

Abstract
Nitric oxide (NO) biology has focused on the tightly regulated enzymatic mechanism that transforms l-arginine into a family of molecules, which serve both signaling and defense functions. However, very little is known of the pathways that metabolize these molecules or turn off the signals. The paradigm is well exemplified in bacteria where S-nitrosothiols (SNO)—compounds identified with antimicrobial activities of NO synthase—elicit responses that mediate bacterial resistance by unknown mechanisms. Here we show that Escherichia coli possess both constitutive and inducible elements for SNO metabolism. Constitutive enzyme(s) cleave SNO to NO whereas bacterial hemoglobin, a widely distributed flavohemoglobin of poorly understood function, is central to the inducible response. Remarkably, the protein has evolved a novel heme-detoxification mechanism for NO. Specifically, the heme serves a dioxygenase function that produces mainly nitrate. These studies thus provide new insights into SNO and NO metabolism and identify enzymes with reactions that were thought to occur only by chemical means. Our results also emphasize that the reactions of SNO and NO with hemoglobins are evolutionary conserved, but have been adapted for cell-specific function.
Three major nitric-oxide synthases (NOSs) and a growing list of alternatively spliced or otherwise modified NOS isoforms regulate the transformation of l-arginine into a family of molecules that are involved in numerous biological processes (1). This wide variety of effects is the reflection of a basic signaling mechanism that is utilized by virtually all mammalian cells and many lower organisms. Specifically, NO groups are introduced into thiol- and transition metal-containing proteins, thereby altering their properties and functions. Among the target proteins known to be physiologically modified by NOSs (or their products) are several classes of ion channels (2, 3), ras protein (4), transcription factors (5–7), multiple enzymes (7, 8), and hemoglobin (9). Potential NO binding/reaction sequences have been identified in some of these target proteins (7). Additional levels of regulation are provided by compartmentalization of the modifying enzymes, by control of NOS substrate or cofactor availability (which may influence product identity), and by the reactivities of small NO-donating molecules that can add NO groups (7, 10, 11).
On the other hand, much less is known of the mechanism(s) that switch off NO signals or detach NO groups from proteins. For example, (S)NO groups in proteins are probably in equilibrium with low-mass SNOs, but the position of the equilibrium in cells and its contribution to NO group removal is not clear. An added peculiarity of redox systems is the dependency on metabolic enzymes, such as superoxide dismutase (SOD) and catalase, that prevent fortuitous damage by oxidant-signaling molecules (12). These enzymes are widely distributed in all cells and are induced by oxidative stresses. By analogy, little is known of the metabolic pathways that limit the reactivity of NO-related molecules in biological systems. For example, oxidation of NO to nitrate by oxyhemoglobin is believed to be the major metabolic route for NO in mammals. In fact, the reaction has not been studied under physiological conditions and its contribution to NO/SNO metabolism has not been elucidated. Additionally, enzymes such as xanthine oxidase and glutathione peroxidase are reported to break down NO-related species (13, 14). However, their physiological relevance to “NO” metabolism is unclear. That is, no such enzyme is known to serve a metabolic function in vivo or is known to be induced specifically by a nitrosative stress.
Signaling pathways responsive to reactive oxygen and nitrogen species are also found in bacteria, where they have been adapted to serve in host defense (6, 15–17). In particular, bacteria (5, 6, 17) have adopted strategies similar to eukaryotic cells (7, 8) for molecular recognition of NO and SNOs. Moreover, inducible pathways recently have been discovered that control the metabolic fate of these molecules (6). That is, Escherichia coli exhibit rapid physiological responses to nitrosative stress that prevent NO/SNO from reaching dangerous levels or exerting untoward effects (5, 6). These observations raise the possibility of novel enzymatic pathways involved in detoxification and/or regulation of NO signal transduction. Here we report both on the presence of constitutive SNO lyase(s) that liberate NO from SNO and the identification of a flavohemoglobin “denitrosolase.” The enzyme, which is known to be induced by NO and SNO (18, 19), metabolizes NO to nitrate. We also offer evidence in favor of a physiological function for the flavohemoglobin in amelioration of a nitrosative stress, in agreement with recent work of others (19).
METHODS
Culture.
Growth of E. coli strain RK4936 in minimal medium, harvest of cells, and lysis were as described (6). Strains YMC10 (wild type) and RB9060 (Δgln, Δhmp) (20) were provided by A. Ninfa, University of Michigan. The flavohemoglobin (HMP) overproducing strain AN1459/pPL757 was provided by N. E. Dixon, Australian National University. The hmp gene is under the control of the bacteriophage λ promoter with a temperature-sensitive repressor; HMP constitutes more than 50% of cellular protein upon a temperature shift (21). To induce HMP, cells were grown from an overnight culture (1% inoculum) to an A600 of 1.0 (2 × 109 cfu/ml) and then diluted 50-fold into fresh medium. When the A600 had reached 0.2, cells were treated with 0.2 mM S-nitrosocysteine (SNO-Cys) for 90 min. To test for inducible resistance, the pretreated cells were diluted (to an A600 of 0.1) into fresh, prewarmed medium and rechallenged with 0.2 mM SNO-Cys. Cell density was then recorded every 15 min for 2 hr. For HMP purification, strain AN1459/pPL757 was grown from an overnight culture (2% inoculum) at 30°C in 4 liters Luria–Bertani medium/50 μg/ml ampicillin to an A600 of 0.5 and then supplemented with 1 mM δ-aminolevulinic acid/1 mM ATP/100 μM riboflavin (22). The temperature then was shifted to 42°C, and the cells were grown for an additional 6 hr in the dark. After harvest by centrifugation, the cell pellet was stored at −20°C.
SNO and NO Metabolism.
A Clark-type NO electrode (Iso-NO, World Precision Instruments, Sarasota, FL) immersed in a stirred glass vial was used to measure NO released or consumed by bacteria or proteins. The meter was used at a setting at which 1 nA corresponds to 1 μM. Cells were suspended in 2 ml minimal medium to an A600 of 1.0. SNO-Cys or an anaerobic solution of NO was then added at final concentrations of 100 μM and 5 μM, respectively. NO consumption by column fractions or purified HMP was measured in 20 mM 1,3-bis[tris(hydroxymethyl)methylamino]propane (Bistris propane), pH 7.0, in the presence of 0.1 mM NADH. For measurement of the aerobic end products of NO reactions, either the NO donor diethylamine-NO (DEANO, 10–100 μM), NO solutions, or SNO-Cys in the presence of 10 μM Cu2+ were added to sealed vials that were filled to capacity. The solution then was assayed for nitrite and nitrate by capillary electrophoresis using a 75 μm × 100 cm CElect amine capillary (Supelco) at −20 kV. The capillary was coated periodically with amine regenerator solution (eCAP, Beckman). NADH stoichiometry for NO consumption was determined in open cuvettes from the absorbance change at 340 nm. To screen for SNO-lyase and NO-metabolizing activity, column fractions were treated with 0.1 mM SNO-Cys or 10 μM NO and assayed for NO2− production and the ability to accelerate NO breakdown with both the Griess assay (6) and NO electrode. Oxygen consumption was measured with a Clark electrode (Yellow Springs Instruments) in a thermostated cell without head space.
Fractionation and Enzyme Purification.
Soluble extracts from wild-type cells in 20 mM Bistris propane, pH 7.0, obtained after centrifugation at 100,000 × g, were separated on a MonoQ HR 10/10 column (Pharmacia) with a linear gradient of from 0 to 1 M NaCl and assayed for SNO-lyase and NO consumption activities. Crude extracts from the HMP overproducing strain were treated with 100 μM hemin and 1 mM DTT (heme reconstitution) and then applied to a 2.5 × 70 cm column of Q Sepharose FF (Pharmacia) and separated with a linear gradient of from 0 to 0.5 M NaCl. Fractions exhibiting an intense brown color were >95% pure HMP as judged by SDS gel electrophoresis.
RESULTS
We observed previously that SNO decomposition in E. coli led to accumulation of nitrite and nitrate (6). Nitrite is a product of NO autooxidation in solution (23, 24). We therefore speculated that SNO is homolytically cleaved to NO and tested for the ability of cells to catalyze this reaction. Fig. 1A shows that cells suspended in growth medium significantly increased the rate of NO release from SNO. Heat inactivation of the reaction in cellular extracts provided suggestive evidence for an enzymatic lyase activity (data not shown). However, the rate of NO decomposition by suspended cells did not obey the third-order kinetics of autooxidation. Rather, the cells accelerated NO decay. This NO metabolizing activity was increased markedly in cells that had been pretreated with SNO (Fig. 1B), an indication that the metabolic activity was inducible. Complementary studies with extracts showed that NO transformation was NADH dependent. We have shown previously that OxyR exerts control over the metabolic fate of SNO and that a mutant strain is highly sensitive to SNO-induced cytostasis (6). However, both the constitutive SNO-lyase and inducible NO-metabolic activities were present in OxyR-deficient cells (not shown). Taken together, these results are consistent with OxyR-independent metabolic pathways that cleave SNO to NO and metabolize NO.
Figure 1.

SNO and NO metabolism in E. coli. (A) E. coli release NO from S-nitrosothiols. S-nitrosocysteine (0.1 mM) was added to growth medium (broken line) or a suspension of E. coli (A 600 = 1; 2 × 109 cfu/ml) in growth medium (solid line), and NO release was measured with an NO electrode. (B) E. coli consume NO in both a constitutive and an inducible manner. A saturated solution of NO was added to growth medium (dotted line), to a suspension of E. coli that had received no treatment (dashed line), or to E. coli pretreated with 0.2 mM SNO-Cys for 90 min (solid line). (C) E. coli contain SNO-lyase activities. A bacterial extract was separated by anion-exchange chromatography, and the column fractions were assayed for the accumulation of nitrite from 0.5 mM SNO-Cys. (D) E. coli contain an inducible NO-metabolizing heme protein. Absorption spectra of column fractions from untreated (dashed line) or SNO-Cys pretreated cells (solid line), which contain the inducible NADH-dependent NO-metabolizing activity.
We attempted to purify and characterize enzymes from extracts of SNO treated and untreated cells by screening chromatographic fractions for SNO-lyase and NADH-dependent NO-metabolic activities. Anion-exchange chromatography separated three major peaks with SNO-lyase activity (Fig. 1C), further purifications of which strongly suggested the presence of several activities (not shown), and one peak with the NO-metabolizing activity. The SNO-lyase activity was not increased by SNO treatment. On the other hand, the NO consumption activity was low in extracts from untreated cells, but strongly induced by SNO treatment (Figs. 1B and 2A). The chromatographic fraction from SNO exposed cells containing this activity exhibited a distinctive hemoglobin spectrum (Fig. 1D). E. coli possess a flavohemoglobin (HMP) of unknown function that reportedly is induced by NO (18). SDS gel electrophoresis (not shown), assays for ferric reductase activity (25) (not shown), and studies of an HMP-deficient mutant identified the NO metabolizing activities with HMP. In particular, extracts from the HMP mutant were unable to catalyze NADH-dependent NO consumption (Fig. 2A), and HMP deficiency markedly increased susceptibility to SNO-induced cytostasis and severely compromised the inducible resistance to nitrosative stress (Fig. 2B).
Figure 2.

HMP is required for NO consumption and resistance to nitrosative stress. (A) NO metabolism. NADH-dependent consumption of 10 μM NO by extracts (1 mg/ml protein) from untreated Δhmp cells (dotted line, top), SNO-Cys-pretreated Δhmp cells (long-dashed line), untreated wild-type cells (short-dashed line), and SNO-Cys-pretreated wild-type cells (solid line). (B) Cytostatic effect of SNO-Cys treatment on isogenic wild-type (wt) and Δhmp mutant strains (hmp). +/+ and −/−, Cells that had or had not been pretreated with 0.2 mM SNO-Cys for 90 min, respectively, and were then rechallenged with the same dose. −/−, Control (wt or Δhmp mutant) cells that were neither pretreated nor rechallenged with SNO-Cys. In both strains, A600 is proportional to cell mass (protein content) at baseline and after treatment. Pretreatments were initiated at A600 = 0.2 for 90 min.
HMP also exerted control on SNO and NO metabolism. Cells pretreated with SNO under aerobic conditions produced increased nitrate and less nitrite from SNO and NO, and this metabolic shift away from nitrite was HMP-dependent (Table 1). Interestingly, the amount of nitrite plus nitrate recovered could not account for the SNO (and to a lesser degree, NO) added, suggesting the existence of an additional (reductive) route of (S)NO decomposition that is, at least partly, HMP-independent. We have described previously an OxyR-dependent pathway, limiting nitrite and nitrate accumulation (6), that is consistent with this finding. While this work was in progress, Crawford and Goldberg reported that HMP somehow affords protection against a variety of nitrosants (19). We conclude that multiple constitutive activities mediate SNO breakdown, among which are lyase(s) that generate NO. However, the HMP-catalyzed breakdown of SNO and NO is essential for acquisition of resistance to nitrosative challenge.
Table 1.
HMP induction increases yield of nitrate in vivo
| Cells | Nitrite, μM | Nitrate, μM |
|---|---|---|
| No pretreatment | ||
| Wild type (SNO-Cys) | 77 | 6.6 |
| Δhmp (SNO-Cys) | 98 | 1.9 |
| SNO pretreatment | ||
| Wild type (SNO-Cys) | 65 | 35 |
| (DEANO) | 143 | 22 |
| Δhmp (SNO-Cys) | 90 | 3.7 |
| (DEANO) | 161 | 3.1 |
Cells either were pretreated or not pretreated with 200 μM SNO-Cys. They were then exposed to either 200 μM SNO-Cys or diethylamine-NO (DEANO), as shown in parentheses above. After 90 min, the growth medium was analyzed for nitrite and nitrate by capillary electrophoresis.
HMP purified from an overexpressing strain (judged to be >95% pure by SDS gel electrophoresis) was used to elucidate the mechanism of NO breakdown. The protein exhibited the same NO-metabolic activity (Fig. 3A) that we had isolated from wild-type cells as well as the NADH-dependent reduction of ferric heme reported by Poole and colleagues (26) (Fig. 3B). The picture that emerged from our studies and analyses of substrate utilization indicated that O2 binds to the heme during aerobic NO turnover (Fig. 3 B and C). Moreover, a nitrosylheme that was formed anaerobically did not turn over rapidly, and the NO ligand was replaced readily by O2 (Fig. 3B). Thus, aerobic NO transformation by the heme domain was seen only with O2 bound. Cyanide significantly inhibited NADH oxidation and attenuated NO consumption, indicating that the site of NO reaction is the oxyheme (Fig. 3D). Product determinations—under conditions where HMP maintained the steady-state NO concentration below 50 nM—revealed that NO was oxidized mostly to nitrate (NO3−); small amounts of nitrite (NO2−) also were formed (Fig. 3E). Product yields were uninfluenced largely by SOD (Fig. 3E), and we did not observe acceleration of dihydrorhodamine oxidation by NO, suggesting that peroxynitrite was not freely formed. The stoichiometry was 2 mol of nitrite/nitrate produced per mol NADH oxidized (not shown). Measurements of oxygen consumption revealed that rates doubled in the presence of NO, and that one molecule of oxygen was consumed per molecule of NO (Fig. 3F). HMP preparations also generated some nitrous oxide anaerobically from NO/SNO (not shown).
Figure 3.

NO oxygenase activities of purified HMP. (A) NO consumption. NO electrode signal after addition of ≈10 μM NO to buffer (solid line) or ≈10 μM NO (dotted line) or ≈35 μM NO (dashed line) to 40 μg/ml HMP + 0.1 mM NADH. (B) Absorption spectra of purified HMP. HMP (450 μg/ml) exhibited an oxidized spectrum under anaerobic conditions (line 1, dashed–dotted); addition of NADH produced a ferrous iron-like spectrum (line 2, dashed); addition of NO saturated solution generated an iron-nitrosyl spectrum (line 3, solid); and air exposure of this iron-nitrosyl (with brief vortexing) resulted in an oxygen-bound (ferrous) iron spectrum (line 4, dotted). (C) Absorption spectra of HMP during aerobic turnover of NO in the presence of NADH. NADH (300 μM) was added to 400 μg/ml HMP in air (dotted line); addition of 100 μM NO from a saturated solution resulted in the consumption of NADH but no loss of the oxygen-bound ferrous iron spectrum (solid line). (D) Aerobic NADH consumption is increased during NO turnover by HMP and is inhibited by cyanide. NO (100 μM) was generated from SNO-Cys/Cu2+ (added to 20 μg/ml HMP at the break) in the absence (solid line) or presence (dotted line) of 1 mM KCN. (E) Product formation by HMP is not significantly influenced by SOD. Nitrite (solid bars) and nitrate (open bars) yields after addition of DEANO to 20 μg/ml HMP in the presence of 0.1 mM NADH and the indicated amounts of SOD. (F) NO oxidation by HMP (solid line) increases oxygen-consumption rate over NO autooxidation (broken line); the stoichiometry is 1 NO per O2 consumed. Where indicated by the asterisks (∗), 100 μM NO was added.
DISCUSSION
The enzymatic mechanism of HMP and functional adaptation of the hemoglobin for NO detoxification are both unprecedented. The heme in the bacterial flavohemoglobin appears to function as a dioxygenase. That is, the heme incorporates an O2 molecule into substrate (NO) in giving rise to product (nitrate) (Eq. 1). We propose a reaction mechanism in which NO binds to Fe(II)O2, forming a nitrosyldioxyl complex (Fe[II]OONO). Release of nitrate then leaves Fe[III]. Alternative production of nitrite might be explained by the reaction of NO with the OONO intermediate (Eq. 2) (24). In either case, electrons from NADH reduce the oxidized iron, regenerating the ferrous heme. A new round of catalysis then is initiated by O2 binding to heme (Eq. 3), which has been shown to occur very rapidly and with high affinity (27).
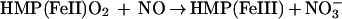 |
1 |
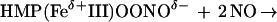 |
2 |
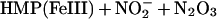 |
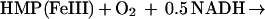 |
3 |
 |
Poole and colleagues originally discovered the propensity of HMP to reduce oxygen to superoxide (18, 27, 28). However, such NADH oxidase activity (28) may not be of physiological relevance because HMP is not highly expressed or clearly functional in the absence of NO/SNO (18, 19). Rather, HMP seems to make use of the superoxide-like reactivity of the O2 ligand to destroy NO. Intriguingly, the reactions of SNO and NO with mammalian hemoglobin are quite different: they produce thionitrosyl (29) and peroxynitrite-related derivatives (30), which have antimicrobial activities (17). Thus, by marrying a reductase module to the globin domain (31), HMP apparently has evolved the means to escape these harmful reactions.
Our studies and the recent work of others (19) reveal an emerging picture of metabolic pathways and tightly regulated detoxification mechanisms for both SNO and NO. A constitutive aerobic-metabolism pathway, which gives rise to nitrite and nitrate, capitalizes on enzymes (lyases) that convert SNO to NO. These lyases are perhaps ubiquitous components of pathways (32) involved either in stress responses, e.g., where they provide the NO substrate for HMP, or in signaling (Fig. 4). The cell also contains NO- and SNO-responsive genes that are induced when reactive nitrogen species exceed a dangerous threshold (5, 6). HMP dioxygenase seems to play a central role in the inducible aerobic-metabolism pathways adapted for detoxification, at least in E. coli (Fig. 2) and Salmonella (19). HMP is not, however, the only determinant of nitrite and nitrate accumulation from (S)NO. Indeed, we reported previously that OxyR influences the metabolic fate of nitrogen oxides under these conditions (6). Crawford and Goldberg have shown further that HMP confers resistance under anaerobic conditions (19). We can confirm that N2O is generated in the anaerobic-metabolism pathway of SNO (unpublished observations), although the mechanism remains to be fully elucidated (Fig. 4).
Figure 4.

Metabolic pathways for SNO and NO. SNO is cleaved to NO by constitutive SNO-lyases. In aerobic metabolism NO is oxidized to nitrate and nitrite (NOx) by the HMP-dioxygenase, the levels of which are up-regulated by nitrosative stress. The accumulation of nitrite is prevented by OxyR-controlled genes (6), although the mechanism is unknown. Under anaerobic conditions, NO/SNO are at least partly metabolized to N2O (not shown).
Aerobic respiration in all cells is characterized by fortuitous generation of reactive oxygen species (12). Their destructive potential is widely believed to play an etiological role in cancer, aging, and degenerative diseases (33). Cells counter this oxidative threat by expressing antioxidant enzymes such as SOD and catalase, which dismutate O2− and H2O2, respectively. Microorganisms must also disarm reactive oxygen species produced by cells of an invaded host, under which condition they up-regulate antioxidant defenses (5, 34). In contrast, anaerobic respiration in bacteria is characterized by fortuitous generation of reactive nitrogen species. Specifically, nitrate respiration or denitrification results in the production of NO as well as nitrosation catalyzed by nitrate reductase (35, 36). This nitrosative threat has been studied extensively because of its carcinogenic potential (36). The focus, however, has been on nitrosation of exogenous amines, when the principle intracellular target is thiols (6, 23, 37). In other words, anaerobic respiration may be viewed as a nitrosative stress characterized by formation of NO and SNO. In addition, bacteria may be exposed to nitrosative stress from environmental sources, such as soil and immune effector cells. Thus, even under aerobic conditions, during which bacteria are not sources of reactive nitrogen, induction of antinitrosative defenses would be called for. Apparently, the cells counter this nitrosative threat by expression of the flavohemoglobin (18, 19), which, we propose, has been adapted to serve a function analogous to SOD/catalase.
Acknowledgments
We thank Irwin Fridovich for discussions. This work was supported by Grants HL52529 and HL59130 from the National Institutes of Health.
ABBREVIATIONS
- SNO
S-nitrosothiol
- SNO-Cys
S-nitrosocysteine
- DEANO
diethylamine-NO
- HMP
flavohemoprotein
- NOS
nitric-oxide synthase
- SOD
superoxide dismutase
References
- 1.Nathan C. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 2.Xu L, Eu J P, Meissner G, Stamler J S. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 3.Lipton S A, Choi Y B, Pan Z H, Lei S Z, Chen H S, Sucher N J, Loscalzo J, Singel D J, Stamler J S. Nature (London) 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 4.Lander H M, Ogiste J S, Teng K K, Novogrodsky A. J Biol Chem. 1995;270:21195–21198. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- 5.Nunoshiba T, DeRojas-Walker T, Wishnok J S, Tannenbaum S R, Demple B. Proc Natl Acad Sci USA. 1993;90:9993–9997. doi: 10.1073/pnas.90.21.9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hausladen A, Privalle C T, Keng T, DeAngelo J, Stamler J S. Cell. 1996;86:719–729. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 7.Stamler J S, Toone E J, Lipton S A, Sucher N J. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 8.Stamler J S. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 9.Gow A J, Stamler J S. Nature (London) 1998;391:169–173. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- 10.Bredt D S. Proc Soc Exp Biol Med. 1996;211:41–48. doi: 10.3181/00379727-211-43950f. [DOI] [PubMed] [Google Scholar]
- 11.Becker K, Savvides S N, Keese M, Schirmer R H, Karplus P A. Nat Struct Biol. 1998;5:267–271. doi: 10.1038/nsb0498-267. [DOI] [PubMed] [Google Scholar]
- 12.Fridovich I. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 13.Sies H, Sharov V S, Klotz L O, Briviba K. J Biol Chem. 1997;272:27812–27817. doi: 10.1074/jbc.272.44.27812. [DOI] [PubMed] [Google Scholar]
- 14.Trujillo M, Alvarez M N, Peluffo G, Freeman B A, Radi R. J Biol Chem. 1998;273:7828–7834. doi: 10.1074/jbc.273.14.7828. [DOI] [PubMed] [Google Scholar]
- 15.Nunoshiba T, DeRojas-Walker T, Tannenbaum S R, Demple B. Infect Immunol. 1995;63:794–798. doi: 10.1128/iai.63.3.794-798.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Groote M A, Testerman T, Xu Y, Stauffer G, Fang F C. Science. 1996;272:414–417. doi: 10.1126/science.272.5260.414. [DOI] [PubMed] [Google Scholar]
- 17.Fang F C. J Clin Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poole R K, Anjum M F, Membrillo-Hernandez J, Kim S O, Hughes M N, Stewart V. J Bacteriol. 1996;178:5487–5492. doi: 10.1128/jb.178.18.5487-5492.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford M J, Goldberg D E. J Biol Chem. 1998;273:12543–12547. doi: 10.1074/jbc.273.20.12543. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Magasanik B. J Bacteriol. 1993;175:7441–7449. doi: 10.1128/jb.175.22.7441-7449.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love C A, Lilley P E, Dixon N E. Gene. 1996;176:49–53. doi: 10.1016/0378-1119(96)00208-9. [DOI] [PubMed] [Google Scholar]
- 22.Seo H G, Fujii J, Soejima H, Niikawa N, Taniguchi N. Biochem Biophys Res Commun. 1995;208:10–18. doi: 10.1006/bbrc.1995.1298. [DOI] [PubMed] [Google Scholar]
- 23.Wink D A, Nims R W, Darbyshire J F, Christodoulou D, Hanbauer I, Cox G W, Laval F, Laval J, Cook J A, Krishna M C, et al. Chem Res Toxicol. 1994;7:519–525. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 24.Wink D A, Cook J A, Kim S Y, Vodovotz Y, Pacelli R, Krishna M C, Russo A, Mitchell J B, Jourd’heuil D, Miles A M, Grisham M B. J Biol Chem. 1997;272:11147–11151. doi: 10.1074/jbc.272.17.11147. [DOI] [PubMed] [Google Scholar]
- 25.Eschenbrenner M, Coves J, Fontecave M. Biochem Biophys Res Commun. 1994;198:127–131. doi: 10.1006/bbrc.1994.1018. [DOI] [PubMed] [Google Scholar]
- 26.Ioannidis N, Cooper C E, Poole R K. Biochem J. 1992;288:649–655. doi: 10.1042/bj2880649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poole R K, Ioannidis N, Orii Y. Microbiology. 1996;142:1141–1148. doi: 10.1099/13500872-142-5-1141. [DOI] [PubMed] [Google Scholar]
- 28.Membrillo-Hernandez J, Ioannidis N, Poole R K. FEBS Lett. 1996;382:141–144. doi: 10.1016/0014-5793(96)00154-8. [DOI] [PubMed] [Google Scholar]
- 29.Jia L, Bonaventura C, Bonaventura J, Stamler J S. Nature (London) 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 30.Wade R S, Castro C E. Chem Res Toxicol. 1996;9:1382–1390. doi: 10.1021/tx9600457. [DOI] [PubMed] [Google Scholar]
- 31.Ermler U, Siddiqui R A, Cramm R, Friedrich B. EMBO J. 1995;14:6067–6077. doi: 10.1002/j.1460-2075.1995.tb00297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordge M P, Addis P, Noronha-Dutra A A, Hothersall J S. Biochem Pharmacol. 1998;55:657–665. doi: 10.1016/s0006-2952(97)00498-x. [DOI] [PubMed] [Google Scholar]
- 33.Sies H. Antioxidants in Disease Mechanisms and Therapy. San Diego: Academic; 1997. [Google Scholar]
- 34.Demple B, Amábile-Cuevas C F. Cell. 1991;67:837–839. doi: 10.1016/0092-8674(91)90355-3. [DOI] [PubMed] [Google Scholar]
- 35.Zumft W G. Arch Microbiol. 1993;160:253–264. doi: 10.1007/BF00292074. [DOI] [PubMed] [Google Scholar]
- 36.Ralt D, Wishnok J S, Fitts R, Tannenbaum S R. J Bacteriol. 1988;170:359–364. doi: 10.1128/jb.170.1.359-364.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis R S, Tamir S, Tannenbaum S R, Deen W M. J Biol Chem. 1995;270:29350–29355. doi: 10.1074/jbc.270.49.29350. [DOI] [PubMed] [Google Scholar]