2-2H,3-19F]alanine dispersed in a solid buffer (bottom). The aromatic 2H spinning sidebands of the paclitaxel analog are much weaker than the sidebands of the deuterated alanine (dotted boxes).
Figure 3.

2H{19F} REDOR spectra of tubulin-bound paclitaxel analog 4 after 64 rotor cycles of dipolar evolution with magic-angle spinning at 8 kHz. The full-echo spectrum is shown in black and the dephased spectrum in red. The time-domain signals were detected synchronously with the rotor so that all sidebands have been folded into the centerbands. The CD3 peak is assigned zero frequency; the aromatic-D peak appears about 5 ppm to low field. The spectra resulted from the accumulation of 640,000 scans.
Table 2.
Interatomic distances for PTX conformations as compared to REDOR-determined separations for PTX on tubulin.
| Distance, Å | |||||||
|---|---|---|---|---|---|---|---|
| Separation | Cmpd | Polar modela | Nonpolar modelb | PTX-NY modelc,d | 1JFFe | T-Taxol modeld,f | REDOR distance |
| R1-R2g | 4 | 7.4 | 8.0 | 7.3 | 6.5 | 7.9 | 7.8h |
| R1-R3g | 5 | 5.5 | 7.2 | 6.4 | 7.2 | 6.6 | 6.3h |
| R2-R3 | 4 | 4.5 | 12.5 | 13.3 | 11.6 | 12.2 | >8h |
| R2-CH | 2 | 9.6 | 8.5 | 9.4 | 9.3 | 9.9 | 10.3i |
| R2-C | 2 | 10.4 | 6.2 | 10.0 | 8.1 | 9.1 | 9.8i |
Mastropaolo, D.; Camerman, A.; Luo, Y.; Brayer, G. D.; Camerman, N. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 6920−6924.
Williams, H. J.; Scott, A. I.; Dieden, R. A.; Swindell, C. S.; Chirlian, L. E.; Francl, M. M.; Heerding, J.; Krauss, N. E. Tetrahedron, 1993, 49, 6545−6560; Ref. 13.
Ref. 18.
Ref. 5.
Ref. 7.
Distances from the 4-OAc methyl group were taken as an average of the three hydrogen positions.
This work, ± 0.5 Å
Ref. 15.
Figure 4.
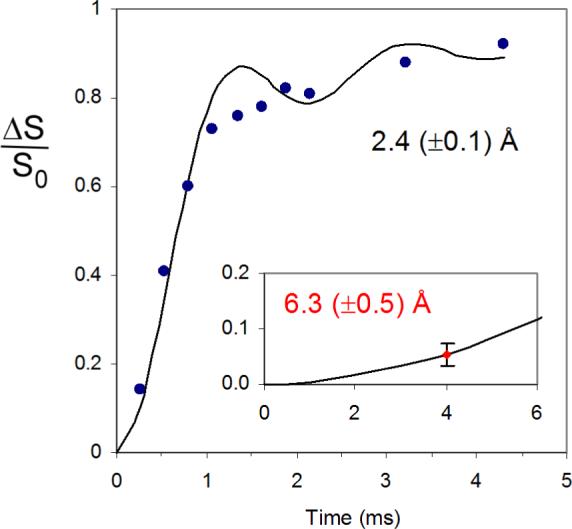
2H{19F} REDOR dephasing (ΔS/S0) for the alanine sample of Figure 2. The experimental dephasing (solid black circles) is consistent with that calculated using SIMPSON (solid black line) assuming a single 2H-19F distance of 2.4 Å. The corresponding REDOR spectra are shown in the Supplemental Material (Figure S2). The inset shows the REDOR dephasing (red) for tubulin-bound paclitaxel analog 5 after 32 rotor cycles of dipolar evolution with magic-angle spinning at 8 kHz. The REDOR spectra are shown in the Supplemental Material (Figure S3). These spectra resulted from the accumulation of 1,056,000 scans. The experimental dephasing is consistent with that calculated using SIMPSON (solid black line) assuming a single 2H3-19F distance of 6.3 Å and the 19F chemical-shift and 2H quadrupolar tensor parameters of Figure S4 of the Supplemental Material. The minor effects of C3 motional averaging were ignored.
The final key distance is the distance between R2 and R3 in tubulin-bound 4. We expected an intensity ratio of methyl to aromatic 2H peaks of 3 to 1 for tubulin-bound 4, but instead observed a ratio of about 14:1 (Figure 3). This made quantitative determination of the dephasing of the aromatic 2H peak difficult and impractical for longer evolution times and even weaker signals. Nevertheless, we are confident that the 100% dephasing predicted by the polar model (Table 2) for an R2-R3 distance of 4.6 Å is not present. The maximum possible dephasing is less than 10%, and is very likely much less than 10%. We conclude that the R2-R3 distance is greater than 8 Å.
REDOR Quantitation
Our conclusions about 2H-19F distances from REDOR experiments depend on the efficiency of the REDOR dephasing pulses. We tested this efficiency using the alanine reference compound of Figure 2. The observed dephasing was close to 100% after 4 msec (Figure 4) and consistent with a 3-bond distance in alanine.
The distance determinations also depend on the stability of the spectrometer under the long-term time averaging required to measure small differences between weak signals. For example, to determine the R1-R3 distance in just 0.1 μmoles of tubulin-bound 5 required more than 1 million scans and 3 weeks of spectrometer time. The stability of the spectrometer for such an extended period was confirmed by performing an identical 2H{19F} experiment on a deuterated alanine sample that had no 19F label. No REDOR difference was observed (Figure S3 of the Supplementary Material). Short-term spectrometer stability in REDOR experiments is guaranteed by scan-to-scan alteration of the collection of dephased-echo (S) and full-echo (S0) signals. Long-term stability arises from active control of the temperature-dependent drift in the power amplifiers.27 This control eliminates fluctuations in pulse amplitudes that result in spectral artifacts. The drive voltages for the power amplifiers were determined by a control circuit that compares the amplitudes of calibration pulses that follow both S and S0 acquisitions to a reference voltage. With this device, the power amplifiers were re-calibrated once a second throughout the entire 3-week data collection period.
The complete cancellation of S and S0 illustrated in Figure S3 (bottom) was also aided by the properties of the transmission-line probe that was used for the measurements. The 19F radiofrequency (470.9 MHz) was introduced to the transmission line at an impedence miminum for the 1H radiofrequency (500.5 MHz). This provided about a 40 dB isolation between the tune-and-match circuits for the two frequencies,28 and a total isolation (including filters and traps) of almost 90 dB. There is no accidental leakage from one radiofrequency channel to another in transmission-line probes of this design.
PTX Conformation Metrics; the Tubulin Bound Conformer
As mentioned above, three contenders for the bioactive bound form of PTX have received high level attention over the past decade: the nonpolar conformer, the polar form and T-Taxol. The present solid-state REDOR-determined interatomic distances for labeled 4 and 5, combined with complementary structural information, permit a distinction among these spatially distinct forms. The polar model proposed as the binding conformation in several reports8,9,10,15 is entirely consistent with the solid state separations of R1-R2 and R1-R3 recorded in Table 2. For all the PTX conformations discussed here, the constancy of R1-R2 is not surprising. The C4 acetate incorporating R1 methyl can adopt three conformations. However, only that with the C=O moiety positioned under the 6-membered C-ring is observed in single crystal X-ray structures. The same conformation is predicted by various computational methods to be lowest in energy. Similarly, while the C2-O bond of the C2 benzoyl group can theoretically adopt two conformations, only one is observed either experimentally (i.e. X-ray structures) or computationally. The second tucks the carbonyl group into the concave cavity of the baccatin core causing severe steric compression. Consequently, adoption of the same conformations at C2 and C4 across the PTX conformational surface ensures a fixed R1-R2 as observed for the nonpolar, polar and T-Taxol rotamers (Table 2). On the other hand, R1-R3 can be expected to vary since the C13 side chain can assume a variety of conformations.17 The polar and nonpolar models present distances that are −0.8 and +0.9 Å, respectively, from the REDOR value (Table 2). While these variations do not disqualify these conformers, they do highlight the fact that T-Taxol is within 0.3 Å of the R1-R3 measurement.
The R2-R3 phenyl-phenyl separation is crucial for rejecting the polar conformation as the bioactive form. The 4.5 Å gap falls far short of the >8 Å measure required by the REDOR experiment. By contrast, T-Taxol and the nonpolar conformers are compatible with this datapoint. In fact both conformations would seem to match the trio of REDOR distances obtained in the present study reasonably well, an observation that might seem to confirm solution phase NMR-based speculations that the nonpolar form is the bioactive one.7 The fundamental difference between them is the absence or presence, respectively, of hydrophobic collapse between the C2 and C3' N-benzoyl phenyl groups. As indicated by the structure associated with Table 2, the latter are unlabeled. As a consequence, potential REDOR dephasing experiments that might provide information on the association of the two C2 and C3' phenyl rings at the binding site (or the lack of it) are silent. On the other hand, our earlier REDOR study15 provides R2-CH and R2-C separations that offer a clear distinction. Since these distances have been discussed recently for other conformers,17 the focus here will be the nonpolar conformer. The latter structure's F2-13CH and F2-13C distances of 8.5 and 6.2 Å fall 1.8 and 3.6 Å, respectively, below the REDOR values (Table 2). The large discrepancy in distances is a consequence of the deep-seated repositioning of the C3' Ph and NHCOPh moieties from polar to nonpolar conformers. Furthermore, the results are in satisfying agreement with previously reported evidence suggesting that the C2 to C3' collapse is prevented by His227 which serves as the centerpiece of a sandwich incorporating the ligand phenyl groups on either side.7,20,29 A particularly compelling corroboration is the synthesis of bridged compounds holding the two phenyl groups in a collapsed orientation. Such compounds are inactive in the microtubule assembly assay and are unable to inhibit PTX binding.30 Other more recent efforts to tether the PTX framework to a nonpolar architecture have likewise led to compounds with low or no activity.31 For these reasons, the nonpolar conformation would appear to be ruled out as a contender for the taxane bioactive conformation.
Two other conformations closely related to T-Taxol deserve mention. The first, the electron crystallographically refined structure 1JFF,5 is a close mimic of T-Taxol.7,17 In its conformational details, however, it is not quite as close a fit as the latter to the REDOR distances. Internal distances differ by 1.0−1.7 Å (Table 2). Thus, while the pdb-deposited low-resolution structure captures the essence of the bound conformation, the modeled refinement (T-Taxol)7,32 provides the best fit to the solid-state measurements. The second conformer, PTX-NY,18 is discussed in more detail below.
Analysis of the PTX-NY Conformation.33
The PTX-NY conformation is a variation of the T-form that differs in the conformation of the C-13 side chain from C1'-C3'.18 It matches all of the REDOR distances well (Table 2), and so it is important to consider its candidacy for the paclitaxel bioactive conformation in the β-tubulin taxane binding site. In previous papers, we ruled out PTX-NY as a candidate since our reconstruction of it strongly suggested that the conformer does not fit the electron crystallographic (EC) density.17,19 Since the publication of these papers, Professor Ojima has kindly provided the original coordinates. This has made it possible to compare our reconstruction with the original and to perform two additional comparisons with it.
PTX-NY Reconstruction
Paclitaxel is a remarkably complex molecular skeleton with four-fused rings (A-D), eleven stereogenic centers and four seemingly flexible side chains at C-2, C-4, C-10 and C-13. Nonetheless, the baccatin core (A-D rings) is rigid. The acetate at C-10 is non-essential for bioactivity. C-2 and C-4 can, in principle, adopt a number of conformations, but in practice each assumes only one in solution and in the solid state of several analogs. That leaves C-13 as the primary flexible element in the molecule. However, apart from rotating phenyl rings, the conformation of this side chain is fully described by the following four dihedral angles: C12-C13-O13-C1', C13-O13-C1'-O1', O13-C1'-C2'-O2' and O2'-C2'-C3'-N3. For PTX-NY the literature gives these angles as −160, +5, +20 and +175 (Ref. 18, Figure 5) Accordingly, the corresponding four torsions in the T-Taxol conformation7 were changed to these values with no further alteration of the molecular structure. Figure 5 pictures PTX-NY (green) overlayed on the 4-angle modified T-Taxol (rust) by a five-point atom-atom superposition (i.e. C-2, C-11, C-13, C-15 and C-3'). The terminal phenyl rings are offset somewhat, particularly at C-2, but the conformation of PTX-NY is faithfully reproduced from C-1' to C-3' as highlighted by the circled atoms in Figure 5. As a result, the previous comparisons of conformations17,19 would appear to be valid.
Figure 5.

PTX-NY (green, Ref. 18) T-Taxol (rust, Ref. 7) modified in four torsions of the C-13 side chain as specified in Fig. 5/Ref. 18) two different views (a and b). The key oxygens at C-1' and C-2' are circled.
C-2 para-Fluorophenyl Orientation
All distances in Table 2 that involve R2 (4 of 5) involve measurement from a fluorine atom at the para position of the C-2 benzoyl phenyl to other atoms in the taxane structure. Accurate atomic separations in a given structure require that the fluorine is correctly located relative to other atoms in the molecule. Given the spatial discrepancy of the C-2 phenyl rings in Figure 5, we decided to examine which of the two structures is responsible and to try to understand the origin of the difference. As standards, we selected the X-ray structures of two landmark taxanes: Taxol34 (PTX, paclitaxel, magenta) and taxotere35 (DTX, docetaxel, orange). These were supplemented with para-F atoms and superposed by aligning the baccatin cores (i.e. atoms C-2, C-11, C-13 and C-15). T-Taxol (yellow) was treated similarly. Figure 6a shows that in all 3 cases, the C-2 ester group and the pendant phenyl rings overlap well; the 3 fluorines clustering within the red sphere with a radius of 1.1 Å. The identical superposition of PTX-NY is shown in Figure 6b. It would appear that while the remainder of the paclitaxel molecule is reasonably optimized, the C-2 side chain in PTX-NY is at odds with the experimental structures. The origin of the displacement is captured in Figure 7 and quantified in Table 3.
Figure 6.

Taxane superpositions within the baccatin core. a) paclitaxel (magenta), docetaxel (orange), T-Taxol (yellow); b) as in (a) with PTX-NY in green. The C-13 side chain has been truncated at C-2' to emphahsize differences in the remainder of the molecule.”
Figure 7.

The geometry around C-2 in the paclitaxel baccatin core: bond angles θ1 (C1-C2-C3), θ2 (C2-O-C(=O)) and θ3. The latter measures from the bisector (b) of C-1 and C-3 to the ester O (i.e. (b-C2-O)). Variable d corresponds to the distance from C-2 to the centroid of C-1, C-3 and O.
Table 3.
Geometry around C-2 for selected taxanes as defined in Figure 7; degrees and Å.
| θ1 | θ2 | θ3 | d | |
|---|---|---|---|---|
| PTX X-ray | 117.4 | 119.6 | 123.5 | 0.49 |
| DTX X-ray | 117.3 | 119.6 | 121.7 | 0.50 |
| T-Taxol | 119.1 | 119.1 | 123.9 | 0.48 |
| PTX-NY | 125.3 | 123.1 | 130.6 | 0.41 |
While the T-Taxol geometry around C-2 is very similar to the experimental structures, PTX-NY exhibits a distortion characterized by a flattening of the C-2 tetrahedral geometry and by an expansion of θ3, the C2-O-C(=O) bond angle, to 131°. Both work together to displace the C-2 side chain towards the concave face of the baccatin core as illustrated by Figures 5b and 6b. The implication is that while the PTX-NY structure closely matches the REDOR distances of Table 2, it is possible that the match for four of the five distances dependent on the location of the C-2 aromatic-F is fortuitous and based on a geometry that deviates significantly from experiment.
Electron Crystallographic Density Fitting
Our attempts to fit PTX-NY into the electron crystallographically refined taxane binding site5,7 without steric congestion have not been completely satisfying. Geney et al.18 apparently faced the same problem, since they report extensive rearrangement of the protein following docking of PTX-NY onto the DTX ligand-placeholder in unrefined 1TUB36 and subsequent protein-ligand minimization. A risk in the latter procedure is that introducing a pre-determined conformation into the binding cleft followed by minimization in order to relieve short non-bonded contacts can cause the system to deviate significantly from the experimental structure. However, in order to make the fairest possible comparison between PTX-NY and T-Taxol, we decided to employ the experimental structure (1JFF5) in which both ligands are docked and superimposed (Figure 8) as previously pictured both by Geney et al.18 and our group.17,19 In this way, the primary difference between them is emphasized; namely the path taken by the C-1' to C-3' segment of the C-13 side chain. Figure 8a illustrates that the trace of the EC density clearly encompasses the C-1'-C-3' atoms in T-Taxol, while the same atoms in PTX-NY are not centered in ligand density. Difference density maps for PTX-NY (Figure 8b) express this result another way. The green difference contour illustrates density unfilled by the ligand, while the red contour represents density that is incorrectly filled. By contrast, the T-Taxol conformer is in complete agreement with the same maps and in accord with very similar visualizations resulting from analysis of reconstructed PTX-NY (Figure 5).17,19
Figure 8.

The structures of T-Taxol (yellow) and PTX-NY18 (green) superposed in the 1JFF tubulin binding site. a) The 2Fobs-Fcalc omit map is shown as a blue 3D contour; b) Difference maps (Fobs-Fcalc) for the PTX-NY structure. Green corresponds to unfilled density, red to incorrectly filled density.
Conclusions and Prospects
Paclitaxel is one of the unique blockbuster anticancer drugs to emerge from natural product screening in the past two decades. Analogs continue to be created and evaluated in the clinic for improved efficacy, reduced toxicity and the ability to elude resistance. Firm knowledge of the conformation of the molecule bound to its tubulin-microtubule target would answer long-standing questions, while contributing to future efforts to remodel taxanes with the baccatin core or to completely re-engineer the molecule's architecture. Accordingly, we have prepared a series of fluoro and deuterio labeled derivatives of PTX suitable for measurement of interatomic distances by solid state 2H{19F} REDOR NMR. Samples of ligand-doped microtubules were subjected to three weeks of spectrometer time in order to measure each of three pairwise distances. The latter were compared to the same quantities for five PTX conformers, each of which has been proposed to be the bioactive form at the tubulin binding pocket.
The outcome definitively eliminates the polar conformer from consideration. The nonpolar conformer is also eliminated by the combination of the present data with our previous REDOR data.15 The PTX-NY structure fits the REDOR data well, though there are irregularities in its molecular geometry as illustrated by Figures 6 and 7 and Table 3. The latter alone, however, do not disqualify the conformer from bioactive candidacy. Critical is the observation that the shape of PTX-NY's C-13 side chain is in conflict with the experimental electron crystallographic density. On the other hand, the T-Taxol structure passes three fundamentally important tests. First, this conformation is within 0.7 Å of four well-defined REDOR distances and fully consistent with the fifth. Second, it is the only conformer in Table 2 that is compatible with the EC density. Third, it is unique in being a template for synthesis of bridged taxanes that match or surpass both the binding affinity and cytotoxicity characteristics of the paclitaxel parent. For these reasons, the T-Taxol conformation proposed by a combination of structure and modeling techniques7 and by synthesis and bioassay20 is confirmed by REDOR measurement of interatomic distances.
Finally, with respect to evaluating prospective bioactive taxane conformations at the binding site of β-tubulin, it must be noted that the electron crystallographic binding model that led to the T-Taxol proposal was derived from Zn+2-stabilized tubulin sheets5,7 and not genuine microtubules. It is thus necessary to determine whether or not this model is suitable as a guide to the 3D-shape of the bioactive form at the tubulin binding site.33 It is no coincidence that virtually every recent synthetic program attempting to prepare more potent and less resistant taxanes based on biostructural principles has employed the EC structure.18,20,29,37 Furthermore, the tubulin dimer structure derived from Zn+2-stabilized sheets has been modeled into the 8 Å resolution microtubule structure determined by cryoelectron microscopy38 and shown to be entirely consistent with the MT structure. Finally, as we have pointed out previously,17,20,29 not only is the REDOR geometry derived from lyophilized MTs coincident with the T-Taxol form (Table 2), but bioactivities in situations where MTs are present as soluble dynamic entities can be significantly improved by exploiting the EC structure and bound ligand from stabilized tubulin sheets. The implication is clear. While the details of tubulin polymerization may vary on the level of protein assembly, the taxane binding site sequestered within β-tubulin appears to be faithfully retained in the two situations. This underscores the necessity that alternative bioactive conformations for paclitaxel pass the three tests outlined in the previous paragraph.
Experimental Section
Synthesis of labeled paclitaxels
Full experimental details of the syntheses of compounds 4 − 5 are provided as Supporting Data.
ED50 Determination
Pure tubulin was obtained from bovine brain and used in polymerization and REDOR experiments. Polymerization activity of paclitaxel and derivatives was evaluated in the following manner. GTP-tubulin (5 μM) in a buffer containing 0.1 M PIPES, 1 mM MgSO4, 2 mM EDTA and 0.1 mM GTP (pH 6.90) was equilibrated to 37 °C in a quartz cuvette in a Hewlett Packard Model 8453 diode array spectrophotometer and a baseline was recorded. Polymerization was initiated by addition of the compound to be tested in DMSO, and the extent of assembly was measured as the change in apparent absorption at 350 nm. In all the solutions the percentage of DMSO was maintained as 4% v/v, to avoid any effect of DMSO on tubulin assembly. A plot of change in apparent absorption at 350 nm versus the concentration of the compound was fit to a rectuagular hyperbola to obtain the ED50 value.
NMR Samples
Paclitaxel analogues 4 (0.7 μmoles) and 5 (0.1 μmoles) were bound 1:1 to tubulin to form microtubule complexes following the protocol described earlier14 with the following modifications: GMPCPP rather than GTP was employed as the nucleotide, and 20 mM sucrose and 1 % PEG (Mn 8,000) were used as the lyo- and cryoprotectants, respectively. A small amount of the freeze dried material was reconstituted in 10 mM phosphate buffer (pH 7.0) and examined by electron microscopy; normal microtubules were observed. The complexes were packed into rotors under a dry atomosphere. A reference sample for testing 2H{19F} REDOR dephasing was prepared by dissolving D-[2-2H,3-19F]alanine (10 mg) with HEPES buffer (100 mg), flash freezing the solution, and then lyophilizing at low temperature. A control sample that contained no 19F was prepared by physically mixing 5 mg of D-[3-2H3]alanine (5 mg) with sulfur powder (100 mg). This mixture was used to confirm that a null REDOR difference signal was observed (Figure S3 of the Supplementary Material) from a 32-Tr 2H{19F} REDOR experiment after 1 million scans.
Spectrometer
Deuterium NMR spectra were obtained using a spectrometer equipped with a four-frequency (HFCD) transmission-line probe operating at a 1H frequency of 500.5 MHz and a 2H frequency of 76.83 MHz. Tune and match circuits for each frequency were external to the magnet. The probe had a 12-mm long, 6-mm inside-diameter analytical coil and a Chemagnetics/Varian magic-angle spinning ceramic stator. Five-mm thin-wall ceramic rotors were spun at 8000 Hz with the speed under active control to within ±2 Hz. A 12-T static magnetic field was provided by an 89-mm bore Magnex superconducting solenoid. The spectrometer was controlled by a Tecmag Apollo pulse programmer. Levels of all radiofrequency pulses were under active control during data acquisition by sampling of test pulses fired on each channel in use immediately after each scan.27
REDOR
The version of REDOR used in these experiments (Figure S1) had a single 2H 180° pulse (5.6 μs) and 16n (n=2,3, ...) 19F 180° pulses (5.8 μs). The phases of the 19F pulses followed the xy8 phase-alternation scheme.39 This combination gave the best echo refocusing and most complete dephasing (Figure S2). Internuclear distances were calculated from the REDOR dephasing by spectral simulations performed using SIMPSON.40 The 1H→2H matched cross-polarization transfer was at 54 kHz for 3 ms, with a 1H 90° pulse of 2.5 μs and 100-kHz dipolar decoupling with TPPM41 phase modulation.
Acknowledgement
The work at Virginia Polytechnic Institute and State University, SUNY Binghamton, and Emory University was supported by the NIH grant CA-69571, and the work at Washington University was supported by NIH grant EB-001964; we are grateful for this support. We thank William Bebout (VPISU) for mass spectroscopic determinations, and Andrew Norris and Peggy Brodie (VPISU) for A2780 cytotoxicity assays. We are likewise grateful to Professor Iwao Ojima for providing the CVFF coordinates to PTX-NY.
Footnotes
Supporting Information Available: Full experimental details of the syntheses of labeled PTX analogs 4 − 5 and S23. Figure S1: Pulse sequence for 2H{19F} REDOR. Figure S2 (2H{19F} REDOR spectra of D-[2-2H,3-19F]alanine dispersed in a solid buffer as a function of the dipolar evolution time). Figure S3 (2H{19F} REDOR spectra of tubulin-bound paclitaxel analog 5 and 2H{19F} REDOR spectra of a control sample of D-[3-2H3]alanine). Figure S4 (Cross-polarization magic-angle spinning 19F and 2H NMR spectra of tubulin-bound paclitaxel analog 4, and simulated spectra using SIMPSON). Complete Ref. 24.
Supplementary Material
References and Notes
- 1.a Rowinsky EK. Ann. Rev. Med. 1997;48:353–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]; b Crown J, O'Leary M. The Lancet. 2000;355:1176–1178. doi: 10.1016/S0140-6736(00)02074-2. [DOI] [PubMed] [Google Scholar]; b Crown J, O'Leary M, Ooi W-S. The Oncologist. 2004;9(Suppl 2):24–32. doi: 10.1634/theoncologist.9-suppl_2-24. [DOI] [PubMed] [Google Scholar]; c Mekhail T, Markman M. Expert Opin. Pharmacother. 2002;3:755–766. doi: 10.1517/14656566.3.6.755. [DOI] [PubMed] [Google Scholar]
- 2.a Kingston DGI. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, editors. CRC Press; Boca Raton, FL: 2005. pp. 89–122. [Google Scholar]; b Geney R, Chen J, Ojima I. Med. Chem. 2005;1:125–139. doi: 10.2174/1573406053175292. [DOI] [PubMed] [Google Scholar]; c Fang W-S, Liang X-T. Mini-Rev. Med. Chem. 2005;5:1–12. doi: 10.2174/1389557053402837. [DOI] [PubMed] [Google Scholar]; d Zefirova ON, Nurieva EV, Ryzhov AN, Zyk NV, Zefirov NS. Russ. J. Org. Chem. 2005;41:315–351. [Google Scholar]
- 3.a Schiff PB, Fant J, Horwitz SB. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]; b Wilson L, Jordan MA. Nat. Rev. Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]; c Horwitz SB. Trends Pharmacol. Sci. 1992;13:134–136. doi: 10.1016/0165-6147(92)90048-b. [DOI] [PubMed] [Google Scholar]; d Jordan MA, Toso RJ, Thrower D, Wilson L. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Blagosklonny MV, Fojo T. Int. J. Cancer. 1999;83:151–156. doi: 10.1002/(sici)1097-0215(19991008)83:2<151::aid-ijc1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 4.Blagosklonny MV, Giannakakou P, El-Deiry WS, Kingston DGI, Higgs PI, Neckers L, Fojo T. Cancer Res. 1997;57:130–135. [PubMed] [Google Scholar]
- 5.Lowe J, Li H, Downing KH, Nogales E. J. Mol. Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 6.Sánchez-Pedregal VM, Reese M, Meiler J, Blommers MJJ, Griesinger C, Carlomagno T. Angew. Chem. Int. Ed. 2005;44:4172–4175. doi: 10.1002/anie.200500503. [DOI] [PubMed] [Google Scholar]
- 7.Snyder JP, Nettles JH, Cornett B, Downing KH, Nogales E. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5312–5316. doi: 10.1073/pnas.051309398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Dubois J, Guenard D, Gueritte-Voeglein F, Guedira N, Potier P, Gillet B, Betoeil J-C. Tetrahedron. 1993;49:6533–6544. [Google Scholar]; b Williams HJ, Scott AI, Dieden RA, Swindell CS, Chirlian LE, Francl MM, Heerding JM, Krauss NE. Can. J. Chem. 1994;72:252–260. [Google Scholar]; c Cachau RE, Gussio R, Beutler JA, Chmurny GN, Hilton BD, Muschik GM, Erickson JW. Supercomput. Appl. High Perform. Comput. 1994;8:24–34. [Google Scholar]
- 9.Vander Velde DG, Georg GI, Grunewald GL, Gunn CW, Mitscher LA. J. Am. Chem. Soc. 1993;115:11650–11651. [Google Scholar]
- 10.Paloma LG, Guy RK, Wrasidlo W, Nicolaou KC. Chem. Biol. 1994;1:107–112. doi: 10.1016/1074-5521(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 11.Ojima I, Chakravarty S, Inoue T, Lin S, He L, Horwitz SB, Kuduk SC, Danishefsky SJ. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4256–4261. doi: 10.1073/pnas.96.8.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ojima I, Kuduk SD, Chakravarty S, Ourevitch M, Begue J-P. J. Am. Chem. Soc. 1997;119:5519–5527. [Google Scholar]
- 13.Snyder JP, Nevins N, Cicero DO, Jansen J. J. Am. Chem. Soc. 2000;122:724–725. [Google Scholar]
- 14.Gullion T, Schaefer J. Adv. Magn. Reson. 1989;13:58–82. [Google Scholar]
- 15.Li Y, Poliks B, Cegelski L, Poliks M, Gryczynski Z, Piszczek G, Jagtap PG, Studelska DR, Kingston DGI, Schaefer J, Bane S. Biochemistry. 2000;39:281–291. doi: 10.1021/bi991936r. [DOI] [PubMed] [Google Scholar]
- 16.Ojima I, Inoue T, Chakravarty S. J. Fluorine Chem. 1999;97:3–10. A related study, which has not yet been reported in full, used the radio frequency driven dipolar recoupling (RFDR) technique. This work indicated a distance of 6.5 Å between the fluorines of 2-(p-fluorobenzoyl)-3'-(p-fluorophenyl)-10-acetyldocetaxel (3), but it is not clear that this result is directly comparable to the REDOR results reported here. [Google Scholar]
- 17.Alcaraz AA, Mehta AK, Johnson SA, Snyder JP. J. Med. Chem. 2006;49:2478–2488. doi: 10.1021/jm051119r. [DOI] [PubMed] [Google Scholar]
- 18.Geney R, Sun L, Pera P, Bernacki RJ, Xia S, Horwitz SB, Simmerling CL, Ojima I. Chem. Biol. 2005;12:339–348. doi: 10.1016/j.chembiol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Johnson SA, Alcaraz A, Snyder JP. Org. Lett. 2005;7:5549–5552. doi: 10.1021/ol051780p. [DOI] [PubMed] [Google Scholar]
- 20.Ganesh T, Guza C, Bane S, Ravindra R, Shanker N, Lakdawala AS, Snyder JP, Kingston DGI. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10006–10011. doi: 10.1073/pnas.0403459101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ojima I, Lin S, Inoue T, Miller ML, Borella CP, Geng X, Walsh JJ. J. Am. Chem. Soc. 2000;122:5343–5353. [Google Scholar]
- 22.Chen S-H, Kadow JF, Farina V, Fairchild CR, Johnston KA. J. Org. Chem. 1994;59:6156–6158. [Google Scholar]
- 23.Holton RA, Zhang Z, Clarke PA, H. N, Procter DJ. Tetrahedron Lett. 1998;39:2883–2886. [Google Scholar]
- 24.Brieva R, Crich JZ, Sih CJ. J. Org. Chem. 1993;58:1068–1075. [Google Scholar]
- 25.Kingston DGI, et al. J. Med . Chem. 1998;41:3715–3726. doi: 10.1021/jm980229d. [DOI] [PubMed] [Google Scholar]
- 26.A reviewer asked whether there is any non-specific binding of the paclitaxel analogs to tubulin. We do not believe that there is any significant non-specific binding in these samples. The molar ratio of the ligand:tubulin dimer was 1:1, and the Kd for paclitaxel binding to microtubules is about 15 nM. Even if the affinities of the REDOR probes for the paclitaxel site on tubulin were 100-fold less than the affinity of paclitaxel for microtubules, >95% of the probe would be bound to the protein under the conditions used to create the samples. Since the REDOR probes are nearly as active as paclitaxel as promoters of tubulin assembly, and since the structural perturbation between paclitaxel and the analogs is very small, it is reasonable to assume that the spectra are obtained on a homogeneous, fully bound sample
- 27.Stueber D, Mehta AK, Chen Z, Wooley KL, Schaefer J. J. Polym. Sci. B: Polym. Phys. 2006;44:2760–2775. [Google Scholar]
- 28.Schaefer J, McKay RA. U.S. Patent 5,861,748. 1999.
- 29.Kingston DGI, Bane S, Snyder JP. Cell Cycle. 2005;4(2):279–289. [PubMed] [Google Scholar]
- 30.Boge TC, Wu Z-J, Himes RH, Vander Velde DG, Georg GI. Bioorg. Med. Chem. Lett. 1999;9:3047–3052. doi: 10.1016/s0960-894x(99)00522-3. [DOI] [PubMed] [Google Scholar]
- 31.a Ojima I, Geng X, Miller ML, Lin S, Pera P, Bernacki RJ. Bioorg. Med. Chem. Lett. 2002;12:349–352. doi: 10.1016/s0960-894x(01)00747-8. [DOI] [PubMed] [Google Scholar]; b Geng X, Miller ML, Lin S, Ojima I. Org. Lett. 2003;5:3733–3736. doi: 10.1021/ol0354627. [DOI] [PubMed] [Google Scholar]; b Querolle O, Dubois J, Thoret S, Dupont C, Guéritte F, Guénard D. Eur. J. Org. Chem. 2003:542–550. [Google Scholar]; c Querolle O, Dubois J, Thoret S, Roussi F, Montiel-Smith S, Gueritte F, Guénard D. J. Med. Chem. 2003;46:3623–3630. doi: 10.1021/jm030770w. [DOI] [PubMed] [Google Scholar]
- 32.Atomic coordinates of the computationally refined T-Taxol-tubulin structure can be found in the Supporting Information of Ref. 17
- 33.We thank a referee for suggesting that we add this section
- 34.Mastropaolo D, Camerman A, Luo Y, Brayer GD, Camerman N. Proc. Natl. Acad. Sci. USA. 1995;92:6920–6924. doi: 10.1073/pnas.92.15.6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guéritte-Voegelein F, Guénard D, Mangatal L, Potier P, Guilhem J, Césario M, Pascard C. Acta Crystallogr. C. 1990;46:781–784. [Google Scholar]
- 36.Nogales E, Wolf SG, Downing KH. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 37.a Boge TC, Wu Z-J, Himes RH, Vander Velde DG, Georg GI. Bioorg. & Med. Chem. Lett. 1999;9:3047–3052. doi: 10.1016/s0960-894x(99)00522-3. [DOI] [PubMed] [Google Scholar]; b He L, Jagtap PG, Kingston DGI, Shen H-J, Orr GA, Horwitz SB. Biochemistry. 2000;39:3972–3978. doi: 10.1021/bi992518p. [DOI] [PubMed] [Google Scholar]; c Giannakakou P, Gussio R, Nogales E, Downing KH, Zaharevitz D, Bollbuck B, Poy G, Sackett D, Nicolaou KC, Fojo T. Proc. Nat. Acad. Sci. USA. 2000;97:2904–2909. doi: 10.1073/pnas.040546297. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Barboni L, Lambertucci C, Appendino G, Vander Velde DG, Himes RH, Bombardelli E, Wang M, Snyder JP. J. Med. Chem. 2001;44:1576–1587. doi: 10.1021/jm001103v. [DOI] [PubMed] [Google Scholar]; e Querolle O, Dubois J, Thoret S, Roussi F, Guéritte F, Guénard D. J. Med. Chem. 2004;47:5937–5944. doi: 10.1021/jm0497996. [DOI] [PubMed] [Google Scholar]; f Hari M, Loganzo F, Annable T, Tan X, Musto S, Morilla DB, Nettles JH, Snyder JP, Greenberger' LM. Mol. Can. Ther. 2006;5:270–278. doi: 10.1158/1535-7163.MCT-05-0190. [DOI] [PubMed] [Google Scholar]
- 38.a Nogales E, Whittaker M, Milligan RA, Downing KH. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]; b Li H, DeRosier DJ, Nicholson WV, Nogales E, Downing KH. Structure. 2002;10:1317–1328. doi: 10.1016/s0969-2126(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 39.Gullion T, Baker DB, Conradi MS. J. Magn. Reson. 1990;89:479–484. [Google Scholar]
- 40.Bak M, Rasmussen JT, Nielsen NC. J. Magn. Reson. 2000;147:296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 41.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. J. Chem. Phys. 1995;103:6951–6958. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.