

Abstract
Glucosamine (GlcN) has been reported to possess several biomedical properties, and currently a great deal of attention has been focused on improving the functional properties of GlcN for different applications. Therefore, this study was conducted to introduce a carboxybutyryl functional group to GlcN and to find out the inhibitory mechanism of a novel GlcN derivative, carboxybutyrylated GlcN (CGlcN), on the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) in bacterial lipopolysaccharide (LPS)-induced mouse macrophages (RAW264.7 cells). In the initial experiments, the production of NO and prostaglandin E2 (PGE2) was inhibited by CGlcN pretreatment and suggested the possibility of down-regulating their respective genes, iNOS and COX-2. Reverse transcription-polymerase chain reaction and Western blot analysis revealed that CGlcN can affect both transcriptional and translational levels of iNOS and COX-2 expression. The data from the nuclear factor-κB (NF-κB) promoter gene transfection experiment supported the idea that inhibition of iNOS and COX-2 is caused by the down-regulation of their transcription factor, NF-κB. Following stimulation with LPS, p38 mitogen-activated protein kinase (p38 MAPK) and c-Jun N-terminal kinase (JNK) present upstream of NF-κB signaling were also inhibited by CGlcN treatment. However, the protein level of another MAPK, extracellular signal-regulated kinase (ERK), remained unaffected. Moreover, following treatment with CGlcN, the protein expression of I-κB kinase (IKK) clearly confirmed that its down-regulation directly inhibited the degradation of IκB and release of NF-κB. Therefore, it can be concluded that CGlcN is capable of inhibiting iNOS and COX-2 expression in LPS-induced RAW264.7 cells via attenuation of NF-κB signaling by p38 MAPK and JNK, but not by ERK.
Keywords: bacterial lipopolysaccharide (LPS), cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), mitogen-activated protein kinase (MAPK), nuclear factor κB (NF-κB)
Introduction
Glucosamine (GlcN) is an N-deacetyl amino sugar derived from the complete hydrolysis of chitosan. Recent advances have provided insights into several health benefits of GlcN, including protective effects against infections and controlling arthritis.1,2 Specially, multiple clinical trials on GlcN have been carried out as a medical therapy for osteoarthritis because it is a precursor for glycosaminoglycans, and glycosaminoglycans are a major component of joint cartilage. Therefore, currently a great deal of attention has been focused on improving the functional properties and biological effects of GlcN for different therapeutic applications.3
Nitric oxide (NO) is an important messenger molecule functioning in vascular regulation, host immune defence, neurotransmission and other systems.4 In particular, diseases such as vascular dysfunction are associated with the impaired production of NO, whereas septic shock, cerebral infarction, diabetes mellitus, neurodegenerative disorders, rheumatoid arthritis and other inflammatory events are associated with NO overproduction.5 NO results from the conversion of l-arginine to l-citrulline by nitric oxide synthase (NOS) in the presence of molecular oxygen and NADPH. At least three main types of NOS isoforms are reported to be present in different tissues.6,7 NOS present in the vascular endothelium (eNOS) and that in central and peripheral neurons (nNOS) are constitutive (cNOS). Continuous release of NO by cNOS plays a role in keeping the vasculature in an active state and in the proper functioning of neuronal signal transduction. NOS in macrophages and hepatocytes, on the other hand, is inducible (iNOS) and becomes active following infections.8,9 During the course of an inflammatory response, large amounts of NO formed by the action of iNOS surpass the physiological amounts of NO, which are usually produced by the action of nNOS or eNOS.10 Therefore, iNOS-derived NO overproduction appears to be a ubiquitous mediator of a wide range of inflammatory conditions and reflects the degree of inflammation which provides a measure of the inflammatory process.11,12 Thus, there is considerable evidence that inhibition of excessive iNOS-derived NO production will be anti-inflammatory.
Similarly to cNOS and iNOS, constitutive cyclooxygenase (COX-1) and inducible cyclooxygenase (COX-2) are also reported to play an important role in immune modulation and pathophysiological events. COX catalyses the conversion of arachidonic acid to prostaglandin H2 (PGH2), the precursor of a variety of biologically active mediators such as PGE2, prostacyclin and thromboxane A2.13,14 Under normal conditions, COX-2 is not or only slightly detectable in most tissues. High amounts of PGE2 derived from COX-2 following induction by many pro-inflammatory mediators, including bacterial lipopolysaccharide (LPS), tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) have been implicated in the pathogenesis of sepsis and inflammation.15
Furthermore, compounds interfering with both iNOS and COX-2 generally act as potential inhibitors of nuclear factor-κB (NF-κB) activation, a transcription factor required for the induction of iNOS and COX-2 expression.16 Specially, the immune cells, macrophages/monocytes, play a crucial role in eliciting the inflammatory response cascades of NF-κB in the acute phase of inflammation.17 After being stimulated, they produce a number of chemokines, enzymes such as cytokines, iNOS and COX-2 for the primary protection of the host.18 However, uncontrolled accumulation of these defence molecules leads to a more severe and acute level of inflammation. NO and prostaglandins, the end-products of major inflammatory enzymes, iNOS and COX-2 have been extensively studied for their activation through NF-κB.19 Both iNOS and COX-2 genes have putative binding sites for NF-κB at their promoter sites to activate gene expression. Therefore, activation and expression of NF-κB during inflammation has a remarkable link with the production of NO and a number of other inflammatory prostaglandins, including PGE2.
The objective of the present work was to modify the functional properties of GlcN by introducing a carboxybutyryl group and to find out the inhibitory effects of a chemically modified GlcN derivative (CGlcN) on the inhibition of COX-2 and iNOS in LPS-stimulated RAW264.7 cells, together with its possible signaling pathway.
Materials and methods
Materials
Chitosan used for the preparation of GlcN was kindly donated by Kitto Life Co. (Seoul, Korea). All chemicals required for synthesis, including succinic anhydride, were obtained from Sigma Chemical Co. (St Louis, MO). Dialysis membranes, with a 100-molecular weight pore size, were purchased from Spectrum Laboratories Inc. (Rancho Dominguez, CA). Dowex-50WX2 cation-exchange resin, used for the purification of CGlcN, was purchased from Dow Chemical Company (Midland, MI). Mouse macrophages, RAW264.7 cells (ATTC no.: TIB-71) were obtained from the American Type Culture Collection (Manassas, VA). Cell culture media [Dulbecco's modified Eagle's minimal essential medium (DMEM)], Trypsin-EDTA, penicillin/streptomycin, fetal bovine serum (FBS) and the other materials required for culturing cells were purchased from Gibco BRL, Life Technologies (Grand Island, NY). The MTT reagent [(3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide)], the Griess reagent and bacterial LPS were purchased from Sigma Chemical Co. Primary and secondary antibodies used for Western blot analysis were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA) and Amersham Pharmacia Biosciences (Piscataway, NJ), respectively. The NF-κB gene promoter reporter vector was purchased from Clontech (Palo Alto, CA).
Instrumental analysis
Infrared spectra were recorded using KBr plates in the Spectrum 2000® Fourier transform infrared (FT-IR) spectrophotometer (Perkin Elmer, Norwalk, CT). 1H NMR and 13C NMR spectra were recorded in a D2O environment using a JNM-ECP-400® (400 MHz) spectrometer (Jeol, Tokyo, Japan) and the optical density was measured using a GENios® microplate reader (Tecan Austria GmbH, Grödig, Austria). Western blot bands were visualized using a LAS3000® Luminescent image analyzer (Fujifilm Life Science, Tokyo, Japan).
Synthesis of carboxybutyrylated glucosamine (CGlcN)
For the preparation of GlcN, 10·0 g of carboxybutyrylated chitooligosaccharides was hydrolyzed by 6 N HCl (50 ml) for 3 hr at 100°. After 3 hr, 50 ml of distilled water and 1·0 g of activated carbon powder was added and stirred for an additional 30 min at 60°. The mixture was filtered, and the filtrate was evaporated. The resultant GlcN (7·8 g) was then rinsed with 100% ethanol. The pH of GlcN (3·0 g) dissolved in methanol (30 ml) was adjusted to between 9·00 and 10·00 by NaHCO3, and succinic anhydride (1·5 g) dissolved in tetrahydrofuran (10 ml) was added drop by drop to the GlcN solution and stirred for 3 hr. After complete evaporation, the resultant CGlcN was dissolved in ethanol and filtered. The filtrate was evaporated and dissolved in tetrahydrofuran to remove the remaining succinic anhydride. The resultant CGlcN (as a solid) was rinsed with ethanol. After evaporation, CGlcN (1·79 g) was dissolved in water and dialyzed using a 100-molecular weight cut-off dialysis membrane. A small amount of non-reacted GlcN in CGlcN was removed following cation-exchange chromatography using a Dowex-50WX2 cation-exchange resin and purified CGlcN (1·73 g) was obtained.
Cell culture
Mouse macrophages (RAW264.7 cells) were cultured as monolayers in DMEM containing 10% FBS, 2 mm glutamine and 100 µg/ml penicillin/streptomycin at 5% CO2 and in a 37° humidified atmosphere. Adherent cells were detached by scraping and then plated onto 10-cm culture dishes, 24- or 96-well plates and used for experiments at ≈ 70–80% confluency.
Assessment of cell viability
Cells were seeded into 96-well plates at a density of 1 × 104 cells/well and incubated with serum-free media in the presence of different concentrations of GlcN or CGlcN. After incubation for 24 hr, 100 µl of MTT (0·5 mg/ml final concentration) was added and incubation was continued for another 4 hr. Mitochondrial succinate dehydrogenase in live cells converts MTT into visible formazan crystals during incubation.20 The formazan crystals were then solubilized in dimethylsulphoxide and the absorbance was measured at 540 nm by using an enzyme-linked immunosorbent assay (ELISA) microplate reader (Tecan Austria GmbH). Relative cell viability was calculated compared with the absorbance of the untreated control group.
Assessment of cellular NO production
RAW264.7 cells were cultured in 96-well plates using DMEM without phenol red and pretreated with different concentrations of CGlcN for 1 hr. Cellular NO production was induced by adding a 1-µg/ml final concentration of LPS and incubation for 48 hr. After incubation, 50 µl of conditioned media containing 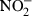 (primary stable oxidation product of NO) was mixed with the same volume of Griess reagent and incubated for 15 min.21 The absorbance of the mixture at 550 nm was measured with an ELISA microplate reader (Tecan Austria GmbH). The values obtained were compared with those of standard concentrations of sodium nitrite dissolved in DMEM, and the concentrations of nitrite in the conditioned media of sample-treated cells were calculated.
(primary stable oxidation product of NO) was mixed with the same volume of Griess reagent and incubated for 15 min.21 The absorbance of the mixture at 550 nm was measured with an ELISA microplate reader (Tecan Austria GmbH). The values obtained were compared with those of standard concentrations of sodium nitrite dissolved in DMEM, and the concentrations of nitrite in the conditioned media of sample-treated cells were calculated.
Assessment of COX-2 production
The production of PGE2, one of the mediators released after activation of COX-2, was used as a marker for the assessment of COX-2.22 For that, RAW264.7 cells were cultured in 24-well plates with serum-free medium and pretreated with different concentrations of CGlcN for 1 hr. The production of PGE2, via COX-2 activation, was stimulated by adding a 1-µg/ml final concentration of LPS and incubation for 24 hr. The conditioned medium was used for PGE2 determination by the Biotrak™ Prostaglandin E2 ELISA assay kit (Amersham Pharmacia Biosciences) according to the manufacturer's instructions. Sample treated conditioned media (50 µl), mouse anti-PGE2 (50 µl) and PGE2 conjugate (50 µl) were added to the anti-mouse-coated wells of 96-well plates and incubated for 1 hr at room temperature. After incubation, the reaction mixture was aspirated and thoroughly washed using a microplate washer (Tecan Austria GmbH). Immediately, 150 µl of 3,3′,5,5′-tetramethylbenzidine (TMB) enzyme substrate was added and incubated for 30 min at room temperature. The reaction was terminated by adding 100 µl of 1 m sulphuric acid and the absorbance was read at 450 nm.
TNF-α immunoassay
The production of TNF-α in RAW264.7 cells was assayed using Biotrak™ ELISA kits (Amersham Pharmacia Biosciences) following the manufacturer's instructions. Cells were treated with different concentrations of CGlcN for 1 h, the production of TNF-α was stimulated by a 1-µg/ml final concentration of LPS and incubation was continued for another 4 hr. Following incubation, conditioned medium was used for the experiment. For that, 50 µl of TNF-α standards (prepared for calibration), or the same volume of CGlcN-treated conditioned medium, was added to the wells of TNF-α antibody-coated 96-well plates in triplicate. Biotinylated antibody reagent (50 µl) was added and incubated for 3 hr at room temperature. The reaction mixture was aspirated and washed using a microplate washer (Tecan Austria GmbH). Streptavidin-horseradish peroxidase conjugate (100 µl) was added and incubated for 30 min at room temperature. After thorough washing, 100 µl of TMB substrate solution was added, incubated for 30 min at room temperature and the reaction stopped by the addition of 100 µl of stop solution. The absorbance was determined at 450 nm using the above-mentioned microplate reader. A TNF-α standard curve was used to quantify the amount of TNF-α released by the cells.
Reverse transcription-polymerase chain reaction
Total RNA was extracted from RAW264.7 cells after treatment with CGlcN for different periods of time. For that, cells were lysed with Trizol® and centrifuged at 12 000 g for 15 min at 25° following the addition of chloroform. Isopropanol was added to the supernatant at a 1 : 1 ratio and the RNA pellet was obtained following centrifugation. After washing with ethanol, extracted RNA was solubilized in diethyl pyrocarbonate-treated RNase-free water and quantified by measuring the absorbance at 260 nm using the GENios® microplate reader (Tecan Austria GmbH).
Equal amounts of RNA (1 µg) were reverse transcribed in a mastermix containing 1 × reverse transcriptase (RT) buffer, 1 mm dNTPs, 500 ng of oligo(dT)15 primers, 140 U of murine Moloney leukaemia virus (MMLV) reverse transcriptase and 40 U of RNase inhibitor, for 45 min at 42°. Polymerase chain reaction was carried out in an automatic Whatman thermocycler (Biometra, Kent, UK) to amplify iNOS, COX-2 and glyceraldehyde-3-phosphate dehydrogenase (G3PDH) mRNA. Primer sequences used to amplify the desired cDNA were as follows: iNOS forward and reverse primers: 5′-CCCTTCCGAAGTTTCTGGCAGCAGC-3′ and 5′-GGCTGTCAGAGCCTCGTGGCTTTGG-3′; COX-2 forward and reverse primers: 5′-GGGGTACCTTCCAGCTGTCAAAATCTC-3′ and 5′-GAAGATCTCGCCAGGTACTCACCTGTATG-3′; and G3PDH forward and reverse primers: 5′-TGAAGGTCGGTGTGAACGGATTTGGC-3′ and 5′-CATGTAGGCCATGAGGTCCACCAC-3′. Polymerase chain reaction (PCR) products electrophoresed on 2% agarose gels were visualized by ethidium bromide staining and quantified using AlphaEase® gel image-analysis software (Alpha Innotech, San Leandro, CA, USA).
NF-κB reporter gene assay
RAW264.7 cells cultured in 10-cm culture dishes were transiently cotransfected with a NF-κB binding site luciferase reporter plasmid (Clontech) and a β-galactosidase expression vector using the Lipofectamine™ 2000 reagent (Invitrogen, San Diego, CA). Transfected cells were subcultured into 24-well plates and treated with different concentrations of CGlcN for 24 hr following stimulation with LPS (1 µg/ml) or TNF-α (6 ng/ml). Cells were washed once with cold phosphate-buffered saline and lysed with 200 µl/well of lysis buffer [25 mm Tri-HCl, pH 8·0, containing 2 mm dithiothreitol (DTT) and 1% Triton-X 100]. Equal amounts (20 µl) of cell lysate and luciferase substrate (luciferin; Promega, Madison, WI) were mixed in a 96-well plate and the luminescence intensity was measured with a luminescence microplate reader (Tecan Austria GmbH). The luciferase activity was normalized to transfection efficiency monitored by the β-galactosidase expression vector in ortho-nitrophenyl-β-d-galactopyranoside (ONPG) buffer. The level of reporter gene expression was determined as a ratio and compared with cells stimulated by LPS or TNF-α alone.
Transfected cells were visualized by the X-Gal staining method. For that, transfected cells were fixed with 0·5% glutaraldehyde and stained with X-Gal solution containing 20 mm K3Fe(CN)6, K4Fe(CN)6 and 1 mm MgCl2. After 24 hr of incubation at 37°, transfected cells were visualized with blue color under a light microscope.
Western blotting
Western blotting was performed according to standard procedures. RAW264.7 cells treated with CGlcN were lysed in lysis buffer containing 50 mm Tris-HCl (pH 7·5), 0·4% Nonidet P-40, 120 mm NaCl, 1·5 mm MgCl2, 2 mm phenylmethylsulfonyl fluoride, 80 µg/ml of leupeptin, 3 mm NaF and 1 mm DTT at 4° for 30 min. Cell lysates (≈ 10 µg of total protein) were resolved on a 4–20% Novex® gradient gel (Invitrogen), electrotransferred onto a nitrocellulose membrane and blocked with 10% skim milk. Different antibodies (Santa Cruz Biotechnology Inc.) were used to detect the respective proteins using a chemiluminescent ECL assay kit (Amersham Pharmacia Biosciences), according to the manufacturer's instructions. Western blots were visualized using an LAS3000® Luminescent image analyzer and protein expression was quantified by multi gauge V3·0 software (Fujifilm Life Science, Tokyo, Japan).
Data analysis
One-way analysis of variance was used for all statistical analyses using independent experiments and data are represented as means ± standard error of the mean. Individual values were compared by Dunnett's test and a P-value of < 0·05 considered as significant, unless otherwise stated.
Results
Synthesis and structural confirmation of CGlcN
During synthesis reactions, GlcN was N-carboxylated to obtain CGlcN (Scheme 1). The reactions were carried out under mild conditions to rule out the possibility of adverse effects on structural change. In particular, maintaining the pH between 9·00 and 10·00 facilitated substitution of the carboxybutyryl group with the amino group. That is because at basic pH, amino groups more actively participate in the reaction than at acidic or neutral pH. Successful synthesis of CGlcN was confirmed by comparing its FT-IR, 1H NMR and 13C NMR spectra with those of GlcN.
Scheme 1.
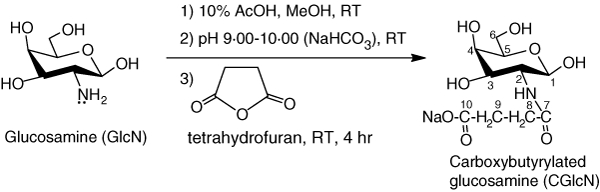
Schematic representation for the synthesis of carboxybutyrylated glucosamine (CGlcN) from glucosamine (GlcN). RT, room temperature (25°).
GlcN: fine white solid; IR (KBr) vmax 3412 (OH), 2864 (CH), 1578 (NH), 1309 (CN), 1108, 1065, 1036 (pyranose) cm−1; 1H NMR (400 MHz, D2O) δ 5·4, 4·9 (1H, H-1α, H-1β), 3·2, 2·9 (1H, H-2α, H-2β), 3·4–3·9 (1H, H-3, 1H, H-4, 1H, H-5 and 2H, H2-6), 4·7 (D2O); 13C NMR (400 MHz, D2O) ä 89·0, 92·1 (C-1α, C-1β), 54·3, 56·2 (C-2α, C-2β), 69·0 (C-3), 76·3 (C-4), 71·2, 72·4 (C-5α, C-5β), 61·1 (C-6).
CGlcN: dark yellow solid; IR (KBr) vmax 3302 (OH), 3123, 2989 (CH), 1657 (amide CO), 1556 (NH), 1414 (carboxyl CO), 1132, 1098, 1045 (pyranose) cm−1; 1H NMR (D2O, 400 MHz) δ 4·8 (1H, H-1), 3·3 (1H, H-2), 3·4–3·7 (1H, H-3, 1H, H-4, 1H, H-5 and 2H, H2-6), 2·6 (2H, H2-8 and 2H, H2-9), 4·7 (D2O); 13C NMR (D2O, 400 MHz) δ 90·0, 93·2 (C-1α, C-1β), 54·3, 57·1 (C-2α, C-2β), 69·9 (C-3), 76·6 (C-4), 71·5, 72·0 (C-5α, C-5β), 62·1 (C-6), 175·2 (C-7), 31·3 (C-8 and C-9), 178·5 (C-10).
New strong peaks appearing in FT-IR spectra of CGlcN at 1657 cm−1 and 1414 cm−1 were assigned for amide CO and carboxyl CO stretches, respectively. Moreover, characteristic chemical shifts appeared in 13C NMR spectra of CGlcN and those were not presented in the spectrum of GlcN at δ 175·2 (CO), 31·3 (-CH2CH2-) or 178·5 (-COO).23 In addition, a new clear chemical shift was observed in the 1H NMR spectrum of CGlcN at δ 2·6, confirming the presence of an -CH2CH2- group in the structure. Taken together, these structural data confirm the successful synthesis of CGlcN.
Cytocompatibile effect of CGlcN
Cytocompatible concentrations of GlcN and CGlcN were tested with the MTT cell viability assay using RAW264.7 cells grown in serum-free media. As depicted in Fig. 1, neither GlcN nor CGlcN exerted any significant (P < 0·01) toxic effect on RAW264.7 cells under the tested concentrations after 24 hr of treatment. Therefore, non-toxic concentrations of CGlcN were used for the experiments in all the experiments.
Figure 1.

Effects of glucosamine (GlcN) and carboxybutyrylated glucosamine (CGlcN) on the viability of RAW264.7 cells. Cells grown in serum-free medium were treated with different concentrations of GlcN or CGlcN for 24 hr and cell viability was assessed by the [(3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide)] (MTT) assay, as described in the Materials and methods. Results of independent experiments were averaged and are shown as percentage cell viability compared with the viability of untreated control cells.
CGlcN inhibited production of NO and PGE2 in RAW264.7 cells
Stimulation of cells with LPS resulted in a significant enhancement of the nitrite concentration in conditioned medium compared with that of LPS non-stimulated blank cells (Fig. 2a). This indicated increased production of NO and release of its stable product, 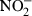 , into the culture medium. However, cells pretreated with CGlcN showed a dose-dependent reduction in the production of NO following stimulation with LPS. Moreover, even at the lowest tested concentration (10 µg/ml) of CGlcN, a significant (P < 0·05) reduction in the level of NO was observed. At 500 µg/ml of CGlcN, the level of NO production was similar to that of non-stimulated cells.
, into the culture medium. However, cells pretreated with CGlcN showed a dose-dependent reduction in the production of NO following stimulation with LPS. Moreover, even at the lowest tested concentration (10 µg/ml) of CGlcN, a significant (P < 0·05) reduction in the level of NO was observed. At 500 µg/ml of CGlcN, the level of NO production was similar to that of non-stimulated cells.
Figure 2.

Effects of carboxybutyrylated glucosamine (CGlcN) on the inhibition of lipopolysaccharide (LPS)-induced nitric oxide (NO) and prostaglandin E2 (PGE2) production in RAW264.7 cells. (a) Cells cultured in phenol red and serum-free media were pretreated with different concentrations of CGlcN for 1 hr and stimulated with a 1-µg/ml final concentration of LPS for 48 hr. Conditioned medium was mixed with an equal amount of the Griess reagent and the absorbance, measured at 550 nm, represented the amount of 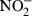 (stable oxidation product of NO) in the medium. The values obtained were compared with those of standard concentrations of sodium nitrite dissolved in Dulbecco's modified Eagle's minimal essential medium (DMEM), and the concentrations of nitrite in conditioned media of sample-treated cells were calculated. (b) Cells growing in serum-free medium were pretreated with different concentrations of CGlcN for 1 hr and stimulated with LPS (1 µg/ml final concentration) for 24 hr. The amount of PGE2 release was determined by the mouse PGE2 enzyme-linked immunosorbent assay (ELISA) kit. BLK, LPS non-stimulated cells; LPS, LPS-stimulated cells. Statistical comparisons, *P < 0·05 and **P < 0·01.
(stable oxidation product of NO) in the medium. The values obtained were compared with those of standard concentrations of sodium nitrite dissolved in Dulbecco's modified Eagle's minimal essential medium (DMEM), and the concentrations of nitrite in conditioned media of sample-treated cells were calculated. (b) Cells growing in serum-free medium were pretreated with different concentrations of CGlcN for 1 hr and stimulated with LPS (1 µg/ml final concentration) for 24 hr. The amount of PGE2 release was determined by the mouse PGE2 enzyme-linked immunosorbent assay (ELISA) kit. BLK, LPS non-stimulated cells; LPS, LPS-stimulated cells. Statistical comparisons, *P < 0·05 and **P < 0·01.
To determine whether CGlcN can inhibit the production of COX-2, RAW264.7 cells were stimulated with LPS and the amount of PGE2 release was assessed using anti-PGE2-coated ELISA plates. Cells stimulated with LPS exhibited a threefold increment in PGE2 release compared with that of LPS non-treated cells (Fig. 2b). This induction was significantly (P < 0·01) reduced by treatment with 100 and 500 µg/ml of CGlcN. These results indicated that CGlcN has the potential to inhibit LPS-induced production of NO and PGE2 in RAW264.7 cells.
CGlcN inhibits mRNA transcription and protein expression of iNOS and COX-2
To identify the gene expression of iNOS that is responsible for LPS-induced NO production, the iNOS mRNA level was studied with reverse transcription–polymerase chain reaction (RT–PCR). Cells pretreated with CGlcN (100 µg/ml) were stimulated with a 1-µg/ml final concentration of LPS, and total RNA was collected at different time-points during incubation. Specific iNOS primers were used to polymerize the reverse transcribed cDNA and were visualized by ethidium bromide staining followed by electrophoretic separation. As shown in Fig. 3(a), RAW264.7 cells treated with LPS alone exhibited a clear time-dependent iNOS mRNA expression after 12 hr of LPS stimulation. The iNOS mRNA level was observed to be maximal during 12–18 hr of stimulation. Therefore, cells pretreated with CGlcN were incubated until 18 hr and tested for the iNOS mRNA levels at different time-points. Interestingly, cells treated with CGlcN inhibited LPS-induced expression of iNOS mRNA clearly during 12–18 hr of incubation. To identify the effects of CGlcN on COX-2 gene expression further, RT-PCR was conducted. As shown in Fig. 3(b), after 12 hr of LPS treatment, expression of the COX-2 mRNA level was attenuated by treatment with higher concentrations of CGlcN. Moreover, the ability of CGlcN to inhibit iNOS mRNA expression was much stronger than the ability to inhibit COX-2 mRNA expression. RNA expression levels of the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (G3PDH), confirmed that the observed results were not an artefact occurring during electrophoresis.
Figure 3.

Effects of carboxybutyrylated glucosamine (CGlcN) on the inhibition of lipopolysaccharide (LPS)-induced mRNA expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) in RAW264.7 cells. (a) Cells pretreated (1 hr) with CGlcN (100 µg/ml) were stimulated with a 1-µg/ml final concentration of LPS and total RNA was collected at different incubation time-points. Specific iNOS primers were used to polymerize the reverse transcribed cDNA and visualized by ethidium bromide staining followed by electrophoretic separation. cDNA bands were compared with those of sample or LPS non-treated blank (BLK) and LPS-alone treated groups. (b) Cells were pretreated with different concentrations of CGlcN for 1 hr and following LPS (1 µg/ml) stimulation, incubation was continued for 12 hr. Total RNA was collected and specific COX-2 primers were used to polymerize the reverse transcribed cDNA, which was visualized by ethidium bromide staining followed by electrophoretic separation. cDNA bands were compared with those of sample or LPS-non-treated blank (BLK) and LPS-alone treated groups. Respective G3PDH mRNA expression levels were used to confirm the equal amounts of RNA used for cDNA synthesis.
To confirm the inhibitory effects of CGlcN on iNOS and COX-2, LPS-treated RAW264.7 cells were assessed for their protein levels, using appropriate antibody, in Western blotting. As shown in Fig. 4, clear dose-dependent reductions in both iNOS and COX-2 protein levels were observed. Several reports have stated that the expression of iNOS and COX-2 is regulated by various stimuli during inflammation in addition to LPS. Moreover, those stimuli, including LPS, are known to induce the expression of iNOS and COX-2 genes through several pathways, finally activating transcription factor NF-κB.
Figure 4.

Inhibition of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) protein expression by carboxybutyrylated glucosamine (CGlcN). RAW264.7 cells were pretreated with different concentrations of CGlcN for 1 hr and stimulated with LPS (1 µg/ml) for another 24 hr. Equal amounts of protein in the cell lysates were electrophoresed and the protein expression levels of iNOS and COX-2 were determined using specific antibodies for iNOS and COX-2. Respective protein levels of actin were used to confirm the equal amounts of protein used for electrophoresis. BLK, LPS non-stimulated; LPS, LPS-stimulated but sample non-treated cells.
Inhibition of iNOS and COX-2 takes place via mitogen-activated protein kinase-mediated NF-κB expression
As iNOS and COX-2 expression are activated by the transcription factor NF-κB, it was assumed that CGlcN may also inhibit NF-κB. Therefore, the promoter activity of the NF-κB gene in RAW264.7 cells was studied following transfection with a NF-κB binding site–luciferase reporter plasmid construct and β-galactosidase expression vector. In this study, NF-κB promoter activity was separately stimulated either by LPS or by TNF-α. Interestingly, after treatment with CGlcN, both LPS and TNF-α-induced NF-κB promoter activities were clearly inhibited (Fig. 5a). NF-κB is composed of two major protein subunits (p50 and p65) and following inhibition of NF-κB activity or expression, one, or both, of these protein subunits can be affected. Therefore, the protein expression level of NF-κB was studied using p50- and p65-specific antibodies and observed that CGlcN treatment inhibited protein expressions of p50 and p65 (Fig. 5b). Therefore, according to the results of the NF-κB reporter gene experiment and Western blot protein determination it was clear that inhibition of iNOS and COX-2 by CGlcN takes place via down-regulation of their transcription factor, NF-κB.
Figure 5.

Inhibitory effects of carboxybutyrylated glucosamine (CGlcN) on nuclear factor-κB (NF-κB) promoter activity and expression in RAW264.7 cells. (a) Cells were transiently cotransfected with NF-κB binding site-luciferase reporter plasmid and β-galactosidase expression vector. Transfected cells were pretreated with different concentrations of CGlcN for 1 hr and stimulated with lipopolysaccharide (LPS) (1 µg/ml) or tumour necrosis factor-α (TNF-α) (6 ng/ml) for another 24 hr. Cells were lysed, and the lysates were tested for luciferase activity. Luciferase activity was normalized to transfection efficiency monitored by the β-galactosidase expression vector in ortho-nitrophenyl-β-d-galactopyranoside (ONPG) buffer. The level of NF-κB promoter activity in CGlcN-treated cells was determined as a ratio, compared with cells stimulated by LPS or TNF-α alone. Statistical comparison, **P < 0·01. (b) RAW264.7 cells were pretreated with different concentrations of CGlcN for 1 hr and then stimulated with LPS (1 µg/ml) for another 24 hr. Equal amounts of protein in the cell lysates were electrophoresed and the levels of NF-κB (p50) and NF-κB (p65) protein expressions were determined using specific antibodies for p50 and p65. The respective protein levels of actin were used to confirm that equal amounts of protein were used for electrophoresis. BLK, LPS non-stimulated; LPS, LPS-stimulated but sample non-treated cells.
To confirm the inhibition of NF-κB in greater detail, the cellular TNF-α level was also assessed using a TNF-α-specific ELISA experiment because NF-κB acts as the major transcription factor for expression of the TNF-α gene following stimulation with LPS. As expected, pretreatment with CGlcN significantly inhibited the expression of TNF-α in LPS-induced RAW264.7 cells (Fig. 6). However at low concentrations, the inhibitory effect was not significant (P < 0·05).
Figure 6.

Effect of carboxybutyrylated glucosamine (CGlcN) on tumour necrosis factor-α (TNF-α) production in lipopolysaccharide (LPS)-stimulated RAW264.7 cells. Cells were pretreated with different concentrations of CGlcN for 1 hr and stimulated with LPS (1 µg/ml) for another 4 hr. Following incubation, the amount of TNF-α release was determined by a TNF-α antibody-coated enzyme-linked immunosorbent assay (ELISA) kit, as described in the Materials and methods.
Under unstimulated conditions, NF-κB remains in the cytoplasm inactively bound to its inhibitor, IκB. LPS-induced activation of NF-κB takes place via activation of I-κB kinase (IKK), resulting in the degradation of IκB and release of active NF-κB. Activation of IKK is mediated mainly via a group of proteins referred to as mitogen-activated protein kinase (MAPKs). Therefore, the effects of CGlcN on the expression of three different MAPK proteins [p38 MAPK, Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK)] were studied. Interestingly, both p38 MAPK and JNK were dose-dependently inhibited by treatment with CGlcN at 100 µg/ml (Fig. 7). However, the ERK protein level was not altered by any concentration of CGlcN. In addition to MAPKs, in the signaling pathway of NF-κB, activation by LPS is regulated by another protein, named NIK. Therefore, the effect of CGlcN on LPS-induced NIK protein expression was also studied. According to the results shown in Fig. 8 there was no inhibitory effect on NIK protein by CGlcN. However, a clear dose-dependent inhibition was observed on the protein levels of IKK and phospho-IκBα. Therefore, these results clearly confirmed that inhibition of iNOS and COX-2 takes place via p38 MAPK- and JNK-mediated down-regulation of NF-κB expression.
Figure 7.

Inhibition of lipopolysaccharide (LPS)-induced protein expression of mitogen-activated protein kinase (MAPK) by carboxybutyrylated glucosamine (CGlcN). RAW264.7 cells were pretreated with different concentrations of CGlcN for 1 hr and stimulated with LPS (1 µg/ml) for another 24 hr. Equal amounts of protein in the cell lysates were electrophoresed and the levels of p38 MAPK, Jun N-terminal kinase (JNK) and extracellular signal regulated kinase (ERK) protein were determined using specific antibodies. Respective protein levels of actin were used to confirm that equal amounts of protein were used for electrophoresis. BLK, LPS non-stimulated; LPS, LPS-stimulated but sample non-treated cells.
Figure 8.

Inhibition of lipopolysaccharide (LPS)-induced protein expression of I-κB kinase (IKK), I kappa B alpha (IκBα) and NIK by carboxybutyrylated glucosamine (CGlcN). RAW264.7 cells were pretreated with different concentrations of CGlcN for 1 hr and then stimulated with LPS (1 µg/ml) for another 24 hr. Equal amounts of protein in the cell lysates were electrophoresed and the expression levels of IKK, IκBα and NIK protein were determined using specific antibodies: Respective protein levels of actin were used to confirm that equal amounts of protein were used for elctrophoresis. BLK, LPS non-stimulated; LPS, LPS-stimulated but sample non-treated cells.
Discussion
In this study, we explain how a novel GlcN derivative (CGlcN) inhibits the LPS-induced expression of major inflammatory mediators, iNOS/NO and COX-2/PGE2, in RAW264.7 cells. Cells treated with CGlcN, even at 500 µg/ml, behaved indistinguishably from controls in terms of cell viability and cytotoxicity. Therefore, CGlcN is cytocompatible and did not adversely interfere with the other assay methods used in this study. Initially, CGlcN was tested for its effects on the inhibition of LPS-induced production of NO and PGE2. The idea behind starting this study came up with the strong radical scavenging properties and some immunosuppressive effects of CGlcN observed in our previous studies. Clear dose-dependent inhibitory effects on NO and PGE2 production suggested that further studies on their respective gene expressions would be of value. Induction of iNOS and COX-2 transcription by LPS was very much higher than in non-treated control cells after ≈ 12–18 hr of incubation and this was significantly reduced following pretreatment with CGlcN. Our observation was in line with previous reports that the maximum iNOS mRNA expression lies at around 18 hr of LPS stimulation.24 This was the first evidence demonstrating that CGlcN is capable of regulating gene transcription. Furthermore, protein expression studies carried out with Western blot experiments revealed that not only the transcription level, but also the translational mechanism, was attenuated by CGlcN. In addition to CGlcN, its starting material, GlcN, was also tested for an inhibitory effect on transcription of those genes. However, the effect was not as clear as for CGlcN (data not shown). In contrast, controversial results have been reported in a few studies identifying the potential effects of GlcN to inhibit iNOS expression.25 Therefore, in this study we focused on CGlcN as a novel material to identify its signaling pathway of inhibiting iNOS and COX-2 expression.
Transcriptional and translational regulations suggested that the effect of CGlcN may affect the transcription factor of those genes. Because compounds interfering with both iNOS and COX-2 generally act as potential inhibitors of NF-κB activation, a transcription factor is required for the induction of iNOS and COX-2.16 The data from transient transfection with a NF-κB promoter vector support this idea because CGlcN could clearly inhibit the LPS- or TNF-α-induced promoter activity of NF-κB. Regulation of iNOS or COX-2 by CGlcN therefore, whether transiently or chronically expressed, is likely to be pretranscriptional, utilizing control mechanisms with NF-κB and its upstream protein(s). Furthermore, this was supported by the inhibition of TNF-α expression by CGlcN following LPS stimulation. However, inhibition of TNF-α by CGlcN, even at 500 µg/ml, was low compared with the inhibition of NF-κB promoter activity. Therefore, the differential response seen in this study may reflect the fact that activation of TNF-α expression may follow some pathways other than NF-κB signaling.26 Down-regulation of NF-κB genes was further confirmed by inhibiting their respective whole-cell proteins (p50 and p65) by CGlcN. Furthermore, the protein levels of p50 and p65 in the nucleus confirmed their down-regulated expression following treatment with CGlcN (data not shown). This interesting possibility further suggested that IKK and other proteins, such as MAPKs and NIK, upstream of the NF-κB signaling pathway, may also be regulated by CGlcN because activation of NF-κB is mainly controlled by its inhibitory complex protein, IκB, and their inhibitor IKK. There are two major signaling pathways that can activate IKK expression. The most important pathway signals via proteins of MAPKs.27 One of the most important finding in this study was, among three MAPKs, that only p38 MAPK and JNK signaling was attenuated by CGlcN, whereas ERK was unaffected. In addition, the other pathway that activated IKK regulation via NIK also remained unaffected following treatment with CGlcN. Activation of IKK enhances degradation of IκB, resulting in the release of NF-κB and its translocation to the nucleolus. Once free NF-κB entered the nucleus it binds with the promoter sequence of genes that regulate their expression, including iNOS and COX-2. According to the results of this study, the expression of IKK was inhibited by CGlcN. Furthermore, the level of the cytoplasmic phospho-IκBα protein was also decreased, in a dose-dependent manner, after treatment with CGlcN. This indicates that IκB binds to NF-κB to make an inhibitory complex. Therefore, protein expression of IKK, following treatment with CGlcN, clearly confirmed that its down-regulation directly inhibited the degradation of IκB and release of NF-κB. Taken together, it can be concluded that iNOS and COX-2 inhibitory effects of CGlcN in RAW264.7 cells takes place via attenuation of NF-κB signaling by p38 MAPK and JNK, but not by ERK. Moreover, CGlcN can be considered as a potential anti-inflammatory material and further studies can be used to identify its effects in animal models.
Acknowledgments
The authors acknowledge the Marine Bioprocess Research Center of the Marine Bio 21 Project, funded by the Ministry of Maritime Affairs and Fisheries Republic of Korea, for the support provided through the research grant P-2004-01.
References
- 1.Distler J, Anguelouch A. Evidence-based practice: review of clinical evidence on the efficacy of glucosamine and chondroitin in the treatment of osteoarthritis. J Am Acad Nurse Pract. 2006;18:487–93. doi: 10.1111/j.1745-7599.2006.00166.x. [DOI] [PubMed] [Google Scholar]
- 2.Zhang GX, Yu S, Gran B, Rostami A. Glucosamine abrogates the acute phase of experimental autoimmune encephalomyelitis by induction of Th2 response. J Immunol. 2005;175:7202–8. doi: 10.4049/jimmunol.175.11.7202. [DOI] [PubMed] [Google Scholar]
- 3.Chen JT, Chen CH, Horng CT, et al. Glucosamine sulfate inhibits proinflammatory cytokine-induced icam-1 production in human conjunctival cells in vitro. J Ocul Pharmacol Ther. 2006;22:402–16. doi: 10.1089/jop.2006.22.402. [DOI] [PubMed] [Google Scholar]
- 4.Paige JS, Jaffrey SR. Pharmacologic manipulation of nitric oxide signaling: targeting NOS dimerization and protein–protein interactions. Curr Top Med Chem. 2007;1:97–114. doi: 10.2174/156802607779318253. [DOI] [PubMed] [Google Scholar]
- 5.Moncada S, Palmer RMJ, Higgs DA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1992;43:109–42. [PubMed] [Google Scholar]
- 6.Gao YT, Panda SP, Roman LJ, Martasek P, Ishimura Y, Masters BS. Oxygen metabolism by neuronal nitric-oxide synthase. J Biol Chem. 2007;282:7921–9. doi: 10.1074/jbc.M609814200. [DOI] [PubMed] [Google Scholar]
- 7.Cunha IW, Lopes A, Falzoni R, Soares FA. Sarcomas often express constitutive nitric oxide synthases (NOS) but infrequently inducible NOS. Appl Immunohistochem Mol Morphol. 2006;14:404–10. doi: 10.1097/01.pai.0000190175.98576.a3. [DOI] [PubMed] [Google Scholar]
- 8.Breitbach K, Klocke S, Tschernig T, van Rooijen N, Baumann U, Steinmetz I. Role of inducible nitric oxide synthase and NADPH oxidase in early control of Burkholderia pseudomallei infection in mice. Infect Immun. 2006;74:6300–9. doi: 10.1128/IAI.00966-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HG, Shrestha B, Lim SY, et al. Cordycepin inhibits lipopolysaccharide-induced inflammation by the suppression of NF-kappaB through Akt and p38 inhibition in RAW 264.7 macrophage cells. Eur J Pharmacol. 2006;545:192–9. doi: 10.1016/j.ejphar.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 10.Xie Q, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem. 1994;269:4705–8. [PubMed] [Google Scholar]
- 11.Sacco RE, Waters WR, Rudolph KM, Drew ML. Comparative nitric oxide production by LPS-stimulated monocyte-derived macrophages from Ovis canadensis and Ovis aries. Comp Immunol Microbiol Infect Dis. 2006;29:1–11. doi: 10.1016/j.cimid.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Farley KS, Wang LF, Razavi HM, Law C, Rohan M, McCormack DG, Mehta S. Effects of macrophage inducible nitric oxide synthase in murine septic lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;290:1164–72. doi: 10.1152/ajplung.00248.2005. [DOI] [PubMed] [Google Scholar]
- 13.Ruan KH, Deng H, So SP. Engineering of a protein with cyclooxygenase and prostacyclin synthase activities that converts arachidonic acid to prostacyclin. Biochemistry. 2006;45:14003–11. doi: 10.1021/bi0614277. [DOI] [PubMed] [Google Scholar]
- 14.Rahman A, Yatsuzuka R, Jiang S, Ueda Y, Kamei C. Involvement of cyclooxygenase-2 in allergic nasal inflammation in rats. Int Immunopharmacol. 2006;6:1736–42. doi: 10.1016/j.intimp.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Kim HS, Ye SK, Cho IH, et al. Hydroxydeoxyguanosine suppresses NO production and COX-2 activity via Rac1/STATs signaling in LPS-induced brain microglia. Free Radic Biol Med. 2006;41:1392–403. doi: 10.1016/j.freeradbiomed.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 16.Bremner P, Heinrich M. Natural products as targeted modulator of nuclear factor-κB pathway. J Pharm. 2002;54:453–72. doi: 10.1211/0022357021778637. [DOI] [PubMed] [Google Scholar]
- 17.Park HJ, Kim IT, Won JH, Jeong SH, Park EY, Nam JH, Choi J, Lee KT. Anti-inflammatory activities of ent-16alphaH,17-hydroxy-kauran-19-oic acid isolated from the roots of Siegesbeckia pubescens are due to the inhibition of iNOS and COX-2 expression in RAW 264.7 macrophages via NF-kappaB inactivation. Eur J Pharmacol. 2007;558:185–93. doi: 10.1016/j.ejphar.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 18.Kim SS, Oh OJ, Min HY. Eugenol suppresses cyclooxygenase-2 expression in lipopolysaccharide stimulated mouse macrophage RAW264.7 cells. Life Sci. 2003;73:337–48. doi: 10.1016/s0024-3205(03)00288-1. [DOI] [PubMed] [Google Scholar]
- 19.Bengmark S. Curcumin, an atoxic antioxidant and natural NFkappaB, cyclooxygenase-2, lipooxygenase, and inducible nitric oxide synthase inhibitor: a shield against acute and chronic diseases. JPEN J Parenter Enteral Nutr. 2006;30:45–51. doi: 10.1177/014860710603000145. [DOI] [PubMed] [Google Scholar]
- 20.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–10. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 21.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal Biochem. 1982;126:131–8. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 22.Ling JJ, Sun YJ, Zhu DY, Chen Q, Han X. Potential role of NO in modulation of COX-2 expression and PGE2 production in pancreatic B-cells. Biochem Biophys Acta Sinica. 2005;37:139–46. [PubMed] [Google Scholar]
- 23.Ronghua H, Yumin D, Jianhong Y. Preparation and anticoagulant activity of carboxybutyrylated hydroxyethyl chitosan sulfates. Carbohydr Polym. 2003;51:431–8. [Google Scholar]
- 24.Chen CW, Lee ST, Wu WT, Fu WM, Ho FM, Lin WW. Signal transduction for inhibition of inducible nitric oxide synthase and cyclooxygenase-2 induction by capsaicin and related analogs in macrophages. Br J Pharmacol. 2003;140:1077–87. doi: 10.1038/sj.bjp.0705533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meininger CJ, Kelly KA, Li H, Haynes TE, Wu G. Glucosamine inhibits inducible nitric oxide synthesis. Biochem Biophys Res Commun. 2000;279:234–9. doi: 10.1006/bbrc.2000.3912. [DOI] [PubMed] [Google Scholar]
- 26.Wu CF, Bi XL, Yang JY, et al. Differential effects of ginsenosides on NO and TNF-alpha production by LPS-activated N9 microglia. Int Immunopharmacol. 2007;7:313–20. doi: 10.1016/j.intimp.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 27.Liang S, Kishore R, McMullen MR, Nagy LE. Lipopolysaccharide stimulation of ERK1/2 increases TNF- production via Egr-1. Am J Physiol Cell Physiol. 2002;282:C1205–11. doi: 10.1152/ajpcell.00511.2001. [DOI] [PubMed] [Google Scholar]