

Abstract
The function of repressor activator protein 1 (Rap1p) at glycolytic enzyme gene upstream activating sequence (UAS) elements in Saccharomyces cerevisiae is to facilitate binding of glycolysis regulatory protein 1 (Gcr1p) at adjacent sites. Rap1p has a modular domain structure. In its amino terminus there is an asymmetric DNA-bending domain, which is distinct from its DNA-binding domain, which resides in the middle of the protein. In the carboxyl terminus of Rap1p lie its silencing and putative activation domains. We carried out a molecular dissection of Rap1p to identify domains contributing to its ability to facilitate binding of Gcr1p. We prepared full-length and three truncated versions of Rap1p and tested their ability to facilitate binding of Gcr1p by gel shift assay. The ability to detect ternary complexes containing Rap1p⋅DNA⋅Gcr1p depended on the presence of binding sites for both proteins in the probe DNA. The DNA-binding domain of Rap1p, although competent to bind DNA, was unable to facilitate binding of Gcr1p. Full-length Rap1p and the amino- and carboxyl-truncated versions of Rap1p were each able to facilitate binding of Gcr1p at an appropriately spaced binding site. Under these conditions, Gcr1p displayed an approximately 4-fold greater affinity for Rap1p-bound DNA than for otherwise identical free DNA. When spacing between Rap1p- and Gcr1p-binding sites was altered by insertion of five nucleotides, the ability to form ternary Rap1p⋅DNA⋅Gcr1p complexes was inhibited by all but the DNA-binding domain of Rap1p itself; however, the ability of each individual protein to bind the DNA probe was unaffected.
Keywords: transcription/cooperative DNA binding/DNA bending
The upstream activating sequence (UAS) elements of glycolytic enzyme genes in Saccharomyces cerevisiae resemble enhancers and proximal promoter elements of higher eukaryotes, in that they are composed of multiple binding sites for several different transcription factors (1–6). Each of the binding sites by themselves displays little UAS activity, but in combination they form powerful activation elements. The center pieces of glycolytic enzyme gene UAS elements are binding sites for the transcription factors repressor activator protein 1 (Rap1p) and glycolysis regulatory protein 1 (Gcr1p). Key to understanding the mechanisms governing high-level glycolytic gene expression is an understanding of the protein–protein and protein–nucleic acid interactions that occur between Rap1p, Gcr1p, and their binding sites. Both Rap1p and Gcr1p have characteristics that raise fundamental questions related to their roles as transcription factors and allow them to serve as models for transcription factors of higher eukaryotes. The activities of Rap1p present a paradox: how can one protein act as both an activator and repressor, depending on sequence context of its binding site (7, 8)? Gcr1p also raises a paradox: how does a DNA-binding protein that binds in vitro with a low degree of sequence specificity recognize and bind at its binding sites in vivo amidst the vast excess of nonspecific binding sites that the rest of the genome comprises (9)?
Much attention has been devoted to analyzing Rap1p and its various activities. Rap1p is a sequence-specific DNA-binding protein composed of 827 amino acid residues (8) that is capable of bending DNA (10). The DNA-binding domain resides in the middle third of the protein between amino acid residues 361 and 596 (11); however the domain responsible for the greatest distortion (≈59°) of DNA lies amino-terminal to the DNA-binding domain, between residues 44 and 247 (12). The crystal structure of the DNA-binding domain of Rap1p in complex with telomeric DNA has been reported (13). The DNA-binding domain itself is also capable of introducing a slight bend (≈20°) in DNA. The silencing domain of Rap1p maps carboxyl-terminal to amino acid residue 662 (14). This domain has been shown to be the site of interaction with the silent information regulatory proteins, Sir3p and Sir4p (15, 16), and with Rap1p-interacting factor, Rif1p (17). A putative trans-activation domain, which partially overlaps the silencing domain, has been mapped between amino acid residues 630 and 695 (18). Unlike the silencing domain, the protein(s) with which the activation domain interacts have yet to be established.
The role of Rap1p in expression of glycolytic enzyme genes is that of an activator (3, 4, 6, 19–23). In the controlling region of these genes, Rap1p-binding sites are found immediately adjacent to Gcr1p-binding sites. In most cases, mutations in Rap1p-binding sites result in greater than a 10-fold reduction in expression of the cognate gene. Rap1p-binding sites, by themselves, are either nonfunctional or function as relatively weak UAS elements (3, 7, 8, 24–26). However, when Rap1p-binding sites are located adjacent to Gcr1p-binding sites a strong synergism is observed between the binding sites and their ability to act as UAS elements (3, 5, 7, 20, 27).
Gcr1p, in contrast to Rap1p, appears to function primarily in the expression of glycolytic enzyme genes (28); however, there are some indications that it may be required for the expression of some other genes (29, 30). The levels of glycolytic enzymes are markedly reduced in gcr1 mutant strains (28, 31, 32), as a result of reduced transcript levels (19, 23, 28, 33, 34). The ability to identify Gcr1p as a DNA-binding protein was hampered by the relatively low degree of specificity that Gcr1p displays for its DNA-binding site in vitro (9). Yet, in vivo Gcr1p-binding sites adjacent to bound Rap1p-binding sites are nearly fully occupied (3, 35, 36). Genetic analysis has implicated CT-boxes, now known to be Gcr1p-binding sites, as important features of the UAS elements of six glycolytic enzyme genes (3–5, 7, 37). In most cases, mutation of the CT-boxes reduced expression greater than 10-fold. Although CT-boxes are essential features of these genes’ UAS elements, they are not functional as UAS elements by themselves (3, 5, 7, 38).
In yeast cells, the DNA-binding ability of Gcr1p at glycolytic enzyme gene UAS elements depends on the presence of Rap1p bound at sites adjacent to the Gcr1p-binding site (39). In vivo binding studies with a rap1–2ts mutant strain demonstrated the binding requirement of Rap1p for continued Gcr1p binding at adjacent sites. Disassociation of Rap1p from its site at nonpermissive temperatures results in the disassociation of Gcr1p from adjacent binding sites (39). Experiments with synthetic UAS elements revealed that the spacing between the individual binding sites is critical for the binding of Gcr1p in vivo.
In terms of in vivo binding and expression, the most dramatic differences observed with the synthetic oligonucleotides were between an oligonucleotide with native spacing between the Rap1p- and Gcr1p-binding sites (known as oligonucleotide N) and an oligonucleotide in which the sites were displaced by five nucleotides from each other (known as oligonucleotide +5). In vivo methylation protection studies showed that Rap1p facilitated the binding of Gcr1p at an appropriately spaced binding site, but not when the binding sites were separated by five nucleotides (39). In this study we used an in vitro system to investigate the requirements for Rap1p-facilitated binding of Gcr1p. For these studies we used the DNA electrophoretic mobility-shift assay to measure and dissect the requirements of Rap1p-facilitated DNA binding of Gcr1p. This system is well suited because it can be used to measure all species of DNA in the reaction.
MATERIALS AND METHODS
Preparation of Gcr1p and Rap1p.
The DNA-binding domain of Gcr1p(631–735) was synthesized in and purified from Escherichia coli as part of a fusion protein between the maltose-binding protein (MBP) and Gcr1p as described previously (9). Coordinates of the Gcr1p DNA-binding domain have been renumbered from 690–844 to 631–735 to reflect the presence of a recently identified intron in GCR1 (40). His-tagged Gcr1p DNA-binding domain was expressed from a derivative of plasmid pET19b in which the sequence encoding the DNA-binding domain for Gcr1p was cloned behind the His-tag at the NdeI site. The fusion protein was purified to near homogeneity by passage over a Ni2+ column. Rap1p and truncated forms were prepared by in vitro transcription and translation in a two-step process as previously described (41).
DNA Electrophoretic Mobility-Shift Assays.
The DNA probes used in this study were described by Drazinic et al. (39) and are shown in Fig. 1A. Oligonucleotide N contained both a Rap1p- and a Gcr1p-binding site, with the native spacing between the sites as found in the PYK1 UAS element. Oligonucleotide +5 had altered spacing between the sites such that the centers of the binding sites were displaced by 5 nucleotides. Oligonucleotide ΔG was identical to oligonucleotide N except that the sequence CTTCC at the core of the Gcr1p-binding site had been deleted. DNA-binding reactions were carried out as described previously (9). Each reaction was carried out in 20 μl of binding buffer [12 mM Hepes, pH 7.5/60 mM KCl/5 mM MgCl2/4 mM Tris⋅HCl/0.6 mM EDTA/0.6 mM DTT/10% (vol/vol) glycerol containing 0.3 μg/μl BSA]. The binding reaction mixtures were electrophoresed through a 5% T (82:1) polyacrylamide gel with 0.5× TBE (1× TBE = 90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3) as running buffer. After electrophoresis, gels were dried, and the positions of free DNA and nucleoprotein complexes were revealed by autoradiography and phosphorimaging.
Figure 1.

Rap1p⋅DNA⋅Gcr1p ternary complex formation requires DNA-binding sites for both Rap1p and Gcr1p. (A) DNA sequence of oligonucleotide probes, N, +5, and ΔG, used in this study. Rap1p- and Gcr1p-binding sites are shown in boldface type. (B) In vitro DNA-binding reactions were carried out under standard conditions as described in the text. Preparations of Rap1p and Gcr1p were allowed to react with the radiolabeled probe DNAs either separately or together as indicated on the figure. Nucleoprotein complexes (Rap1p⋅DNA⋅Gcr1p, Rap1p⋅DNA, Gcr1p⋅DNA) were resolved from free DNA by nondenaturing polyacrylamide gel electrophoresis and were revealed by autoradiography of the dried gel. Lanes 1–4 utilized oligonucleotide N, a probe with native spacing between Rap1p- and Gcr1p-binding sites; lanes 5–8 utilized oligonucleotide +5, a probe with out-of-phase spacing between Rap1p- and Gcr1p-binding sites; and lanes 9–12 utilized oligonucleotide ΔG, a probe with a Rap1p-binding site but not a Gcr1p-binding site.
Quantifying DNA Electrophoretic Mobility-Shift Assays.
The ability and the extent to which Rap1p and truncated forms of it facilitated the binding of Gcr1p were determined by DNA electrophoretic mobility-shift assay. Facilitated binding was assessed in the following manner. Radiolabeled probe DNA was allowed to react with each of the various forms of Rap1p and Gcr1p alone. The probe was also allowed to react with mixtures of Rap1p and Gcr1p together. The various forms of the probe (free DNA, Rap1p⋅DNA, Gcr1p⋅DNA, Rap1p⋅DNA⋅Gcr1p) were then resolved from one another on nondenaturing polyacrylamide gels. The amount of probe in each position of the dried gels was determined by PhosphorImager (Molecular Dynamics). The amount of probe present in the ternary complex bound by both Rap1p and Gcr1p was quantified and compared with the amount expected in the ternary complex if each protein bound the probe independently. If Rap1p and Gcr1p bind the probe independently, as would be the case in the absence of cooperative binding, then the amount of probe in the ternary complex, bound by both proteins, is predicted as the product of the proportions of the probe bound by each protein alone. The fraction of probe bound by each protein alone was determined from the appropriate control reactions. Obtaining observed values greater than expected values indicates cooperative binding between the two proteins.
RESULTS AND DISCUSSION
Formation of Rap1p⋅DNA⋅Gcr1p Ternary Complex Requires a Functional Gcr1p–DNA-Binding Site.
It has been suggested that, under certain circumstances, Gcr1p can be localized to UAS elements in vivo in the absence of Gcr1p–DNA-binding sites by direct protein–protein interactions with Rap1p (42). Thus, we were interested in determining whether we could detect ternary Gcr1p⋅Rap1p⋅DNA complexes by DNA electrophoretic mobility-shift assays in the absence of Gcr1p-binding sites on the probe DNA. Fig. 1 shows the results of a DNA electrophoretic mobility-shift assay using three different probe DNAs. Lanes 1–4 show the results with oligonucleotide N, harboring both Rap1p- and Gcr1-binding sites with native spacing between them as found in the UAS element of the PYK1 gene. With this probe, approximately 3-fold more material was found in the ternary complex in lane 4 than would be expected by Rap1p and Gcr1p each binding independently at their respective binding sites. Lanes 5–8 show the results obtained with oligonucleotide +5, which contains both binding sites, but in which they have been displaced 5 nucleotides from one another. Substantially less material was found in the ternary complex (lane 5) with this probe than would be expected by the independent binding of Rap1p and Gcr1p. Lanes 9–12 show the results with oligonucleotide ΔG, an otherwise identical probe except the Gcr1p site was mutated. With probe ΔG, Rap1p was still capable of binding; however, Gcr1p was unable to bind directly to the probe. Moreover, no ternary complex was detected in lane 12. Thus, the ternary complexes observed in lanes 4 and 5 required functional Gcr1p–DNA-binding sites and were not solely the result of protein–protein interactions between Rap1p and Gcr1p.
Domains of Rap1p Contributing to Facilitated Binding of Gcr1p.
There are two attractive hypotheses for the mechanism(s) by which Rap1p facilitates the binding of Gcr1p (39). According to one hypothesis, protein–protein interactions between Rap1p and Gcr1p stabilize Gcr1p on its binding site. The other hypothesis states that Rap1p-induced DNA bending alters the topology of the adjacent Gcr1p–DNA-binding site, thereby creating a high-affinity binding site for Gcr1p. Identification of domains of Rap1p required for Rap1p-facilitated binding of Gcr1p may provide insight into the mechanism of facilitated binding. Several domains in Rap1p may be involved (Fig. 2A). The DNA-binding domain of Rap1p resides in the middle third of the protein (11) and is distinct from Rap1p’s putative activation domain, which lies toward the carboxyl terminus (18). The DNA-bending domain of Rap1p is associated with a region of the polypeptide in the amino terminus of the molecule (12), although the DNA-binding domain itself is capable of introducing a slight bend in DNA (13). According to the protein–protein interaction model, one might predict that the activation domain of Rap1p may be required for facilitated binding of Gcr1p. Alternatively, according to the bending model, one might predict that the asymmetric DNA-bending domain of Rap1p may be required for facilitated binding of Gcr1p. If both models are operating, then contributions from both the asymmetric bending domain and putative activation domain may be observed.
Figure 2.

Domains at both the amino and carboxyl termini of Rap1p participate in Rap1p-facilitated binding of Gcr1p at appropriately spaced binding sites, but they hinder ternary complex formation when the binding sites are out-of-phase by five nucleotides. (A) Cartoon of the functional domain structure of Rap1p [after Müller et al. (12)]. (B) Cartoon of the functional domain structure of Gcr1p. (C) Truncated forms of Rap1p used in the in vitro DNA-binding studies. (D) Portion of Gcr1p in the MBP-Gcr1p fusion protein used in the DNA-binding studies. (E) DNA-binding studies with truncated versions of Rap1p to map domains in Rap1p that influence the ability of Gcr1p to bind at an appropriately spaced in-phase binding site. In vitro DNA-binding reactions with oligonucleotide N were carried out under standard conditions as described in the text. Preparations of Rap1p and Gcr1p were allowed to react with the radiolabeled probe DNAs either separately (lanes 2, 3, 5, 7, and 9) or together (lanes 4, 6, 8, and 10) as indicated on the figure. Nucleoprotein complexes were resolved from free DNA by nondenaturing polyacrylamide gel electrophoresis and were revealed by autoradiography of the dried gel. (F) DNA-binding studies with truncated versions of Rap1p to map domains in Rap1p that influence the ability of Gcr1p to bind at an adjacent out-of-phase binding site. In vitro DNA-binding reactions with oligonucleotide +5 were carried out under standard conditions as described in the text and for E. ← denotes positions of free DNA or the various bimolecular nucleoprotein complexes containing either Rap1p⋅DNA or Gcr1p⋅DNA as indicated on the right. ★ denotes positions of the ternary complexes present in lanes 4, 6, 8, and 10 of E and F.
To test the hypothesis that domains in addition to the DNA-binding domain of Rap1p (Rap1p(361–596)) are required for facilitated binding of Gcr1p, we subjected Rap1p to molecular dissection. In addition to full-length Rap1p we prepared three truncated forms of Rap1p, shown in Fig. 2C, and tested them in a gel electrophoretic mobility-shift assay for their ability to facilitate the binding of Gcr1p at an appropriately spaced Gcr1p-binding site (Fig. 2E). As a control an identical set of binding studies were carried out with the probe in which the Rap1p- and Gcr1p-binding sites were out of phase with respect to one another (Fig. 2F). An analysis of the data presented in Fig. 2E showed that full-length Rap1p(1–827) and two of the three truncated forms of Rap1p facilitated the binding of Gcr1p. Surprisingly, both Rap1p(1–569) and Rap1p(361–827) facilitated the binding of Gcr1p. However, the DNA-binding domain of Rap1p(361–596) by itself, while able to bind the probe, was unable to facilitate the binding of Gcr1p. These results were in marked contrast to those observed with oligonucleotide +5, in which the Rap1p- and Gcr1p-binding sites were displaced by 5 nucleotides (see Fig. 2F). With this oligonucleotide, Rap1p(1–827), Rap1p(1–596), and Rap1p(361–827) did not facilitate the binding of Gcr1p, they actually hinder the formation of a ternary complex with Gcr1p (lanes 6, 8, and 10). In each case there was less material present in the ternary complex than would be expected on the basis of independent binding of Rap1p and Gcr1p to the oligonucleotide. Unlike the other forms of Rap1p, the DNA-binding domain alone, Rap1p(361–596), did not facilitate or hinder the binding of Gcr1p on oligonucleotides N and +5, respectively.
The results presented in Fig. 2 suggest that both protein–protein interaction and DNA-bending may be contributing to Rap1p’s ability to facilitate the binding of Gcr1p. The observation that molecules of Rap1p that facilitate Gcr1p binding to oligonucleotide N hinder Gcr1p binding to oligonucleotide +5 suggests that when the Rap1p- and Gcr1p-binding sites are displaced by 5 nucleotides, Rap1p either sterically hinders the binding of Gcr1p or places the Gcr1p-binding site in an unfavorable conformation for Gcr1p binding. Consistent with this view is the related observation that Rap1p(361–596), which harbors neither the asymmetric DNA-bending domain nor the putative activation domain, does not facilitate or hinder the binding of Gcr1p on either oligonucleotide tested.
Regardless of the mechanism(s) by which Rap1p facilitates the binding of Gcr1p, the results suggest that multiple domains of Rap1p are involved. The ability of either the amino- or the carboxyl-terminal domain of Rap1p along with its DNA-binding domain to facilitate the DNA binding of Gcr1p suggests that the amino- and carboxyl-terminal domains of Rap1p may be functionally redundant with respect to facilitated binding of Gcr1p. In view of these results, it is interesting to note that Rap1p can function in vivo with deletions that remove much of its amino terminus (between residues 44 and 274) or truncations that remove much of its carboxyl terminus (12, 43); however, RAP1 is an essential gene (8). Although the reason for Rap1p’s essentiality is unknown, mutations that prevent Rap1p from binding DNA are lethal to haploid cells (44). Thus, the ability of Rap1p to bind DNA is essential for its function. The apparent dispensability of Rap1p’s amino or carboxyl terminus in terms of viability may be because they provide partially redundant function to the protein that is needed in addition to its DNA-binding activity for cell viability.
Measurements of Rap1p-Facilitated Binding of Gcr1p.
The degree to which Rap1p facilitates the binding of Gcr1p was determined by comparing the relative differences in the apparent dissociation constants (Kd) obtained for Gcr1p with its binding site to the apparent Kd obtained for Gcr1p with its binding site when Rap1p was bound at an adjacent site. The relative affinities of Rap1p and Gcr1p for oligonucleotide N, or any other oligonucleotide, can be measured through quantitative DNA electrophoretic mobility-shift experiments. The amount of protein needed to shift 50% of the probe is a measure of the apparent Kd when the concentration of the probe is far below the Kd value because of the following relationships (45):
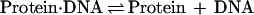 |
1 |
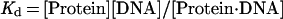 |
2 |
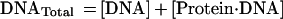 |
3 |
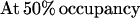 |
4 |
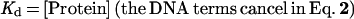 |
When one considers Rap1p-facilitated binding of Gcr1p, two sets of equations operate, one for Gcr1p binding to free DNA and one for Gcr1p binding to Rap1p-bound DNA (Rap1p⋅DNA), but in each case the Kd relationship of Eq. 4 holds, namely at 50% occupancy Kd = [Gcr1p]. The two sets of equations are shown below:
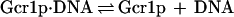 |
5 |
vs.
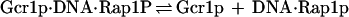 |
5a |
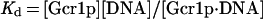 |
6 |
vs.
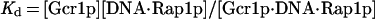 |
6a |
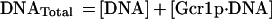 |
7 |
vs.
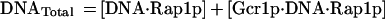 |
7a |
If Rap1p facilitates the binding of Gcr1p, then Gcr1p will show a higher affinity for the DNA substrate of Eq. 5a (DNA⋅Rap1p) than for the DNA substrate of Eq. 5 (DNA). Accordingly, Gcr1p will tend to bind Rap1p-bound DNA preferentially over free DNA. Thus a ternary complex of Rap1p⋅DNA⋅Gcr1p would form preferentially over Gcr1p⋅DNA complex. DNA electrophoretic mobility-shift assays are ideally suited for this analysis because all species of the DNA probe can be detected and quantified, thereby allowing Gcr1p’s affinity for free DNA (DNA) and Rap1p-bound DNA (DNA⋅Rap1p) to be assessed simultaneously in the same assay.
In Fig. 3 we show a representative series of experiments where we measured the degree to which Rap1p and truncated forms of it facilitate the binding of Gcr1p(631–785). In each case, the probe DNA, oligonucleotide N, was incubated with a constant amount of Rap1p and titrated with increasing amounts of Gcr1p(631–785). The various species of probe DNA were then resolved from one another by gel electrophoresis. All species of the probe were resolved on the gel. Once again we observed that Rap1p(1–827), Rap1p(1–569), and Rap1p(361–827) were capable of facilitating the binding of Gcr1p; in each case the ternary complex consisting of Rap1p⋅DNA⋅Gcr1p was obtained with lower concentrations of Gcr1p than the Gcr1p⋅DNA complex (see Fig. 3). From these assays, and replicates not shown, the affinity of Gcr1p(631–785) for Rap1p(1–827)-bound DNA (Rap1p⋅DNA) was approximately 4-fold greater than it was for identical DNA that was not bound by Rap1p. Fig. 3D shows that unlike the other versions of Rap1p, the DNA-binding domain of Rap1p(361–596) did not facilitate the binding of Gcr1p. This result again indicates that domains in addition to the DNA-binding domain of Rap1p are required to facilitate the binding of Gcr1p.
Figure 3.

Gcr1p titration series demonstrating Rap1p-facilitated binding of Gcr1p by Rap1p(1–827), Rap1p(1–596), and Rap1p(361–827) but not by Rap1p(361–596). (A) Titration of Gcr1p(631–785) in vitro binding reactions with Rap1p(1–827). (B) Titration of Gcr1p(631–785) in vitro binding reactions with Rap1p(1–596). (C) Titration of Gcr1p(631–785) in vitro binding reactions with Rap1p(361–827). (D) Titration of Gcr1p(631–785) in vitro binding reactions with Rap1p(361–596). The in vitro DNA-binding reactions were carried out under standard conditions with oligonucleotide N as described in the text. Preparations of Gcr1p and Rap1p were allowed to react with the radiolabeled probe DNAs either separately (lanes 12 and 13, respectively, in each gel) or together (lanes 2–11 in each gel) as indicated on the figure. The concentration of Rap1p was held constant in the binding reactions and the concentration of Gcr1p was titrated by using a 2-fold dilution series from a nearly homogeneous purified stock of MBP-Gcr1p. Nucleoprotein complexes were resolved from free DNA by nondenaturing polyacrylamide gel electrophoresis and were revealed by autoradiography of the dried gel. ← denotes positions of the various species of the probe as indicated on the figure.
In addition to the experiments shown in Fig. 3, we carried out similar experiments where we used His-tagged versions of Gcr1p DNA-binding domain to ensure that the results we observed were not artifacts of the MBP moiety fused to the Gcr1p DNA-binding domain. Visual inspection of the results obtained with the His-tagged versions of Gcr1p(631–785) were essentially identical to those obtained with MBP-Gcr1p(631–785) (data not shown). Thus, it is unlikely that the increased affinity that Gcr1p displays for Rap1p-bound DNA in vitro was an artifact of the fusion and not because of intrinsic properties of Rap1p and Gcr1p.
If domains of Gcr1p in addition to its DNA-binding domain (Gcr1p(631–785)) participate in cooperative binding reactions with Rap1p, then a greater degree of facilitated binding would be expected with full-length Gcr1p than that observed in the experiments of Fig. 3. To address this possibility we carried out a similar series of experiments with full-length versions of Gcr1p. Inspection of Fig. 4 shows that Rap1p facilitates the binding of full-length Gcr1p(1–785) to the same extent that it facilitates the binding of Gcr1p(631–785). Thus, domains of Gcr1p outside of its DNA-binding domain do not appear to be involved in Rap1p-facilitated binding of Gcr1p.
Figure 4.

Gcr1p titration series demonstrating Rap1p-facilitated binding of full-length Gcr1p. Titrations of Gcr1p(1–785) in vitro binding reactions with Rap1p(1–827) were carried out as described in the text and in the legend to Fig. 3.
In vivo expression studies have shown that there is a strong synergism between Rap1p- and Gcr1-binding sites (3, 5, 7, 20, 27). In vivo footprinting experiments suggest that Rap1p binding is essential for occupancy of adjacent Gcr1p-binding sites (39). Yet, in vitro measurements of Rap1p-facilitated binding of Gcr1p demonstrated that Rap1p increased the affinity of the Gcr1p DNA-binding site by a relatively modest 4-fold. One variable affecting the occupancy state of the Gcr1p-binding site in vivo is the concentration of Gcr1p itself inside the cell. At low concentrations, the fold difference in Gcr1p’s binding to Rap1p-bound DNA versus free DNA is substantially greater than 4-fold. The possibility also exists that there may be other proteins in the cell that contribute to Rap1p-facilitated binding of Gcr1p that were not present in our in vitro binding assays. The degree to which Rap1p facilitates the binding of Gcr1p is similar to the degree to which the human SWI/SNF complex facilitates the binding of the Gal4p DNA-binding domain on nucleosome cores (46).
Other Binding Partners?
We have searched the yeast genome (47) for matches to the degenerate consensus binding sites proposed for Rap1p (7) and Gcr1p (35), and we have found 315 and 837 matches, respectively. In only a few cases are Rap1p and Gcr1p sites located adjacent to one another, primarily at glycolytic enzyme gene UAS elements. We have previously shown, in the context of a glycolytic enzyme gene UAS element, that unless Rap1p is bound at an adjacent site Gcr1p is unable to bind its site in vivo (39). Thus, the number of bona fide Gcr1p-binding sites is likely to be substantially smaller than the number identified on the basis of sequence identity to the degenerate consensus sequence considering Gcr1p’s DNA-binding characteristics. However, the numbers suggest the intriguing possibility that both Gcr1p and Rap1p have other binding partners with whom they interact. Consistent with this idea, Uemura et al. (48) recently presented genetic evidence that Gcr1p itself may be able to facilitate its own binding at appropriately spaced Gcr1p-binding sites in vivo. In yeast we envision that Rap1p plays a role similar to that of Max of higher cells in that it has the capacity of interacting with several different proteins to mediate either repression or activation.
Conclusions.
We have demonstrated that domains of Rap1p in addition to its DNA-binding domain are required to facilitate the binding of Gcr1p at an adjacent binding site. Both Rap1p(1–569) and Rap1p(361–827) in addition to Rap1p(1–827) were competent to facilitate the binding of Gcr1p. This observation indicates that domains on either side of Rap1p’s DNA-binding domain participate in facilitated DNA-binding reactions with Gcr1p. The precise mechanistic details of how Rap1p facilitates the binding of Gcr1p are yet to be elucidated but are likely to be shared with other proteins.
Acknowledgments
This work was supported by Grant MCB-9404721 from the National Science Foundation.
ABBREVIATIONS
- UAS
upstream activating sequence
- Rap1p
repressor activator protein 1
- Gcr1p
glycolysis regulatory protein 1
- MBP
maltose-binding protein
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Chambers A, Stanway C, Tsang J S, Henry Y, Kingsman A J, Kingsman S M. Nucleic Acids Res. 1990;18:5393–5399. doi: 10.1093/nar/18.18.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brindle P K, Holland J P, Willett C E, Innis M A, Holland M J. Mol Cell Biol. 1990;10:4872–4885. doi: 10.1128/mcb.10.9.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott E W, Baker H V. Mol Cell Biol. 1993;13:543–550. doi: 10.1128/mcb.13.1.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willett C E, Gelfman C M, Holland M J. Mol Cell Biol. 1993;13:2623–2633. doi: 10.1128/mcb.13.4.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bitter G A, Chang K K, Egan K M. Mol Gen Genet. 1991;231:22–32. doi: 10.1007/BF00293817. [DOI] [PubMed] [Google Scholar]
- 6.Nishizawa M, Araki R, Teranishi Y. Mol Cell Biol. 1989;9:442–451. doi: 10.1128/mcb.9.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchman A R, Lue N F, Kornberg R D. Mol Cell Biol. 1988;8:5086–5099. doi: 10.1128/mcb.8.12.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shore D, Nasmyth K. Cell. 1987;51:721–732. doi: 10.1016/0092-8674(87)90095-x. [DOI] [PubMed] [Google Scholar]
- 9.Huie M A, Baker H V. Yeast. 1996;12:307–317. doi: 10.1002/(sici)1097-0061(19960330)12:4<307::aid-yea912>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 10.Vignais M L, Sentenac A. J Biol Chem. 1989;264:8463–8466. [PubMed] [Google Scholar]
- 11.Henry Y A, Chambers A, Tsang J S, Kingsman A J, Kingsman S M. Nucleic Acids Res. 1990;18:2617–2623. doi: 10.1093/nar/18.9.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Müller T, Gilson E, Schmidt R, Giraldo R, Sogo J, Gross H, Gasser S M. J Struct Biol. 1994;113:1–12. doi: 10.1006/jsbi.1994.1027. [DOI] [PubMed] [Google Scholar]
- 13.Konig P, Giraldo R, Chapman L, Rhodes D. Cell. 1996;85:125–136. doi: 10.1016/s0092-8674(00)81088-0. [DOI] [PubMed] [Google Scholar]
- 14.Sussel L, Shore D. Proc Natl Acad Sci USA. 1991;88:7749–7753. doi: 10.1073/pnas.88.17.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, Mao X, Lustig A J. Genetics. 1994;138:1025–1040. doi: 10.1093/genetics/138.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moretti P, Freeman K, Coodly L, Shore D. Genes Dev. 1994;8:2257–2269. doi: 10.1101/gad.8.19.2257. [DOI] [PubMed] [Google Scholar]
- 17.Hardy C F, Sussel L, Shore D. Genes Dev. 1992;6:801–814. doi: 10.1101/gad.6.5.801. [DOI] [PubMed] [Google Scholar]
- 18.Hardy C F, Balderes D, Shore D. Mol Cell Biol. 1992;12:1209–1217. doi: 10.1128/mcb.12.3.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butler G, Dawes I W, McConnell D J. Mol Gen Genet. 1990;223:449–456. doi: 10.1007/BF00264453. [DOI] [PubMed] [Google Scholar]
- 20.Chambers A, Tsang J S, Stanway C, Kingsman A J, Kingsman S M. Mol Cell Biol. 1989;9:5516–5524. doi: 10.1128/mcb.9.12.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Machida M, Jigami Y, Tanaka H. Eur J Biochem. 1989;184:305–311. doi: 10.1111/j.1432-1033.1989.tb15020.x. [DOI] [PubMed] [Google Scholar]
- 22.McNeil J B, Dykshoorn P, Huy J N, Small S. Curr Genet. 1990;18:405–412. doi: 10.1007/BF00309909. [DOI] [PubMed] [Google Scholar]
- 23.Scott E W, Allison H E, Baker H V. Nucleic Acids Res. 1990;18:7099–7107. doi: 10.1093/nar/18.23.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goncalves P M, Griffioen G, Minnee R, Bosma M, Kraakman L S, Mager W H, Planta R J. Nucleic Acids Res. 1995;23:1475–1480. doi: 10.1093/nar/23.9.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Devlin C, Tice Baldwin K, Shore D, Arndt K T. Mol Cell Biol. 1991;11:3642–3651. doi: 10.1128/mcb.11.7.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woudt L P, Smit A B, Mager W H, Planta R J. EMBO J. 1986;5:1037–1040. doi: 10.1002/j.1460-2075.1986.tb04319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogden J E, Stanway C, Kim S, Mellor J, Kingsman A J, Kingsman S M. Mol Cell Biol. 1986;6:4335–4343. doi: 10.1128/mcb.6.12.4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clifton D, Fraenkel D G. J Biol Chem. 1981;256:13074–13078. [PubMed] [Google Scholar]
- 29.Ciriacy M, Freidel K, Lohning C. Curr Genet. 1991;20:441–448. doi: 10.1007/BF00334769. [DOI] [PubMed] [Google Scholar]
- 30.Turkel S, Liao X B, Farabaugh P J. Yeast. 1997;13:917–930. doi: 10.1002/(SICI)1097-0061(199708)13:10<917::AID-YEA148>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 31.Clifton D, Weinstock S B, Fraenkel D G. Genetics. 1978;88:1–11. doi: 10.1093/genetics/88.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker H V. Mol Cell Biol. 1986;6:3774–3784. doi: 10.1128/mcb.6.11.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holland M J, Yokoi T, Holland J P, Myambo K, Innis M A. Mol Cell Biol. 1987;7:813–820. doi: 10.1128/mcb.7.2.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodicio R, Heinisch J J, Hollenberg C P. Gene. 1993;125:125–133. doi: 10.1016/0378-1119(93)90319-x. [DOI] [PubMed] [Google Scholar]
- 35.Huie M A, Scott E W, Drazinic C M, López M C, Hornstra I K, Yang T P, Baker H V. Mol Cell Biol. 1992;12:2690–2700. doi: 10.1128/mcb.12.6.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanway C A, Gibbs J M, Kearsey S E, López M C, Baker H V. Mol Gen Genet. 1994;243:207–214. doi: 10.1007/BF00280318. [DOI] [PubMed] [Google Scholar]
- 37.Chambers A, Stanway C, Kingsman A J, Kingsman S M. Nucleic Acids Res. 1988;16:8245–8260. doi: 10.1093/nar/16.17.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanway C A, Chambers A, Kingsman A J, Kingsman S M. Nucleic Acids Res. 1989;17:9205–9218. doi: 10.1093/nar/17.22.9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drazinic C M, Smerage J B, López M C, Baker H V. Mol Cell Biol. 1996;16:3187–3196. doi: 10.1128/mcb.16.6.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tornow J, Santangelo G. Genetics. 1994;138:973–974. doi: 10.1093/genetics/138.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker H V. Proc Natl Acad Sci USA. 1991;88:9443–9447. doi: 10.1073/pnas.88.21.9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tornow J, Zeng X, Gao W, Santangelo G M. EMBO J. 1993;12:2431–2437. doi: 10.1002/j.1460-2075.1993.tb05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kyrion G, Boakye K A, Lustig A J. Mol Cell Biol. 1992;12:5159–5173. doi: 10.1128/mcb.12.11.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurtz S, Shore D. Genes Dev. 1991;5:5264–5268. doi: 10.1101/gad.5.4.616. [DOI] [PubMed] [Google Scholar]
- 45.Riggs A D, Suzuki H, Bourgeoss S. J Mol Biol. 1970;48:67–83. doi: 10.1016/0022-2836(70)90219-6. [DOI] [PubMed] [Google Scholar]
- 46.Schnitzler G, Sif S, Kingston R E. Cell. 1998;94:17–27. doi: 10.1016/s0092-8674(00)81217-9. [DOI] [PubMed] [Google Scholar]
- 47.Cherry J M, Adler C, Ball C, Chervitz S A, Dwight S S, Hester E T, Jia Y, Juvik G, Roe T, Schroeder M, Weng S, Botstein D. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uemura H, Koshio M, Inoue Y, López M C, Baker H V. Genetics. 1997;147:521–532. doi: 10.1093/genetics/147.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]