

Abstract
Previous studies of the annexin family of Ca2+ binding proteins identified a soluble monomer in the absence of Ca2+ and a trimer adsorbed on the membrane surface in the presence of Ca2+. On the basis of site-directed spin-labeling studies of annexin XII at low pH, we now report a membrane-inserted form of the protein with a dramatically different structure. The data suggest that upon insertion a continuous transmembrane α-helix is reversibly formed from a helix–loop–helix motif in the solution structure. Other regions with similar membrane-insertion potential were identified in the amino acid sequence, and we propose that the corresponding helices come together to form an aqueous pore that mediates the ion channel activity reported for several annexins.
Annexins are a structurally conserved family of proteins characterized by reversible Ca2+-dependent membrane binding. Membrane association is of critical importance for their proposed functions, including vesicular trafficking, membrane fusion, and ion-channel formation (1). High-resolution crystal structures of the soluble forms of several different annexins have been solved, and all structures share a common fold as exemplified by the annexin XII crystal structure in Fig. 1 (2). The fold consists of four domains, each of which contains five helices connected by short loops. The loop regions between helices A and B and between helices D and E on the convex side of the protein are believed to account for the Ca2+-dependent binding of the protein to membranes by jointly coordinating Ca2+ with phosphatidylserine.
Figure 1.

Crystal structure of hydra annexin XII (2). Helices A and B (domain 3) and helices D and E (domain 2) are highlighted in blue and red, respectively. Sites of the spin-labeled residues 138–163 are indicated by residue labels and solid spheres on the α-carbons.
Previous studies of this Ca2+-bound state found that annexin XII forms trimers on the surface of bilayers containing phosphatidylserine under physiological conditions. The fold of this membrane-surface-bound protein appears to be similar to that found in crystals of the water-soluble form (3). This structure, however, does not provide an obvious explanation for the ion-channel formation demonstrated for several annexins (4, 5). Annexin V has recently been shown to bind to membranes at low pH in the absence of Ca2+ (6). However, no information is available on the structure or topography of this membrane-bound state. In the present study, we examine the structure of annexin XII bound to membranes at low pH and demonstrate that a member of the annexin gene family can undergo a major conformational change and adopt a transmembrane configuration, a topology that can readily account for ion-channel formation.
The experimental approach is site-directed spin labeling, a powerful tool for determination of secondary, tertiary, and quaternary structure of both soluble and membrane-bound proteins (7, 8). The strategy of site-directed spin labeling requires the introduction of a paramagnetic nitroxide side chain in the protein through the selective modification of substituted cysteine residues. The nitroxide-containing side chain used in the present study is designated R1. 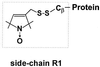
The electron paramagnetic resonance (EPR) spectrum of R1 in a protein provides information on the mobility and solvent accessibility of the side chain, the former being measured by the resonance line shape and the latter being inferred from an accessibility parameter (Π) proportional to the collision rate of the nitroxide with paramagnetic reagents in solution (9, 10). The mobility and/or accessibility of R1 are often sufficient to identify a given site as a tertiary contact, buried, loop, or helix surface site (9). Data obtained through a continuous sequence (an R1 “scan”) reveals regular secondary structure by virtue of the periodic variation in mobility and accessibility (7). Magnetic interactions between two R1 side chains can be used to estimate interspin distances in the range of 10–25 Å (11–15). For membrane proteins, the additional feature of topography with respect to the bilayer can be explored using the differential accessibility of R1 to nonpolar (O2) and polar [nickle(ethylenediaminediacetic acid), NiEDDA] paramagnetic reagents (10 16). Thus R1 side chains exposed to the hydrophobic interior of the bilayer have a relatively high Π(O2), whereas those exposed to water have a relatively high Π(NiEDDA). For residues exposed to the lipid chains, the function Φ = ln Π(O2)/Π(NiEDDA) has been shown to be a linear function of depth within the membrane (10, 16). In the present study, we applied the above features of site-directed spin labeling to map the structure of membrane-bound annexin XII.
MATERIALS AND METHODS
Protein Expression and Purification.
Specific mutations were introduced into the annexin XII pSE420-mrp33H plasmid (17) by use of the single-site mutagenesis kit from CLONTECH. First, two native Cys residues in annexin XII were replaced with Ala (C113A/C303A) as described (3). The C113A/C302A mrp33H.09 plasmid was used as the starting point for all the mutations reported herein. The preparation of single (K132C) and double (R16C/E163C and K27C/D188C) mutants suitable for the evaluation of multimer formation were as described (3). To scan a continuous region for evaluation of membrane insertion, a series of 26 single cysteine substitution mutants were prepared in the sequence of residues 138–163. All mutations were confirmed by DNA sequencing by using a Sequenase 2.0 kit (Amersham). The annexin XII mutants were expressed in recombinant bacteria and purified by reversible Ca2+-dependent binding to phospholipid vesicles followed by column chromatography (3).
Spin Labeling and EPR Measurements.
The methods used to modify the introduced cysteines with (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methanethiosulfonate (a gift from K. Hideg, University of Pecs, Hungary) were as described (3). Spin-labeled mutants of annexin XII are designated by giving the sequence position of the cysteine substitution followed by the code for R1, the nitroxide side chain. For example, K132C labeled with R1 is designated 132R1. Spin-labeled proteins were stored in dilute buffer at pH 7.4.
EPR experiments were performed by adding spin-labeled annexin XII mutants (≈30 μg of protein) to buffer (either 20 mM Hepes/100 mM NaCl, pH 7.4, or 100 mM sodium acetate at pH 4.0) in either the presence or absence of phospholipid vesicles. The molar ratio of protein to lipid was 1:500 or 1:1,000, at neutral or acidic pH, respectively. In certain experiments at neutral pH, binding of annexin XII to the vesicles was induced by Ca2+ (1 mM). EPR spectra were recorded approximately 5 min after preparing the sample. At both neutral pH in the presence of Ca2+ or at acidic pH in the absence of Ca2+, binding to phospholipid vesicles was virtually quantitative as judged by cosedimentation assays of the vesicles with the bound protein.
Large vesicles were made by the Reeves/Dowben method (18) using phosphatidylserine (bovine brain) and phosphatidylcholine (egg) at a 2:1 molar ratio. EPR spectra were obtained by using a Varian model E-109 spectrometer fitted with a loop gap resonator (19). All spectra were obtained at 2-mW incident microwave power and a field modulation of ≈1 G (1 G = 0.1 mT).
For the accessibility measurements, the oxygen concentration was that of oxygen in equilibrium with air and the NiEDDA concentration was 100 mM. The immersion depth, d, for lipid-exposed residues was calculated from the Φ parameter (10, 16). The calibration of Φ in terms of depth was obtained with the use of 1-palmitoyl-2-DOXYL-stearoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids) with the spin-label attached at the 5, 7, 10, and 12 positions on the acyl chains as described (10, 16). For the spin-labeled phosphatidylcholines in bilayers containing annexin under the conditions described above, d[Å] = 4.8Φ + 8.5.
RESULTS
Annexin XII Binding to Vesicles at Low pH.
By using a standard cosedimentation assay (20), we found that annexin XII reversibly bound to phosphatidylserine-containing vesicles in the absence of Ca2+ at low pH (data not shown). Binding was nearly complete at pH 4 and was half-maximal at approximately pH 5.8. Thus, annexin XII appears to assume three distinct states: a soluble state, a membrane-surface-bound state in the presence of Ca2+, and a membrane-associated state at low pH in the absence of Ca2+.
Differences Between the Ca2+- and H+-Induced Membrane-Bound Forms of Annexin XII.
A recent site-directed spin labeling study (3) showed that at neutral pH in the presence of Ca2+ annexin XII binds to the surface of bilayers and assumes a configuration generally similar to that in crystals (2). Trimer formation was readily detected by strong spin–spin interactions between double mutants 27R1/188R1 and 16R1/163/R1 containing R1 side chains placed at positions that were far apart within the monomer but in close proximity at the contact face within trimers. The single mutant 132R1 also efficiently detected trimer formation because three of these symmetry-related residues are located within about 7 Å of each other in the center of the trimer. The normalized EPR spectra for those constructs are given in Fig. 2. At neutral pH strong spin–spin interaction causes a dramatic broadening of the spectrum and concomitant decrease in signal amplitude after the monomer in solution (blue trace) forms a trimer after Ca2+-dependent binding to bilayers (red trace). In contrast, the change in spectral line shape and signal amplitude upon membrane binding at pH 4.0 in the absence of Ca2+ (green traces) was much smaller. This data clearly establishes that the trimeric state [or the related hexameric state (2)] does not form under these conditions. Furthermore, the line shapes of the EPR spectra of the mutants bound to membranes at pH 4.0 (Fig. 2) are different form those obtained for protein bound to membranes in the presence of Ca2+ at pH 7.4 when the spin–spin coupling is reduced or eliminated (compare green traces in Fig. 2 with the data in figure 3 of reference 3). Thus, significant changes in the tertiary fold appear to occur when annexin XII binds to membranes at low pH in the absence of Ca2+.
Figure 2.

EPR spectra for annexin XII mutants 132R1, 16R1/163R1, and 27R1/188R1 in solution at pH 7.4 (blue trace), membrane-bound at pH 7.4 with 1 mM Ca2+ (red trace), and membrane-bound at pH 4.0 (green trace). The reference spectra at pH 7.4 were reproduced from ref. 3 with permission from the Journal of Biological Chemistry. The amplitudes of certain spectra were increased by a scaling factor shown on the right. The scan width is 100 G.
Structural Features of the Sequence of Residues 138–163 in Solution and in the Low-pH Membrane-Bound Form.
To investigate pH-dependent structural changes in annexin XII, residues 138–163 in domain II (Fig. 1) were selected for a nitroxide scanning experiment, in which each of the native side chains was sequentially replaced with R1. These residues are located in the D helix (residues 138–141), the D–E connecting loop (residues 142–145), the E helix (residues 146–156), and a loop that connects domain II and III (residues 157–163). The loop connecting the D and E helix contains a putative Ca2+ coordination site (Glu-142).
The secondary structure and location of tertiary contact sites in the sequence of residues 138–163 in solution are revealed by the pattern of R1 side-chain mobility reflected in the EPR spectral line shapes shown in Fig. 3. Qualitatively, the spectra at neutral pH in the absence of vesicles and Ca2+ are consistent with the crystal structure of the annexin XII monomer (Fig. 3, black line). For example, the spectra of R1 residues in loop regions (residues 142–145 and residues 157–163) reflect high mobility, and are similar to those for R1 in loop domains of other proteins (9). The spectra of R1 residues on the buried surface of the E helix (residues 147, 150, 151, 154, and 155) are characteristically broad, indicating the expected immobilization of R1, whereas those for R1 on the exposed surface of the helix (residues 148, 149, 153, and 156) reflect a relatively higher mobility and are similar to those observed for R1 at comparable sites in T4 lysozyme (9). Fig. 4 Upper (solid line) summarizes these qualitative observations by using the inverse second moment of the spectra (〈H2〉) as a crude but convenient measure of side-chain mobility (7, 9). The periodic dependence of 〈H2〉 with sequence position is clearly seen for the regions corresponding to the short D helix (residues 138–143) and the E helix (residues 145–156) but not in the extended loop (residues 157–163). It is interesting to note that the average mobility of residues in the D helix is higher than that for the E helix, suggesting that the former has additional dynamic modes, presumably backbone fluctuations.
Figure 3.

EPR spectra of annexin XII containing the R1 nitroxide side chain at the indicated sites in solution at pH 7.4 (black line) and pH 4.0 (red line). The spectra within a pair are normalized to the same number of spins. For convenience of presentation, the amplitudes of some spectra were reduced by the scaling factor shown to the right of the spectra. The dashed vertical lines are drawn to aid in comparison of spectral features. The scan width is 100 G.
Figure 4.

Inverse second moment (〈H2〉) for the EPR spectra of R1 as a function of sequence position. (A) Annexin XII in solution at pH 7.4 (solid line) and at pH 4.0 (dashed line). (B) Annexin XII bound to vesicles at pH 4.0 in the absence of Ca2+. The gray boxes on the left and right correspond to residues in the D and E helices, respectively, in the crystal structure of annexin XII (2).
At pH 4 in the absence of vesicles, the EPR spectra of R1 along the sequence of residues 138–163 are qualitatively similar to those recorded at neutral pH (Fig. 3, red traces), and the helix–loop–helix–loop structure of the protein at neutral pH is apparently retained. This is clearly revealed in the periodicity of H2 with sequence position (Fig. 4A, dashed line). After binding to membranes at pH 4 in the absence of Ca2+, the spectra were strikingly different (Fig. 5, black trace), indicative of major changes in the tertiary fold in this region. At many sites the overall mobility pattern reverses upon membrane binding. For example, the majority of the most buried residues in the solution structures (residues 139, 147, 151, 154, and 155) become the most mobile residues on membrane binding. Conversely, the highly mobile residues of the short D–E loop in solution show a distinct decrease in apparent mobility upon membrane binding at low pH, a result that indicates the formation of an ordered structure. Fig. 4B summarizes the mobility data for the membrane-bound state at low pH. From the plot, it is apparent that the mobility in the membrane-bound state is periodic in position throughout the sequence, up to about residue 159, consistent with the existence of an extended helical structure in the membrane. Remarkably, the period is the same as that for the solution structure, but the two differ in phase by 180° (Fig. 4). Thus the structure of the sequence of residues 138–156 is largely helical in both the solution and membrane-bound states, but the membrane-bound structure is essentially inverted relative to the solution structure. That is, residues buried in the protein interior in the solution structure find themselves exposed on a helical surface in the membrane-bound state and vice versa.
Figure 5.

EPR spectra of annexin XII containing the R1 nitroxide side chain at the indicated sites in solution at pH 4.0 (red line) and bound to membranes at pH 4.0 (black line). The spectra for pH 4.0 in solution are reproduced from Fig. 3 for comparison of the changes that accompany membrane binding. The spectra within a pair are normalized to the same number of spins. For convenience of presentation, the amplitudes of some spectra at pH 4.0 in solution were reduced by the scaling factor shown to the right of the spectra. The dashed vertical lines are drawn to aid in comparison of spectral features. The scan width is 100 G.
To investigate further the underlying secondary structure of this form of annexin XII, the R1 accessibility to collision with paramagnetic reagents was examined. The paramagnetic reagents O2 and NiEDDA have high and low solubility, respectively, in the interior of bilayers and both are excluded from the interior of proteins (10, 16). Fig. 6A shows the accessibility parameters Π(O2) (circles) and Π(NiEDDA) (triangles) in the sequence of residues 138–163 in the membrane-bound state of annexin XII at pH 4.0. Both Π(O2) and Π(NiEDDA) show a striking oscillatory behavior with a period of 3 or 4 residues, indicating an α-helical structure, consistent with the conclusion reached above based on side-chain mobility. Moreover, the oscillations in Π(O2) and Π(NiEDDA) are 180° out of phase, characteristic of a helix asymmetrically solvated on one surface by water and on the opposing surface by the hydrophobic interior of the bilayer (16). Residues at local maxima of Π(O2) (residues 139, 143, 147, 151, 154, and 158) cluster on one face of the putative helix (Fig. 7A, yellow circles), and residues at local maxima of Π(NiEDDA) (residues 141, 145, 149, 152, and 156) cluster on the opposite face (Fig. 7A, orange circles). Residues of intermediate accessibility to either reagent lie on surfaces separating these extremes (Fig. 7A, white circles).
Figure 6.

Accessibility parameters and immersion depths for R1 side chains in membrane-bound annexin XII at pH 4.0 and in the absence of Ca2+. (A) The accessibility parameters for O2 (circles) and NiEDDA (triangles) as a function of sequence. (B) The immersion depth in the bilayer of lipid-exposed R1 residues with maximal Φ values (residues 139, 143, 147, 151, 154, and 158).
Figure 7.

(A) Helical net representation of annexin XII residues 138–158 at low pH. Residues corresponding to maxima in Π(O2) are in yellow and to maxima in Π(NiEDDA) are in orange. Flanking basic residues (K137 and R159/K160) are proposed to stabilize the transmembrane topography by interacting with the head groups of the acidic phospholipids. (B) Model for the pH-triggered membrane insertion of helices D–E in annexin XII monomer. At neutral pH in solution, or adsorbed to the membrane in the presence of Ca2+, helices D (green/yellow) and E (blue) form a helical hairpin with a short connecting loop (red). In the surface-bound state, Ca2+ is jointly coordinated by Glu-142 and phosphatidylserine (Left). When the pH is lowered, the carboxylate groups are protonated, making it possible for them to be inserted into the bilayer interior as a transmembrane helix (Right). The gray helices are speculative (see text) and are included to represent the remaining portion of the annexin monomer. The number of gray helices in the model is arbitrary.
The accessibility data (Fig. 6A) showing that the sequence of residues 138–158 forms an amphipathic α-helix is entirely consistent with the mobility data (Figs. 4 and 5) for the membrane-bound state at low pH. The hydrophobic residues that are the most accessible to oxygen (Fig. 6A) are also the most mobile (Figs. 4 and 5) as would be expected for a spin label facing the acyl chains of the bilayer. Thus, these data firmly establish that the hydrophobic face of the helix (Fig. 7A, yellow circles) faces the interior of the bilayer and not the interior of the protein.
Topography of the Sequence of Residues 138–163 in Membrane-Bound Annexin XII at Low pH.
Two structural models can account for the asymmetric solvation in the membrane-bound state at low pH. In one model, the helix is transmembrane, lining a water-filled pore (see Discussion for a model of the putative pore). In the other model, the helix lies adsorbed parallel to the bilayer surface. These two cases can be distinguished experimentally by determining the immersion depth of the lipid-exposed residues (10, 16). If the helix is parallel to the bilayer surface, residues facing the bilayer may vary in depth in an irregular fashion depending on the location of the side chain. On the other hand, if the helix is transmembrane, the immersion depth increases regularly with sequence from either membrane surface to the center of the bilayer. As shown (10, 16), Φ may be used to estimate the nitroxide immersion depth for lipid-exposed residues after proper calibration. Fig. 6B shows the immersion depth of residues with maximal Φ values for spin-labeled annexin XII mutants bound to bilayers at pH 4.0. As can be seen, residue 147 is clearly at the deepest position in the bilayer and is located approximately 18 Å from the surface. The immersion depth on either side of this residue decreases linearly with approximately 4 Å per turn, close to the expected value from an idealized transmembrane helix. Thus, the data strongly favor a transmembrane orientation of the helix formed from residues 138 to 158.
The Low-pH-Induced Structural Changes Are Reversible.
To determine whether or not the low pH-induced membrane association and structural changes are reversible, advantage was taken of the large EPR spectral changes of R1 at some sites that accompany membrane binding. For example, the spectrum of 144R1 broadens considerably and is reduced in amplitude upon membrane association at low pH (Fig. 5; note scale factor of 0.5). Figs. 8 A and B reproduce the spectra of 144R1 in solution at pH 7.4 and bound to membranes at pH 4.0, respectively, with the same vertical scale. Fig. 8C shows the spectrum obtained after shifting the pH of the sample of Fig. 8B back to pH 7.4. Evidently, the low pH-triggered insertion is reversible, because the spectra of Fig. 8 A and C are essentially identical. Similar experiments using other spin labeled sites gave equivalent results (data not shown).
Figure 8.

Reversibility of structural changes that occur when annexin XII undergoes membrane insertion at low pH. The EPR spectrum of the same sample of annexin XII mutant 144R1 was sequentially recorded in solution at pH 7.4 (A) and then in the membrane-bound form at pH 4.0 (B) as described in Fig. 3. The membrane-bound protein then was extracted with buffer at pH 7.4, the vesicles were removed by centrifugation and the EPR spectrum of the protein in solution was determined (C).
DISCUSSION
Previous studies defined two forms of annexins at neutral pH—a soluble monomer in the absence of Ca2+ and trimers on the surface of membranes in the presence of Ca2+ (3). For both the monomer and trimer at neutral pH, the EPR data at the sites investigated were consistent with the structure of these forms found in the crystal (ref. 3 and Figs. 3 and 4). The results presented herein on membrane-bound annexin XII in the absence of Ca2+ at low pH indicate a strikingly different structure. Under these conditions the protein did not form the trimer (Fig. 2), and the side-chain mobility of R1 in a scan from residues 138 to 163 revealed the apparent formation of a continuous helix with the concomitant loss of the disordered D–E loop of the solution structure (Figs. 3–5). Furthermore the 180° phase shift between the periodic functions of 〈H2〉 in the solution and membrane-bound states indicates that the structure rearranges dramatically by an “inside-out” refolding mechanism upon membrane binding to expose residues previously buried in the core of the protein structure to the surrounding solvent (Fig. 4). The nature of the solvent for these highly mobile exposed residues in the membrane-bound state is suggested by the accessibility data of Fig. 6A. The high oxygen accessibility and low NiEDDA accessibility at those sites are typical for residues facing the fluid hydrophobic interior of the phospholipid bilayer (21, 22). Finally, the depth profile for these lipid-exposed residues and the accessibility data argue in favor of a transmembrane helical structure (Fig. 6B).
Fig. 7B summarizes schematically the structural transition that apparently occurs upon membrane binding at pH 4. The D and E helices are conserved in the membrane-bound state (despite the completely altered tertiary packing interaction), but the D–E interhelical loop of the solution structure is apparently recruited to form a continuous helical segment that inserts into the bilayer and extends across its thickness. The amino acid sequence of the protein is in fact compatible with both the helix–turn–helix motif of the solution structure and the proposed membrane-inserted helix at low pH. For example, the hydrophobic residues immobilized in the core of the soluble protein (residues 139, 143, 147, 151, 154, and 155; Fig. 1) remain in a hydrophobic environment provided by the bilayer interior in the membrane-bound state. The predominantly hydrophilic residues facing water in the soluble protein remain solvated by water in the pore of a putative channel (see Discussion below). The transmembrane topography is additionally stabilized by interaction of the flanking basic residues (Lys-137 and Arg-159/Lys-160) with the head groups of the acidic phospholipid bilayer (Fig. 7A).
It is likely that the pH dependence of the membrane insertion is a consequence of protonation or deprotonation of charged residues. Although there are many such residues in the protein, it is interesting to note that the scanned region contains two Glu residues (Fig. 7B). These two carboxylate residues, Glu-138 and Glu-142, are located within the D helix and the D–E loop of domain II. In the membrane-bound state, these residues are located near the hydrophobic face of the putative transmembrane helix (Fig. 7B). Thus protonation of these residues in solution at low pH would reduce the unfavorable free energy of transfer to the hydrophobic interior of the bilayer. Moreover, Glu-138 is involved in a salt bridge in the protein core of the solution structure, and protonation of this site is expected to destabilize the solution structure relative to the membrane-bound state. Therefore, protonation of Glu-138 and Glu-142 may constitute the pH switch in this domain. Interestingly, Glu-142 may be a ligand for joint Ca2+ coordination with phosphatidylserine in the membrane-bound trimer state (Fig. 7B). If so, Ca2+ would be expected to antagonize the effect of H+ and favor the surface-bound trimer state relative to the membrane-inserted state. Preliminary experiments support this hypothesis, because the presence of Ca2+ raises the concentration of H+ required to produce membrane insertion (data not shown).
Inspection of the amino acid sequences around Ca2+ binding A–B and D–E loops other than the one scanned herein revealed additional regions with putative transmembrane helices containing ≈20 amino acids that were each flanked by either positively charged or good turn-forming residues. All helices have amphipathic character, and all helices have the important distinction that, as for the sequence of residues 138–158, Glu and Asp residues are located at or near the hydrophobic face. Also most of these carboxylate residues are either engaged in salt bridge formation in the solution structure or are likely Ca2+ ligands. The general similarity of these sequences to residues 138–158 suggests that they may also show a similar H+-triggered membrane insertion. These additional helices (the gray helices in Fig. 7B) may form the remainder of the pore structure in which residues 138–158 apparently participate. If this is the case, the lack of totally immobilized residues (Fig. 5) suggests that the helix packing is loose.
The demonstration that the transmembrane 138–158 helix is asymmetrically solvated with one side facing water (Fig. 6A) clearly indicates the presence of a water-filled channel. Further studies will be required to determine whether sequences other than residues 138–158 form transmembrane helices and participate in formation of the aqueous pore illustrated in Fig. 7B. Regardless of which helices are involved in pore formation, this model provides a structural framework for explaining previous reports that annexins form ion channels. In fact, preliminary studies clearly show that annexin XII mediates ion flux across bilayers in a pH-dependent manner (Y. Sokolov, J.M.I., H.T.H., and J. Hall, unpublished results).
Although it is clear that annexin XII assumes a transmembrane form in vitro, we can only speculate concerning whether protons might be a physiological trigger for membrane insertion of annexins in intact cells. The half-maximal association of annexin XII with bilayers occurs at pH ≈5.8, significantly more acidic than the cytosol. Annexins are primarily cytosolic proteins and have not been found inside low pH compartments such as synaptosomes and endosomes, although they are known to associated with the membranes of these organelles (1). At first glance it would appear that the cytosolic pH is not low enough to induce membrane insertion of annexins. However, annexins and a number of other proteins with affinities for Ca2+ well below those of the Ca2+ concentration of the bulk cytosol respond to Ca2+ signals. It is widely assumed that these responses are possible because localized areas of high Ca2+ concentration occur near sites of Ca2+ influx. By analogy, it is possible that reversible pH-induced membrane association of annexins is a transient response to local pH changes.
Annexins are major cellular proteins and significant physiological effects could occur if even a very small percent of the total cellular pool formed transmembrane ion channels. With this in mind, it is important to remember that formation of transmembrane annexin XII is a reversible process thereby indicating that there is an equilibrium between the inserted and other forms. Thus, even at neutral pH a small, but perhaps physiologically significant, fraction of annexin XII will exist in the form of a transmembrane channel. One could also imagine a mechanism in which annexins assume the transmembrane form by a pH-independent mechanism. In intact cells, the carboxylate “switch” residues may have their pKa values shifted upward or their negative charges shielded by other cellular components so that annexins can insert into membranes at neutral pH. In fact, approximately 20% of annexin XII is associated with hydra cellular membranes in the absence of Ca2+ (20) and several reports of Ca2+-independent forms of mammalian annexins have been published (1). Additional studies will be required to determine whether these forms of annexins are related to the transmembrane form of annexin XII defined herein.
In summary, the above results demonstrate that the structure of the low pH form of annexin XII bound to membranes has a transmembrane topography that is very different from the Ca2+-induced trimer adsorbed on the membrane surface. In fact, annexin XII can be reversibly converted from a soluble protein to either a peripheral or transmembrane protein, depending on the concentrations of H+ and Ca2+. The mechanism of H+-induced membrane insertion involves the formation of a continuous elongated helix from a helix–turn–helix motif in the solution structure. The structural changes in annexin XII are reminiscent of the pH-induced structural reorganization of certain proteins such as hemagglutinin in aqueous solution (23, 24) but differ in one important point. These other proteins appear to be in a metastable state at neutral pH so that the low pH-induced changes are irreversible (25). In contrast, the pH-induced changes in annexin XII are fully reversible. Although the biological implications of this membrane insertion mechanism are not yet fully appreciated, the discovery of a transmembrane helix in annexin that apparently lines an aqueous pore can explain existing biochemical and electrophysiological data on the formation of ion channels.
Acknowledgments
The excellent technical assistance of Elizabeth Baer is gratefully acknowledged. This work was supported by the National Institute of Health (W.L.H and H.T.H) and the Jules Stein Eye Institute Professorship Endowment (W.L.H.). R.L. is the recipient of a National Research Service Award from the National Eye Institute. Support from the Bundy Foundation (W.L.H) is gratefully acknowledged.
ABBREVIATION
- NiEDDA
nickle(ethylenediaminediacetic acid)
References
- 1.Seaton B A, editor. Annexins: Molecular Structure to Cellular Function. New York: Landes; 1996. [Google Scholar]
- 2.Luecke H, Chang B T, Mailliard W S, Schlaepfer D D, Haigler H T. Nature (London) 1995;378:512–515. doi: 10.1038/378512a0. [DOI] [PubMed] [Google Scholar]
- 3.Langen R, Isas J M, Luecke H, Haigler H T, Hubbell W L. J Biol Chem. 1998;273:22453–22458. doi: 10.1074/jbc.273.35.22453. [DOI] [PubMed] [Google Scholar]
- 4.Demange P, Voges D, Benz J, Liemann S, Göttig P, Berendes R, Burger A, Huber R. Trends Biochem Sci. 1994;19:272–276. doi: 10.1016/0968-0004(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 5.Pollard H B, Rojas E. Proc Natl Acad Sci USA. 1988;85:2974–2978. doi: 10.1073/pnas.85.9.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohler G, Hering U, Zschornig O, Arnold K. Biochemistry. 1997;36:8189–8194. doi: 10.1021/bi9703960. [DOI] [PubMed] [Google Scholar]
- 7.Hubbell W L, Mchaourab H S, Altenbach C, Lietzow M A. Structure. 1996;4:779–783. doi: 10.1016/s0969-2126(96)00085-8. [DOI] [PubMed] [Google Scholar]
- 8.Hubbell, W. L., Gross, A., Langen, R. & Lietzow, M. A. Curr. Opin. Struct. Biol., in press. [DOI] [PubMed]
- 9.Mchaourab H S, Lietzow M A, Hideg K, Hubbell W L. Biochemistry. 1996;273:810–822. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 10.Altenbach C, Greenhalgh D A, Khorana H G, Hubbell W L. Proc Natl Acad Sci USA. 1994;91:1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hustedt E J, Smirnov A I, Laub C F, Cobb C E, Beth A H. Biophys J. 1997;72:1861–1877. doi: 10.1016/S0006-3495(97)78832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mchaourab H S, Oh K J, Fang C J, Hubbell W L. Biochemistry. 1997;36:307–316. doi: 10.1021/bi962114m. [DOI] [PubMed] [Google Scholar]
- 13.Rabenstein M D, Shin Y K. Proc Natl Acad Sci USA. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farrens D L, Altenbach C, Yang K, Hubbell W L, Khornana H G. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 15.Steinhoff H J, Radzwill N, Thevis W, Lenz V, Brandenburg D, Antson A, Dodson D, Wollmer A. Biophys J. 1997;73:3287–3298. doi: 10.1016/S0006-3495(97)78353-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oh K J, Zhan H, Cui C, Hideg K, Collier R J, Hubbell W L. Science. 1996;273:810–812. doi: 10.1126/science.273.5276.810. [DOI] [PubMed] [Google Scholar]
- 17.Mailliard W S, Luecke H, Haigler H T. Biochemistry. 1997;36:9045–9050. doi: 10.1021/bi970749v. [DOI] [PubMed] [Google Scholar]
- 18.Reeves J, Dowben R M. J Cell Physiol. 1969;73:49–60. doi: 10.1002/jcp.1040730108. [DOI] [PubMed] [Google Scholar]
- 19.Hubbell W L, Froncisz W, Hyde J S. Rev Sci Instr. 1987;58:1879–1886. [Google Scholar]
- 20.Schlaepfer D D, Fisher D A, Brandt M E, Bode H R, Jones J M, Haigler H T. J Biol Chem. 1992;267:9529–9539. [PubMed] [Google Scholar]
- 21.Altenbach C, Marti T, Khorana H G, Hubbell W L. Science. 1990;248:1088–1092. doi: 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- 22.Altenbach C, Yang K, Farrens D L, Farahbakhsh Z T, Khorana H G, Hubbell W L. Biochemistry. 1996;35:12470–12478. doi: 10.1021/bi960849l. [DOI] [PubMed] [Google Scholar]
- 23.Carr C M, Kim P S. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 24.Bullough P A, Hughson F M, Skehel J J, Wiley D C. Nature (London) 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 25.Carr C M, Chaudhry C, Kim P S. Proc Natl Acad Sci USA. 1997;94:14306–14313. doi: 10.1073/pnas.94.26.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]