

SUMMARY
HCN1 hyperpolarization-activated cation channels act as an inhibitory constraint of both spatial learning and synaptic integration and long-term plasticity in the distal dendrites of hippocampal CA1 pyramidal neurons. However, as HCN1 channels provide an excitatory current, the mechanism of their inhibitory action remains unclear. Here we report that HCN1 channels also constrain CA1 distal dendritic Ca2+ spikes, which have been implicated in the induction of LTP at distal excitatory synapses. Our experimental and computational results indicate that HCN1 channels provide both an active shunt conductance that decreases the temporal integration of distal EPSPs and a tonic depolarizing current that increases resting inactivation of T-type and N-type voltage-gated Ca2+ channels, which contribute to the Ca2+ spikes. This dual mechanism may provide a general means by which HCN channels regulate dendritic excitability.
INTRODUCTION
Hippocampal activity-dependent long-term synaptic plasticity is widely thought to be a key cellular substrate for spatial learning and memory (Morris et al., 2003). Due to the cooperative and associative nature of such forms of plasticity, the individual postsynaptic potentials from a large number of synaptic inputs must be integrated by neuronal dendrites to elicit a postsynaptic response sufficient to induce plastic changes. Over the past several years it has become clear that dendrites are endowed with a wide array of voltage-gated ion channels that shape the integration of synaptic inputs and enable the active processing of synaptic information (London and Hausser, ■ ■ ■ ; Magee and Johnston, ■ ■ ■). Although much is now known about the molecular mechanisms underlying synaptic plasticity, we understand less about how the active integrative properties of neuronal dendrites influence the induction of synaptic plasticity to regulate learning and memory.
Here we focus on the role of the hyperpolarization-activated cation current (Ih), encoded by the HCN channel gene family (HCN1-4; Robinson and Siegelbaum, 2003), in the regulation of dendritic excitability and long-term synaptic plasticity in hippocampal CA1 pyramidal neurons. HCN1 is highly expressed in the apical dendrites of the CA1 neurons in a gradient of increasing density with increasing distance from the soma (Lorincz et al., 2002; Magee, 1998; Notomi and Shigemoto, 2004; Santoro et al., 2000). Mice with a forebrain-restricted deletion of HCN1 show an increase in spatial learning and an increase in temporal integration and long-term potentiation (LTP) of EPSPs generated at the perforant path (PP) inputs to the CA1 neurons (Nolan et al., 2004). These inputs, which arise from layer 3 neurons of entorhinal cortex, terminate on the distal CA1 dendrites in stratum lacunosum moleculare (SLM), the site of greatest HCN1 channel density. Thus, HCN1 channels exert an inhibitory constraint on dendritic integration and synaptic plasticity at the PP inputs to CA1 pyramidal neurons and constrain hippocampal-dependent spatial learning.
The inhibitory effect of HCN1 revealed by its genetic deletion is consistent with previous studies on the role of Ih in dendritic integration. Application of the organic Ih antagonist ZD7288 enhances the magnitude of the voltage change during an EPSP and slows the time course of EPSP decay, increasing temporal integration in both CA1 pyramidal neurons (Magee, 1998, ■ ■ ■) and neocortical layer 5 pyramidal cells (Stuart and Spruston, ■ ■ ■ ;Williams and Stuart, ■ ■ ■ ; Berger et al., ■ ■ ■). Blockade of Ih also facilitates the firing of local spikes in CA1 dendrites in stratum radiatum (Magee, ■ ■ ■ ; Poolos et al., 2002) and lowers the threshold for the activation of dendritic Ca2+ spikes triggered by back-propagating action potentials in layer 5 neurons (Berger et al., ■ ■ ■). Conversely, upregulation of Ih in CA1 neurons by the anticonvulsant lamotrigine (Poolos et al., 2002) and in entorhinal cortex neurons by dopamine (Rosenkranz and Johnston, 2006) inhibits firing of dendritic action potentials.
Previous studies have ascribed the inhibitory actions of Ih to its being partially active at the resting potential, providing a shunt conductance that decreases input resistance, membrane time constant, and temporal integration, which decreases the depolarization during an EPSP (Magee, 1998; Stuart and Spruston, ■ ■ ■ ; Poolos et al., 2002). However, since the Ih reversal potential (~−30 mV) is positive to the threshold for spike firing (−50 to −40 mV), Ih generates an excitatory current at subthreshold voltages. As a result, blockade of Ih hyperpolarizes the resting membrane by 5–10 mV, which counteracts any increase in the magnitude of a subthreshold EPSP due to the increase in input resistance. In a simple model containing only Ih, a leak conductance, and an excitatory synaptic input, the absolute peak EPSP voltage should actually be closer to threshold in the presence of Ih than in its absence. Indeed, the excitatory effect of Ih underlies its contribution to spontaneous rhythmic firing in both the heart and in central neurons (Robinson and Siegelbaum, 2003). Thus, despite the general finding that Ih exerts a potent inhibitory control on dendritic excitability, the mechanism underlying this inhibitory action and its constraint on the induction of LTP remains unclear.
In this study we have investigated the possibility that HCN channels produce their inhibitory effects on synaptic plasticity by nonlinear interactions with other dendritic voltage-gated channels. In particular, we have used Ca2+ imaging to investigate the effects of Ih on local regenerative calcium spikes in CA1 neuron distal dendrites (Golding et al., 2002; Wei et al., 2001), as such spikes have been suggested to be important for the induction of PP LTP (Golding et al., 2002). We find that either genetic deletion of HCN1 or pharmacological blockade of Ih enhances the amplitude and duration of distal nonlinear dendritic Ca2+ events evoked by a brief tetanic burst of perforant path synaptic stimulation. Our computational and experimental data indicate that this effect is due to the combined action of the reduction in Ih to hyperpolarize the membrane and enhance dendritic integration through an increase in input resistance. The hyperpolarization reduces resting inactivation of T-type and N-type voltage-gated calcium channels (VGCCs), which contribute to Ca2+ influx during the dendritic spikes; the increase in input resistance increases the amplitude of the EPSP, helping to offset the inhibitory effects of hyperpolarization. This mechanism is likely to contribute to the inhibitory effects of Ih on dendritic excitability seen in a wide variety of neurons. Moreover, regulation of HCN channel function by modulatory transmitters may provide an important physiological mechanism to dynamically control Ca2+-dependent processes at distal dendrites.
RESULTS
Whole-cell recordings were obtained from CA1 pyramidal neurons in acute hippocampal slices from wild-type (WT) and HCN1 KO (−/−) mice (Nolan et al., ■ ■ ■). Neurons were loaded with a low-affinity Ca2+-indicator dye (Oregon Green BAPTA-5N) and a Ca2+-independent dye (Alexa Fluor 594) to monitor dendritic structure (Figure 1A; Experimental Procedures). Local PP inputs were activated by a glass bipolar stimulating patch electrode placed within 50–100 μm of the targeted branch under visual guidance (Figure 1A). The somatic voltage response was recorded under current-clamp conditions, with inhibitory synaptic transmission blocked using both GABAA and GABAB receptor antagonists. At the same time we used two-photon microscopy to measure the Ca2+ signals in the distal CA1 dendrites in stratum lacunosum-moleculare (SLM), at least 50 μm past the main branching point of the distal dendritic tuft (>400 μm from the soma).
Figure 1. Burst Stimulation of PP Inputs Elicits Nonlinear Distal Ca2+ Events.
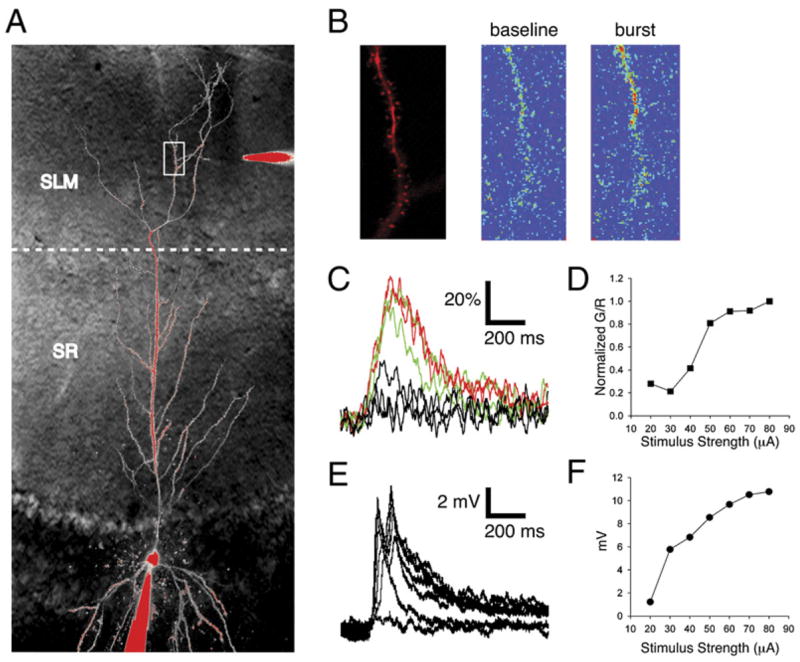
(A) Schematic of experiment. Whole-cell recordings of CA1 pyramidal neurons were obtained using a patch pipette (bottom of image) filled with a Ca2+-independent red fluorescent dye (25 μM Alexa 594; signal R) and a Ca2+-indicator green fluorescent dye (500 μM OGB-5N; signal G). An extracellular stimulating electrode (top of image) filled with Alexa 594 was placed ~50 μm from a dendrite of interest (white box) in stratum lacunosum-moleculare (SLM) under two-photon visual guidance.
(B) Visualization of SLM dendrite segment using a 2D scan. (Left) Image of dendrite morphology. (Right) Ca2+-sensitive fluorescence before (left) and during (right) a Ca2+ spike induced by synaptic stimulation (ten stimuli at 100 Hz). Pseudocolor image in which warmer colors indicate brighter Ca2+ dye fluorescence. Differences in resting fluorescence reflect degree to which dendrite region is in focal plane.
(C) Ca2+ signal transients with burst stimulation using 40, 50, and 60 μA currents measured in linescan mode through dendritic shaft, expressed as percent change in G/R (defined as ΔS/S0 × 100%, where S = G/R; see Experimental Procedures).
(D) Plot of normalized peak Ca2+ signal versus stimulus current. Distal Ca2+ signals show a nonlinear sigmoidal dependence on stimulus current.
(E) Voltage responses to PP burst stimulation corresponding to Ca2+ signals in panel (D).
(F) Plot of peak depolarization (from panel [E]) versus stimulus current.
Burst Stimulation of PP Inputs Elicit Nonlinear Dendritic Calcium Events in SLM Dendrites
We first characterized the Ca2+ events induced in distal dendrites in response to a brief burst of PP synaptic stimulation (ten stimuli at 100 Hz) similar to that used during induction of PP LTP (Nolan et al., 2004). The burst stimulus elicited a large transient increase in the Ca2+ fluorescence signal in a continuous segment of SLM dendrite during 2D scanning (~8 Hz) (Figure 1B). Ca2+ increases were observed in both spines and dendrite shafts. However, segments proximal to the active portion of a dendrite or on adjacent branches showed little or no Ca2+ increase, indicating that the Ca2+ event was localized to the distal dendritic branch.
Previous studies in rat hippocampal slices found that local glutamate application to distal CA1 neuron dendrites (Cai et al., 2004; Wei et al., 2001) or burst stimulation of distal synapses (Golding et al., 2002) can elicit local dendritic Ca2+ action potentials. To determine whether the signals we observed in mouse hippocampal slices also represented dendritic Ca2+ spikes, we asked whether the responses displayed properties consistent with regenerative activity. The peak Ca2+ signal in the distal dendritic shaft (measured by the change in Oregon Green/Alexa 594 fluorescence ratio, normalized to baseline, ΔS/S0; see Experimental Procedures) was quantified on a fast timescale using linescan mode (500 Hz sampling). There was a steep sigmoidal relationship between current stimulus intensity and peak fluorescence change (Figures 1C and 1D), indicative of a nonlinear regenerative response. At low stimulus intensities (typically <40 μA) there was little dendritic fluorescence change despite a measurable somatic depolarization (Figure 1E), consistent with the need for recruitment of a threshold number of distal inputs. The peak Ca2+ change reached a maximum value of 100.4% ± 8.9% at high stimulus intensities (Figure 1D; n = 22). In contrast, the peak somatic voltage response continued to increase with increasing stimulus strength (Figure 1F), perhaps reflecting the further recruitment of synapses and/or Ca2+ spikes at different dendritic branches.
The Ca2+ events had a remarkably long duration, with a mean half-width of 315.5 ± 23.6 ms. Often a plateau phase was observed that was followed by a faster decay to baseline, with an 80%–20% decay time of 258.8 ± 31.7 ms (Figure 2A, black). In contrast, the Ca2+ signals rose more rapidly (77.8 ± 4.84 ms; 20%–80% rise time), comparable to the duration of the stimulus burst. The time course of these Ca2+ signals is similar to that of the distal Ca2+ spikes observed in rat slice cultures (Cai et al., 2004; Wei et al., 2001) but is much longer than the dendritic Ca2+ spikes observed in the distal apical trunk of CA1 neurons in stratum radiatum (Golding et al., 2002; Schiller et al., 1997). Such long events may reflect unique properties of voltage-gated channels at more terminal apical dendrites (Cai et al., 2004) and/or the large NMDA/AMPA receptor ratio found at perforant path synapses (Nicholson et al., 2006; Otmakhova et al., 2002).
Figure 2. HCN1 Deletion Increases Magnitude and Prolongs Duration of Distal Ca2+ Events.
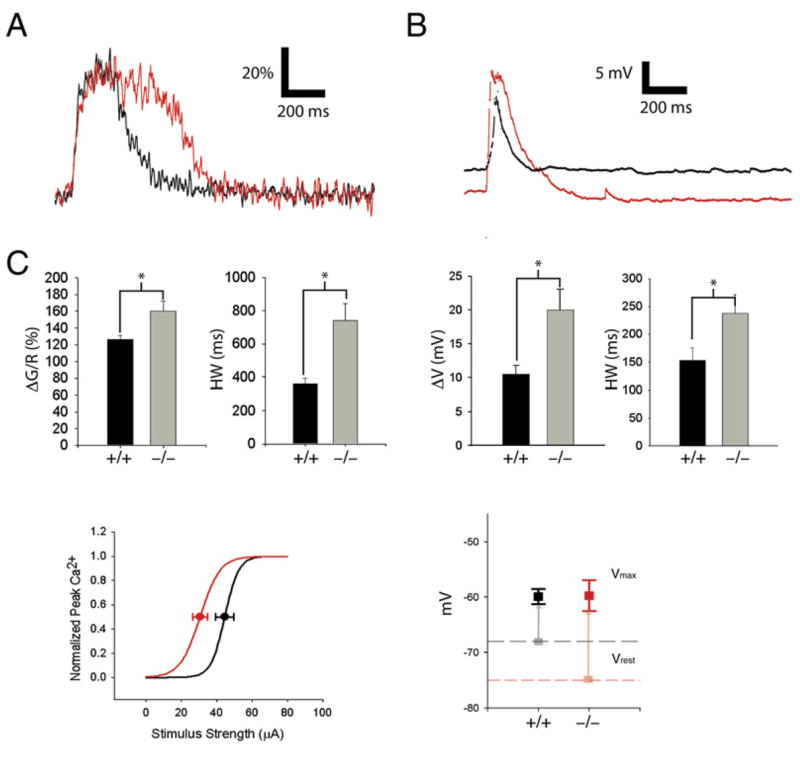
(A) Distal Ca2+ transient in SLM dendrites in HCN1 knockout mouse (KO; red trace) and wild-type littermate (WT; black). Vertical scale bar represents fluorescence signal (ΔS/S0 × 100%).
(B) Somatic voltage in response to PP burst stimulation in KO and WT mice. Note that resting potential in KO mice was hyperpolarized relative to WT mice by 7 mV.
(C) Mean peak Ca2+ signal and duration in KO mice (−/−) versus WT littermates (+/+). (ΔS/S0: WT = 131.8% ± 8.5%, n = 7; KO = 174.8 ± 15.0, n = 7; p < 0.03; unpaired t test; Half-width: WT = 340 ± 33.7 ms; KO: HW = 768 ± 93.3 ms; p < 0.01).
(D) Mean somatic voltage responses in KO versus WT mice. Peak depolarization (ΔV): WT = 8.6 ± 0.7 mV; KO = 16.3 ± 1.9 mV. Half-width: WT = 224.8 ± 21.9 ms; KO = 367.9 ± 52.3 ms. (E) Effect of HCN1 deletion on peak Ca2+ response versus stimulus current relation. Average sigmoid relationship between peak Ca2+ and stimulus strength in KO (red) and WT (black) littermates from fits of Boltzmann relation to individual curves. The KO mice exhibit a slight left-shifted half-maximal stimulus value (I1/2: WT = 45.2 ± 3.0 μA [n = 5]; KO = 35.2 ± 3.8 μA [n = 5]; p = 0.075). The slopes (k) of the relationships were not significantly different (k: WT = 3.9 ± 1.2; KO = 5.6 ± 1.9; p = 0.45).
(F) Effect of HCN1 deletion on resting potential (light squares, dashed lines) and peak EPSP voltage (solid squares). Resting potentials (Vrest) were more negative in KO mice relative to WT mice (WT: Vrest = −68.1 ± 1.3 mV, n = 6; KO: Vrest = −74.9 ± 2.4 mV, n = 6; p < 0.05). The peak potential during EPSP elicited by PP burst stimulation (Vpeak) was similar in WT and KO neurons (WT: Vpeak = −59.9 ± 1.4 mV [n = 6]; KO: Vpeak = −59.7 ± 2.8 mV [n = 6]; p = 0.96). Vertical lines indicate the magnitude of depolarization due to the burst stimulus.
Given the long duration and large amplitude of these Ca2+ signals, it was important to verify that the dye was not saturated. We therefore measured the maximal Ca2+ signal under saturating conditions in response to steady-state bath application of the Ca2+ ionophore ionomycin in the presence of 2 mM external Ca2+ (Figure S1 available online). The peak Ca2+ signal elicited by a synaptic burst was equal to 44.9% ± 7.9% of the maximum signal with ionomycin, indicating that slightly less than half of the dye molecules were complexed with Ca2+. Based on our measured value for the Kd of the dye of 13.4 μM (Figure S1), we calculate that the fluorescence change elicited by a synaptic burst corresponded to a peak free [Ca2+] value of ~11 μM, a postsynaptic level sufficient for induction of LTP (Yang et al., 1999).
Despite the relatively strong synaptic stimulation, the somatic voltage responses were usually subthreshold (Figure 1E, F), with axonal spiking observed in only 8 out of 22 cells. On average, the absolute somatic depolarization reached 12.0 ± 0.8 mV, with a fast rise (60.14 ± 7.3 ms) and slower decay (80%–20%) time (490 ± 40.7 ms). These results are consistent with previous findings that SLM inputs are typically weak and inefficient at driving somatic spikes (Golding and Spruston, 1998; Jarsky et al., 2005) but can elicit long-lasting distal Ca2+ spikes (Cai et al., 2004; Wei et al., 2001). It is likely that the local depolarization in the distal dendrite is several-fold greater than that observed in the soma and sufficient for triggering regenerative activity, given the attenuation of voltage signals along apical CA1 dendrites (Golding et al., 2005).
Deletion of HCN1 Enhances Both Peak Amplitude and Duration of Distal Dendritic Ca2+ Events
To examine the influence of HCN1 on distal dendritic excitability, we compared the dendritic Ca2+ events in HCN1 knockout (KO) mice to those in wild-type (WT) littermates. Similar to previous results (Nolan et al., 2004), we found that the somatic resting potential of the KO mice (−74.9 ± 2.4 mV, n = 6) was significantly more negative than that of wild-type mice (−68.1 ± 1.3 mV, n = 6). Despite this hyperpolarization, PP stimulation elicited a Ca2+ transient in the distal dendrites of the KO mice that was both larger in peak amplitude and longer in duration than the corresponding signals from WT littermates (Figures 2A and 2C).
Deletion of HCN1 increased the peak Ca2+ signal (ΔS/S0) by ~30%, from 131.8% ± 8.5% (n = 7) in WT mice to 174.8% ± 15.0% (n = 7) in KO mice (p < 0.03, Student’s t test). The effect on duration was even more dramatic, with half-width increasing over 2-fold, from 340 ± 33.7 ms in the WT mice to 768 ± 93.3 ms in the mutants (p < 0.01). The somatic voltage responses to PP burst stimulation displayed a similar trend (Figures 2B and 2D), with the KO mice exhibiting a larger peak depolarization (16.3 ± 1.9 mV) and longer half-width (367.9 ± 52.3 ms) compared to the values in WT mice (8.6 ± 0.7 mV and 224.8 ± 21.9 ms). There was no significant difference in either Ca2+ rise times (WT: 51 ± 4.7 ms versus KO: 68 ± 8 ms; p > 0.05) or 80%–20% decay times (WT: 300 ± 38.7 ms, KO: 442.3 ± 82.8 ms; p > 0.05) between genotypes, suggesting that the prolonged time course of the distal events was not due to altered Ca2+ buffering and/or extrusion.
The similarity of decay times of the Ca2+ transients indicates that the major effect of the KO was to prolong the duration of the plateau phase and increase its amplitude. Based on the dye Kd, the 30% increase in peak fluorescence change upon HCN1 deletion represents a 70% increase in peak [Ca2+], to a value of 18.9 μM, consistent with the increase in PP LTP in the HCN1 KO mice (Nolan et al., 2004).
To determine whether deletion of HCN1 lowers the threshold current for generating Ca2+ spikes, we compared the relationship between peak Ca2+ and stimulus intensity in WT versus KO mice (Figure 2E). HCN1 deletion showed a trend to produce a small decrease in the stimulus current needed to elicit distal Ca2+ events, with KO mice exhibiting a half-maximum stimulus current (I1/2 = 35.2 ± 3.8 μA, n = 5) that was ~22% lower than the value in WT mice (I1/2 = 45.2 ± 3.0 μA, n = 5), although the difference did not attain statistical significance (p = 0.075). The peak somatic voltage during the burst EPSP was also relatively unchanged by HCN1 deletion (Figure 2F; peak voltage = −59.9 ± 1.4 mV in WT versus −59.7 ± 2.8 mV in KO mice; p = 0.96), consistent with a relatively unchanged threshold. The lack of change in absolute peak EPSP voltage is likely due to the offsetting effects of enhanced integration (due to the increase in input resistance) versus membrane hyperpolarization upon HCN1 deletion.
Pharmacological Blockade of Ih Enhances Distal Dendritic Calcium Events Specifically in the Perforant Path
The dramatic prolongation and enhancement of the distal Ca2+ transients in the HCN1 KO mice is surprising given that the HCN1 channels generate a depolarizing current and, moreover, should rapidly shut off within ~20–50 ms during depolarizing events (Magee, 1998). Since gene deletions can result in reactive changes in expression of other gene products, we tested the effects of acute pharmacological blockade of Ih in WT mice with the organic antagonist ZD7288. To minimize nonspecific actions of this compound on synaptic transmission (Chen, 2004; Chevaleyre and Castillo, 2002) and other channels (Felix et al., 2003), we used relatively low concentrations of drug (10 μM) and short times of exposure (10–15 min) that were just sufficient to eliminate the characteristic Ih-dependent depolarizing sag in voltage in response to a somatic hyperpolarizing current step (Figure 3A).
Figure 3. Pharmacological Blockade of Ih with ZD7288 Increases Amplitude and Duration of Distal Ca2+ Events.
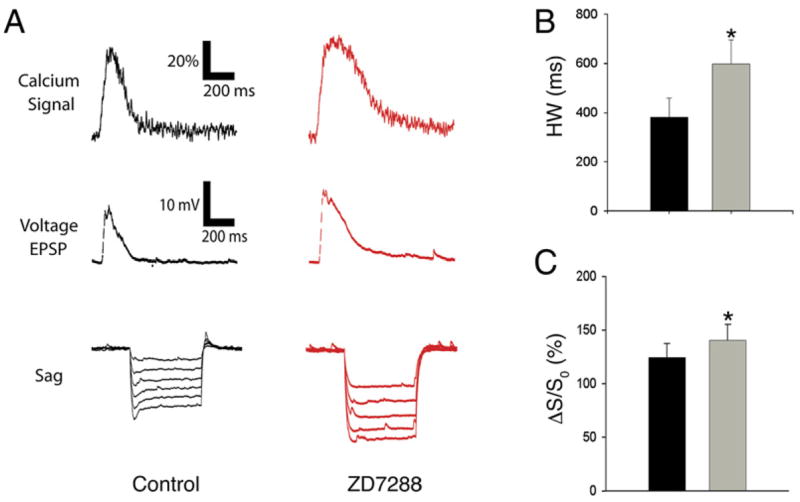
(A) Example Ca2+ (top) and somatic voltage (middle) responses from a WT mouse to PP burst stimulation in the absence (black) and presence (red) of ZD7288 (10 μM). (Bottom) Hyperpolarization-induced voltage sag due toactivation of Ih in response to current steps from −20 to −120 pA was blocked with ZD7288.
(B) Average half-width of distal Ca2+ events before and after application of ZD7288. Duration of Ca2+ transients was significantly prolonged (+57%) by ZD7288 (Control: HW = 345.1 ± 74.6 ms; ZD7288: HW = 541.7 ± 99.2 ms; p < 0.01).
(C) Average peak Ca2+ before and after application of ZD7288. Peak Ca2+ levels were increased by ~17% in drug (Control: ΔS/S0 = 109.4% ± 18.6%; ZD7288: ΔS/S0 = 127.9% ± 17.8%; n = 7; paired t test, p < 0.01).
Acute application of ZD7288 under these conditions produced similar changes in the distal dendritic Ca2+ transient to those seen upon deletion of HCN1 (Figure 3). Thus we observed an ~17% increase in peak Ca2+ signal, from 109.4% ± 18.6% in the absence of drug to 127.9% ± 17.8% in the presence of ZD7288 (n = 7; p < 0.01). We also found an ~57% prolongation in the Ca2+ signal duration, from 345.3 ± 74.6 ms in the absence of drug to 541.7 ± 99.2 ms in the presence of blocker (p < 0.01). Ca2+ rise and decay times in control conditions (τr = 72.33 ± 9.07 ms, τd = 275 ± 97.01) and in the presence of ZD7288 (τr = 109.75 ± 6.38 ms, τd = 464.33 ± 103.12 ms) showed no statistically significant differences, similar to results with HCN1 deletion. The similarity in effects of reducing Ih either by pharmacological or genetic means suggests that the enhancement in dendritic Ca2+ events reflects a specific effect due to the loss of Ih, rather than a nonspecific effect or compensatory change in other channels.
If the enhancement in distal dendritic Ca2+ signals is indeed causally related to the loss of local Ih, then ZD7288 should produce much less of a change in Ca2+ signals observed in stratum radiatum (SR) in response to SC stimulation, as the levels of HCN1 and Ih are much lower in proximal dendrites than in distal dendrites. Indeed, burst stimulation of SC inputs identical to that used in the PP experiments elicited a proximal Ca2+ response in SR that showed little change following blockade of Ih with ZD7288 (Figure 4A1 traces). There was, if anything, a small, statistically insignificant, decrease in Ca2+ signal duration (Figure 4B; Control HW = 150.67 ms ± 4.64 ms; ZD7288 HW = 144 ± 2.26 ms, n = 6, p = 0.14) and peak amplitude (Figure 4C; Control ΔS/S0 = 292.59% ± 29.8%, ZD7288 ΔS/S0 = 226.36% ± 35.1%, p = 0.08). The lack of augmentation of the Ca2+ signal elicited by SC stimulation was not due to use of the higher-affinity Ca2+ dye, Fluo-4, in these experiments (needed to measure the lower-amplitude proximal Ca2+ signals), as Fluo-4 detected a large prolongation in Ca2+ event duration at the distal dendrites in response to PP stimulation (Figures 4A2 and 4B; Control HW = 558.6 ms ± 118.11, ZD7288 HW = 995 ± 245.53 ms, p < 0.01). However, the peak Fluo-4 Ca2+ signal in the distal dendrites did not show an augmentation following Ih blockade (Control 146.33% ± 17.7%, ZD7288 145.27% ± 14.1%; p = 0.92), most likely due to saturation of the higher-affinity dye by the very large distal Ca2+ transient. The selective ability of ZD7288 to enhance the distal but not proximal Ca2+ signal supports the view that the calcium enhancement induced by HCN channel blockade was due to the local reduction of Ih.
Figure 4. Blockade of Ih Prolongs Ca2+ Signals Elicited by Perforant Path but Not Schaffer Collateral Stimulation.

Dendritic Ca2+ signals in proximal or distal CA1 dendrites in response to 100 Hz burst stimulation (10 stimuli) of SC or PP inputs, respectively, under control conditions (black) or in the presence of 10 μM ZD7288 (red). Ca2+ signals were measured with higher affinity Ca2+ dye, Fluo-4 (400 μM).
(A1) Effects of ZD7288 on proximal dendritic Ca2+ signals (top) and somatic voltage response (bottom) elicited by stimulation of SC synapses. (A2) Effects of ZD7288 on distal dendritic Ca2+ signals and somatic voltage response induced by burst stimulation of PP inputs. Distal but not proximal Fluo-4 Ca2+ signals are prolonged by ZD7288.
(B) Effects of ZD7288 on Ca2+ signal half-width duration in distal dendrites in response to PP stimulation or in proximal dendrites in response to SC stimulation. Grey symbols and lines show half-width durations for each individual experiment. Black symbols and lines show mean values. Values for distal dendrites in response to PP stimulation: Control: HW = 558.6 ± 118.11 ms; ZD7288: HW = 995 ± 245.53 ms; n = 10; p < 0.01, paired t test. Values for SR dendrites with SC stimulation: Control: HW = 150.67 ± 4.64 ms; ZD7288: HW = 144 ± 2.26 ms; n = 6, p = 0.14.
(C) Summary of effects of ZD7288 on HW and peak Ca2+ signals (ΔS/S0) elicited by PP (Control: ΔS/S0 = 146.3% ± 17.7%, ZD7288: ΔS/S0 = 145.3% ± 14.1%; p = 0.92) and SC stimulation (Control ΔS/S0 = 292.59% ± 29.8%, ZD7288 ΔS/S0 = 226.36% ± 35.1%, p = 0.08).
Does the prolongation of the distal Ca2+ signal reflect a prolongation of the distal voltage response to synaptic stimulation? We examined this question by obtaining whole-cell dendritic voltage recordings in mid to distal SR (85 and 200 μm from the soma) (Figure 5A). Consistent with our imaging data, PP burst stimulation elicited large, long-lasting depolarizing responses in the dendrites, and these responses were significantly prolonged by ZD7288 (10 μm) (Figures 5B1–2 and C1–2). This prolongation was primarily due to an increase in the afterdepolarization following the end of stimulation, indicating an increase in local excitatory dendritic current. In contrast, ZD7288 produced little prolongation of the dendritic voltage response to SC stimulation, consistent with a specific effect of Ih blockade (Figures 5D1 and 5D2).
Figure 5. Acute Blockade of Ih Prolongs Duration of the Long-Lasting Dendritic Depolarization in Response to PP but Not SC Stimulation.
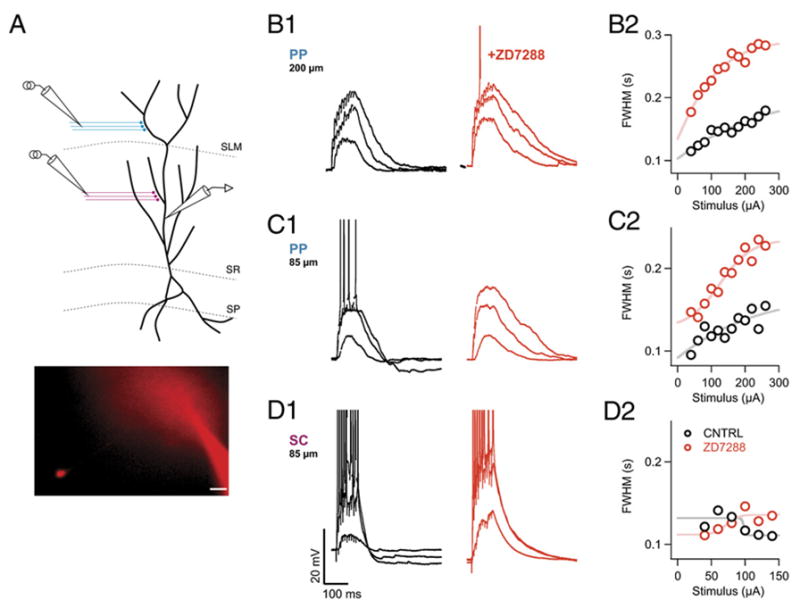
(A) Schematic of the recording configuration. Whole-cell current-clamp voltage recordings were obtained from the apical trunk of a CA1 pyramidal neuron. Extracellular electrodes were positioned to stimulate either PP inputs in SLM or SC inputs in SR. (Inset) Brightfield Alexa 594 fluorescent image of a representative recording. Soma of patch-clamped dendrite is visible in the CA1 pyramidal cell layer. Scale bar, 20 μm.
(B1) Voltage responses to low, intermediate, or strong PP stimulation from a whole-cell dendritic recording obtained ~200 μm from the center of the soma, in the absence (black traces) or presence (red traces) of ZD7288 (10 μM).
(C1) Voltage responses in absence or presence of ZD7288 to low, intermediate, or strong PP stimulation from a second dendritic recording ~85 μm from soma.
(D1) Dendritic voltage responses to SC stimulation from same recording shown in (C) in the absence and presence of ZD7288.
(B2–D2) Plots of half-width duration of voltage responses as function of stimulus intensity in the absence (black circles) or presence (red circles) of ZD7288 for experiments shown in (B1)–(C1).
Pharmacological Analysis of Distal Ca2+ Events in SLM
Which ion channels and receptors mediate the distal Ca2+ events and regulate their properties by interacting with Ih? We first examined the importance of ionotropic glutamate receptors, which have been previously shown to trigger distal Ca2+ spikes (Golding et al., 2002; Wei et al., 2001). Bath application of APV (50 μM) or CNQX (10 μM) to block NMDA receptors or AMPA receptors, respectively, caused a substantial reduction in both the peak somatic voltage response and amplitude of the distal Ca2+ signal in response to a burst of PP stimulation (Figures 6A, 6B, and 6G). Thus, application of APV reduced the Ca2+ signal (ΔS/S0) to 11.9% ± 2.3% of its control value (p < 0.01, n = 5) and decreased peak somatic voltage amplitude to 62.5% ± 5.0% of control (p < 0.05, n = 5). Similarly, CNQX reduced the Ca2+ signal to 24.5% ± 8.8% (n = 4, p < 0.05) and voltage amplitude to 26.5% ± 10.0% (n = 4, p < 0.05) of their initial levels, indicating the importance of both AMPA and NMDA receptors.
Figure 6. Ca2+ Signals in Distal Dendrites Are Initiated by Activation of NMDA and AMPA Receptors, and Mediated by Ni2+-Insensitive T-Type and N-Type Voltage-Gated Ca2+ Channels.

(A–F) Examples of effects of pharmacological agents on distal Ca2+ signals (top) and somatic voltage responses (bottom) elicited by burst stimulation of PP inputs. Black, control traces. Red, traces obtained in the presence of the following blockers: (A) 50 μM D-APV, an NMDAR antagonist; (B) 20 μM CNQX, an AMPAR antagonist; (C) combination of 50 μM Ni2+ (CaV3.2 T-type and R-type VGCC antagonist) and 20 μM nimodipine (L-type VGCC antagonist); (D) 20 μM mibefradil, general T-type channel antagonist; (E) 1 μM ω-conotoxin GVIA, N-type VGCC antagonist; and (F) 30 μM cyclopiazonic acid, a blocker of the SERCA pump.
(G) Summary of effects of pharmacological inhibitors on distal Ca2+ signal (black) and somatic voltage response (gray). Asterisks denote statistical significance comparing drug versus control (p < 0.05). D-APV: Ca2+ signal (ΔS/S0) reduced to 11.9% ± 2.3% of control (p < 0.01, n = 5); ΔV = 62.5% ± 5.0% of control (p < 0.05, n = 5). CNQX: ΔS/S0 = 32.7% ± 8.7% of control (p < 0.01, n = 4); ΔV = 26.5% ± 10.0% of control (p < 0.05, n = 4). Ni2+ + nimodipine: ΔS/S0 = 99.3% ± 3.6% of control; ΔV = 86.1% ± 4.6% of control (p > 0.1 for both; n = 4). Mibefradil: ΔS/S0 = 53% ± 11.4% of control, p < 0.05; ΔV = 80% ± 9.9% of control (p > 0.1; n = 4). ω-CTX GVIA: ΔS/S0 = 57.3% ± 9.6% of control, p < 0.05; ΔV = 90.8% ± 14.5% of control, p > 0.1 (n = 4). CPA: ΔS/S0 = 110.7% ± 14.4% of control (p = 0.42; n = 6).
Are NMDA receptors sufficient for generating the local nonlinear Ca2+ signals, as found for Ca2+ spikes in basal dendrites of neocortical pyramidal neurons (Schiller et al., 2000), or are other sources of Ca2+ also required? We first examined the involvement of Ni2+-sensitive and L-type voltage-gated Ca2+ channels, which have been implicated in distal LTP (Golding et al., 2002; Remondes and Schuman, 2003). However, combined application of 50 μM Ni2+ (an inhibitor of R- and T-type VGCCs) and 20 μM nimodipine (a dihydropyridine L-type VGCC blocker) had little effect on either the dendritic Ca2+ signal (ΔS/S0 = 99.3% ± 3.6% of control) or on the somatic EPSP (ΔV = 86.1% ± 4.6% of control, n = 4, p = 0.89) in response to a burst of PP stimuli (Figures 6C and 6G). This finding is in contrast to the sensitivity of Ca2+ transients in SR to these antagonists, as previously reported (Christie et al., 1995) and confirmed by our own experiments (Figure S4).
We next focused on the CaV3.3 T-type channels and N-type channels, two types of Ni2+- and nimodipine-insensitive VGCCs that are expressed in distal CA1 den-drites (McKay et al., 2006; Mills et al., 1994; Westenbroek et al., 1992). Indeed, the peak Ca2+ transients were substantially reduced by application of either the general T-type channel antagonist mibefradil (Figures 6E and 6G; ΔS/S0 = 53% ± 11.4% of control, p < 0.05; n = 5) or by the N-type channel antagonist ω-conotoxin GVIA (Figures 6D and 6G; ΔS/S0 = 57.3% ± 9.6% of control, p < 0.05; n = 5). However, the peak EPSP at the soma was only slightly reduced by either mibefradil (EPSP = 80% ± 9.9% of control, n = 5) or ω-conotoxin GVIA (EPSP = 90.8% ± 14.5% of control, n = 4) (Figures 6D, 6E, and 6G). Importantly, neither agent caused a significant change in the PP field EPSP (Figure S2), indicating that they did not alter transmitter release from the PP terminals (or cause significant block of AMPA or NMDA receptors). Thus, the distal Ca2+ spikes appear to recruit both CaV3.3 T-type and CaV2.2 N-type voltage-gated Ca2+ channels. Although mibefradil can also block L-type and R-type VGCCs (Randall and Tsien, 1997), our finding that the distal Ca2+ events are insensitive to Ni2+ and high concentrations of nimodipine indicates that the effects of mibefradil do indeed reflect T-type channel blockade. We also think it unlikely that the effects of mibefradil are due to block of voltage-gated Na+ channels (Eller et al., 2000), since we find that low concentrations of TTX (30 nM) that preferentially block dendritic Na+ spikes (Gasparini et al., 2004) do not reduce the amplitude of the local distal Ca2+ signal (Figure S5). Finally, we found that Ca2+ release from intracellular stores does not contribute to the distal Ca2+ signals, because blockade of the smooth endoplasmic reticulum Ca2+ (SERCA) pump with 30 μM cyclopiazonic acid (CPA) had no effect on the distal Ca2+ transient (Figures 6F and 6G; ΔS/S0 = 110.7% ± 14.4% of control, n = 6, p = 0.42).
A Computational Model for the Enhancement in the Distal Ca2+ Transient upon Ih Blockade
To gain further insight into the mechanism by which Ih blockade alters the distal Ca2+ transients, we developed a computational model representing an active distal dendritic branch with voltage-gated conductances, including T-type and N-type VGCCs (see Supplemental Section 2). In the presence of Ih, a simulated burst of synaptic activation of NMDA and AMPA receptors on the dendritic branch evoked a strong depolarization that triggered a nonlinear long-lasting spike (Figure 7A1). Removal of Ih hyperpolarized the resting membrane by ~6 mV, similar to the hyperpolarization observed in HCN1 KO mice (Nolan et al., 2004). Strikingly, the same synaptic stimulation elicited a local Ca2+ spike whose duration was enhanced nearly 2-fold, with a smaller increase in peak amplitude. In agreement with our somatic voltage recordings, blockade of Ih did not affect the peak absolute voltage reached during the burst of EPSPs prior to the spike, indicating that the hyperpolarization upon loss of Ih is offset by an increase in EPSP amplitude and temporal summation due to the increase in input resistance. Interestingly, when we examined a morphologically realistic model of a CA1 neuron, Ih blockade produced changes in somatic voltage responses similar to our experimental data (Figure S3).
Next we focused on the single-compartment model to gain insight into how removal of Ih altered the local Ca2+ transient in the distal dendrites. Blockade of Ih nearly doubled the peak Ca2+ current carried by the N- and T-type VGCCs during a dendritic spike (95% increase) and markedly prolonged the duration of Ca2+ influx (Figure 7A2). Furthermore, when we included a Ca2+ dye and endogenous Ca2+ buffering and transport to the model to calculate the Ca2+ fluorescence signal (Figure 7A3; see Supplemental Material Section 2), removing Ih produced a large (90%) increase in the peak Ca2+ signal and a 2-fold prolongation of its duration (116% increase in HW) (Figure 7A3), similar to our experimental findings (Figures 2 and 3). Importantly, these modeling results were robust, as they were observed over a wide range of parameter space for the voltage-gated Ca2+ channel kinetics (data not shown).
Figure 7. Computational Model of Distal Ca2+ Events and Effects of Ih Blockade.
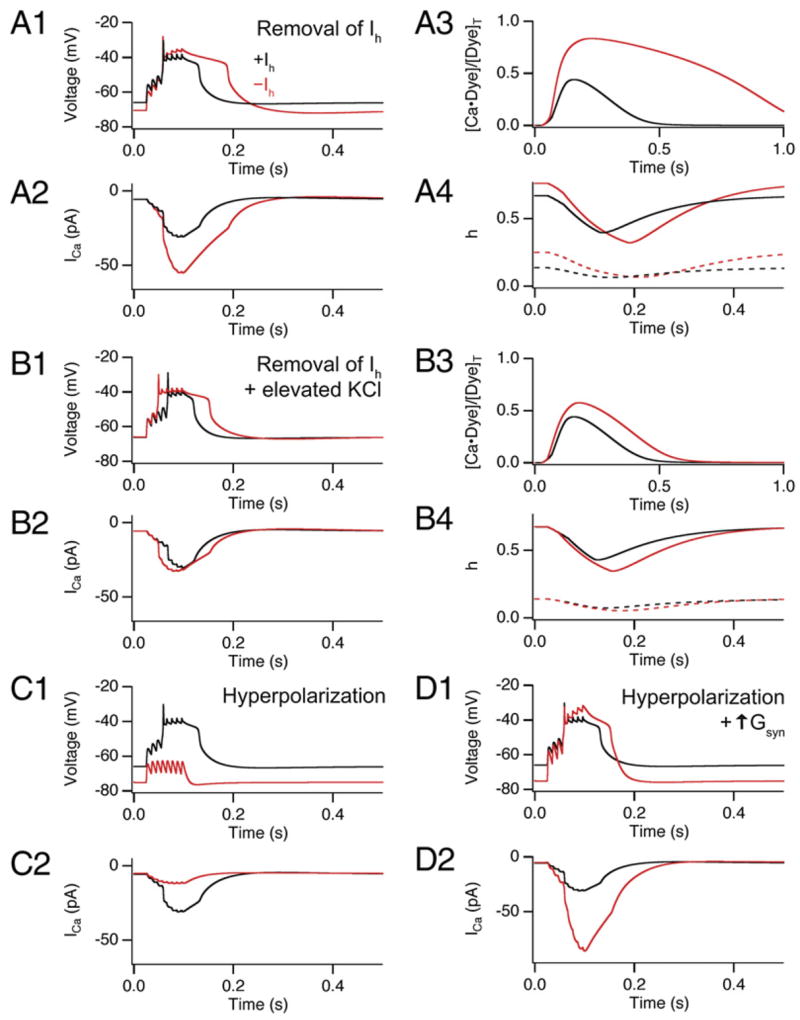
(A and B) Results from a single-compartment model for Ca2+ spike elicited by PP burst stimulation showing distal dendritic membrane voltage (panels labeled 1), total VGCC current (panels labeled 2), Ca2+-bound fraction of dye (panels labeled 3), and T-type and N-type VGCC inactivation gating variable (h, panels labeled 4). Simulations shown in the presence (black traces) or absence (red traces) of Ih. (A) Effects of Ih removal. (A1) Burst of 100 Hz synaptic stimulation evoked a nonlinear voltage response (Ca2+ spike). Removal of Ih hyperpolarized the resting membrane and enhanced the duration of Ca2+ spike. (A2) The sum of T-type and N-type Ca2+ current during Ca2+ spike shows enhanced Ca2+ influx upon removal of Ih. (A3) The fraction of Ca2+-bound indicator dye is enhanced and prolonged upon removal of Ih, similar to our experimental results. (A4) Plot of inactivation gating variable h for T-type and N-type VGCCs before (starting values) and during Ca2+ spike (solid lines, N-type; dashed lines, T-type), showing decreased inactivation upon removal of Ih. Note: h = 1 when all inactivation gates are open.
(B) Effects of Ih removal when membrane hyperpolarization is prevented by shifting the reversal potential of the leak conductance from −80.0 to −72.9 mV. Parameters defined as in (A).
(C) Effects of membrane hyperpolarization alone in maintained presence of Ih. Black traces, control. Red traces, during injection of a −0.011 nA hyperpolarizing current. (D) Effect of membrane hyperpolarization in maintained presence of Ih but with a 60% increase in excitatory synaptic conductance to trigger a dendritic Ca2+ spike. Black and red traces in the absence and presence of −0.011 nA current.
What is responsible for the pronounced increase in Ca2+ influx during the dendritic spikes in response to removal of Ih? Analysis of the T-type and N-type VGCCs gating parameters revealed that the voltage-dependent activation of both channels was similar during the Ca2+ spikes in the presence and absence of Ih. However, the fraction of inactivated channels both before and during the spikes was significantly less when Ih was removed (Figure 7A4), due to the hyperpolarization of the resting membrane. This effect produced an 84% increase in availability of T-type channels and a 14% increase in availability of N-type channels prior to the spike.
Since loss of Ih both hyperpolarizes the resting membrane and increases the input resistance, we used the model to dissect the relative importance of these two effects. We first asked whether hyperpolarization is necessary for the enhancement in the Ca2+ signal. Following removal of Ih, the membrane was depolarized back to its original potential by adjusting the K+ reversal potential (EK) to a more positive value (EK = −72.9 mV). Under these conditions where Ih was absent but resting potential was unchanged, the model generated a nearly normal-sized Ca2+ influx and Ca2+ signal even though there was still a significant increase in input resistance due to the lack of Ih (Figure 7B). Thus, membrane hyperpolarization is necessary for the enhancement.
We then tested whether hyperpolarization alone is sufficient to enhance the distal Ca2+ spike. A normalsized Ih was maintained in the simulation while the membrane was hyperpolarized to a negative potential equal to that normally reached upon Ih removal. Although steady-state availability of T- and N-type channels was increased to a similar extent as seen upon removal of Ih, the burst of EPSPs failed to trigger a Ca2+ spike because the peak EPSP voltage was now subthreshold (Figure 7C). This is in contrast to our experimental results where loss of Ih had little effect on the threshold for eliciting a dendritic spike (Figure 2). To counteract the inhibitory effect of the hyperpolarization, we increased the size of the synaptic conductance to reach a peak voltage similar to that seen when EPSPs are elicited from the normal resting potential. Under these conditions, the model dendrite generated a Ca2+ signal with a significantly enhanced amplitude and duration, similar to that observed in the absence of Ih (Figure 7D). Thus, spike prolongation does not require the increase in input resistance associated with the removal of Ih, as long as the EPSP is large enough to reach threshold.
These simulation results indicate that the effect of Ih removal to enhance the Ca2+ spikes depends on two synergistic actions. First, the hyperpolarization of the resting membrane removes resting inactivation of T- and N-type VGCCs, which enhances subsequent Ca2+ influx during a spike. Second, the increase in input resistance increases temporal integration and allows an EPSP to reach threshold to trigger a Ca2+ spike despite the hyperpolarization.
Counteracting the Hyperpolarization Induced by Ih Blockade Occludes the effects on Calcium Enhancement
To provide an experimental test of the importance of membrane hyperpolarization for the enhancement in the Ca2+ spike, we counterbalanced the normal hyperpolarization upon blockade of Ih with ZD7288 by elevating external K+, similar to the simulation results with an altered EK. Distal Ca2+ signals were measured before and after bath application of ZD7288 (10 μM) in elevated (6.25 mM) KCl. This concentration of KCl was calculated to be appropriate for maintaining the distal dendritic resting potential at its normal level upon blockade of Ih (see Supplemental Data).
Application of ZD7288 in elevated KCl was still effective at blocking HCN channels, as evident by the block of the hyperpolarization-induced voltage sag normally caused by Ih activation (Figure 8A). Importantly, the addition of KCl to the ZD7288 solution was also effective in preventing the hyperpolarization of the somatic resting potential. Whereas application of ZD7288 alone hyperpolarized the membrane from its normal value of −68.1 ± 1.3 mV to −75.5 ± 0.45 mV, coapplication of ZD7288 plus 6.25 mM KCl resulted in a slight depolarization of the resting membrane to −63.6 ± 0.4 mV. Although the somatic membrane potential is 4–5 mV more positive than its original resting potential in 2.5 mM KCl in the absence of ZD7288, the KCl concentration of 6.25 mM is what we calculated as being necessary to maintain the resting potential of the distal dendritic membrane at its normal level upon blockade of Ih, due to its high density of Ih (see Supplemental Data).
Figure 8. Coapplication of ZD7288 with Elevated KCl Prevents the Hyperpolarization Induced by Ih Blockade and Inhibits Ca2+ Spike Enhancement.
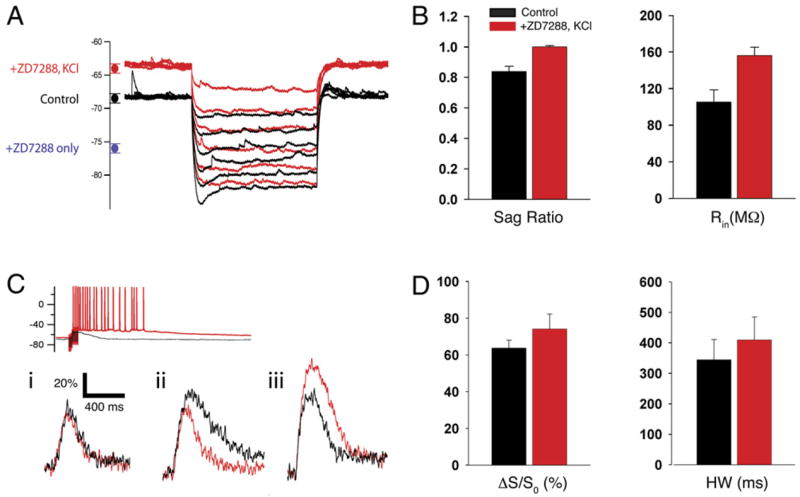
(A) Somatic voltage responses to hyperpolarizing current injections in normal KCl (black traces, 2.5 mM) and after application of 10 μM ZD7288 plus 6.25 mM KCl (red traces). The circles on the left show mean (±SE) values of resting potential: Control (black), Vm = −68.2 ± 0.4 mV (n = 22); ZD7288 + 6.25 mM KCl (red), Vm = −63.6 ± 0.4 mV (n = 8); ZD7288 only (blue), Vm = −75.5 ± 0.45 mV (n = 7).
(B) Mean effect of ZD7288 plus 6.25 mM KCl on depolarizing sag (left) and input resistance (right). Input resistance (Rin): Control, 105.4 ± 13.2 MΩ; ZD7288 plus 6.25 mM KCl, 156.1 ± 9.1 MΩ. Sag ratio (steady-state/peak voltage response to hyperpolarizing current): Control, 0.84 ± 0.03; ZD7288 plus KCl, 1.00 ± 0.008 (p < 0.01, paired t test; n = 7).
(C) Example somatic voltage (top) and distal Ca2+ signals (bottom) in response to ZD7288 plus 6.25 mM KCl. Black traces, control (2.5 mM Ca2+ without ZD7288). Red traces, in the presence of 10 μM ZD7288 plus 6.25 mM KCl. Application of ZD7288 plus elevated KCl did not alter or slightly decreased peak Ca2+ in 6 of 8 cells (examples i and ii). In 2 of 8 cells, ZD7288 plus elevated KCl increased the Ca2+ signal (iii). Increase in KCl enhanced excitability in all 8 cells, leading to prolonged firing in response to PP stimulation.
(D) Pooled data for effects of ZD7288 plus 6.25 mM KCl on mean distal Ca2+ signal amplitude (ΔS/S0) and duration (HW). Control: ΔS/S0 = 66.4% ±5.8%, HW = 344.6 ± 66.3 ms; ZD7288 + KCl: ΔS/S0 = 74.2% ± 8.1% (p = 0.34, n = 7), HW = 409.7 ± 75.3 ms (p = 0.27, n = 7).
When we delivered a burst of distal PP stimulation in the presence of ZD7288 plus 6.25 mM KCl, the normal effect of Ih blockade to prolong the distal Ca2+ spikes was largely eliminated (Figures 8C and 8D). In 6 out of 8 cells examined, application of ZD7288 plus KCl either had no effect on Ca2+ spike duration and amplitude or caused a small decrease (Figure 8C). However, in 2 of the 8 cells, there was still a substantial increase in Ca2+ signal peak amplitude and duration, similar to what we observed when ZD7288 was applied alone. This may reflect variability in the distal dendritic resting potential (Kole et al., 2006). Alternatively, it may reflect a small hyperpolarization-independent component of the effect of ZD7288 plus 6.25 mM KCl, due to the increase in input resistance and decrease in K+ driving force, as observed in the computational model (Figure 7B). On average over all eight experiments, application of ZD7288 plus 6.25 mM KCl produced an ~12% increase in the peak Ca2+ signal (Control: ΔS/S0 = 66.4% ± 5.8%; ZD7288 plus KCl: ΔS/S0 = 74.2% ± 8.1%, p = 0.34) accompanied by an ~19% increase in half-width (Control: 344.6 ± 66.3 ms; ZD7288 plus KCl: 409.7 ± 75.3 ms, p = 0.27) (Figure 8D). These effects were significantly smaller than the 17% (p < 0.01) increase in peak and 57% (p < 0.01) increase in duration of the Ca2+ signals when ZD7288 was applied alone (see Figure 3). Thus, in agreement with the model, our experimental results are consistent with the view that membrane hyperpolarization makes an important contribution to the enhancement in the distal dendritic Ca2+ signal upon blockade of Ih.
DISCUSSION
This study describes a mechanism by which HCN channels constrain nonlinear Ca2+ events at distal dendrites of CA1 pyramidal neurons, providing an explanation for the widespread finding that blockade of the excitatory Ih enhances dendritic excitability. We find that the unique effects of Ih to decrease input resistance while depolarizing the resting membrane exert a powerful inhibitory influence on Ca2+ spikes generated in distal dendrites. As a result, reduction of Ih, either due to HCN1 deletion or pharmacological blockade, leads to an increase in the peak amplitude and a dramatic prolongation in duration of the distal Ca2+ signals elicited by a short burst of perforant path synaptic stimulation.
According to our pharmacological results, the distal Ca2+ events are triggered by synaptic activation of both AMPA and NMDA receptors, which generates an initial depolarization that then activates both Ni2+-insensitive T-type and N-type voltage-gated Ca2+ channels. Immunocytochemical studies show that the Ni2+-insensitive CaV3.3 subunit is the major T-type isoform expressed in the distal CA1 neuron dendrites (McKay et al., 2006), suggesting that this isoform participates in the distal Ca2+ spikes. Interestingly CaV3.3 channels display significantly slower inactivation kinetics than the other T-type channel isoforms (CaV3.1 and CaV3.2), with time constants of inactivation >50 ms, appropriate for generating long-lasting Ca2+ spikes (Lee et al., 1999). The N-type channels display even slower inactivation kinetics, with time constants > 100 ms (Fox et al., 1987; Nowycky et al., 1985). These two VGCCs also show relatively slow activation kinetics and require relatively strong depolarizations to activate, with midpoint activation voltages of ~21 mV for CaV3.3 (Lee et al., 1999) and +10 mV for N-type channels (Fox et al., 1987; Nowycky et al., 1985). Thus, the triggering of regenerative events mediated by such channels will require large, long depolarizations, such as those elicited by bursts of synaptic stimulation. Finally, the steady-state inactivation of both CaV3.3 and N-type VGCCs shows a steep dependence on voltage near typical resting membrane potentials. As a result, steady-state availability of these channels will be responsive to small changes in resting potential.
Our computational and experimental findings indicate that the voltage-dependent properties of Ih are finely tuned to interact with the voltage-dependent gating of the T- and N-type Ca2+ channels to regulate the distal Ca2+ spikes. The effect of Ih to depolarize the resting membrane increases the steady-state inactivation of these VGCCs, whereas the effect of Ih to decrease the input resistance reduces the size of the EPSP, thus decreasing the voltage-dependent activation of the calcium channels. These combined effects of Ih therefore exert a powerful inhibitory effect on T- and N-type calcium opening, thus inhibiting Ca2+ influx during the distal dendritic Ca2+ spikes. As dendrites contain an array of voltage-gated channels in addition to Ih, other ionic mechanisms are also likely to regulate the distal Ca2+ spikes (e.g., Wei et al., 2001; Cai et al., 2004).
One surprising result from our study is that neither Ni2+-sensitive VGCCs (including CaV3.2 T-type and CaV2.3 R-type) nor dihydropyridine-sensitive L-type VGCCs appear to contribute to the distal Ca2+ spikes. These results were unexpected, as previous studies have found that either the combined application of Ni2+ and nimodipine (Golding et al., 2002) or the application of a high concentration of nifedipine (a dihydropyridine similar to nimodipine) alone partially inhibits perforant path LTP (Remondes and Schuman, 2003). Recently, Ni2+-sensitive and L-type VGCCs channels have been found to contribute to Ca2+ influx into CA1 neuron dendritic spines in stratum radiatum in response to local uncaging of glutamate (Bloodgood and Sabatini, 2007). Thus, the Ni2+-sensitive or L-type channels may participate in perforant path LTP by directly contributing to Ca2+ influx in spines on distal dendrites.
The inhibitory role of Ih in regulating the long-lasting Ca2+ spikes in CA1 dendrites in stratum lacunosum moleculare is in general agreement with previous findings that Ih inhibits other forms of dendritic excitability, such as the firing of briefer spikes in proximal CA1 dendrites in stratum radiatum (Fan et al., 2005; Magee, 1998; Poolos et al., 2002), distal dendrites in neocortical layer V neurons (Williams and Stuart, ■ ■ ■ ; Berger et al., ■ ■ ■ ; Kole et al., ■ ■ ■), and dendrites of entorhinal cortex layer V neurons (Rosenkranz and Johnston, 2006; Shah et al., 2004). The surprisingly long duration of the Ca2+ spikes in the SLM dendrites that we observe is in agreement with the Ca2+ waveforms seen in distal CA1 dendrites of rat hippocampal slice cultures (Wei et al., 2001; Cai et al., 2004). However, briefer Ca2+ spikes from distal dendrites of rat CA1 neurons have also been reported (Golding et al., 2002), suggesting that there may be different modes of regenerative activity induced by slightly different stimulus protocols. The long-lasting Ca2+ spikes in SLM dendrites may depend on the unique distribution of voltage-gated channels in these membranes. For example, the Kv4.2 A-type K+ channel, which exerts a powerful inhibitory effect on excitability, is restricted to the SR region of CA1 dendrites (Rhodes et al., 2004), where it prevents the invasion of the SR dendrites by distal Ca2+ spikes generated in SLM (Cai et al., 2004). Despite these differences in local ion channels, the genesis of dendritic action potentials in many neuronal cell types is typically mediated by voltage-dependent Ca2+ channels; thus, the inhibitory mechanism that we have identified in CA1 SLM dendrites is likely to extend to other areas of the nervous system with high densities of Ih.
Recent studies have correlated the modulatory role of Ih in dendritic integration to performance in memory tasks, both for hippocampal-dependent spatial memory in mice (Nolan et al., 2004) and working memory mediated by the prefrontal cortex (Wang et al., 2007). Here we purposefully used stimulation protocols similar to those used to induce LTP at PP synapses (Golding et al., 2002; Nolan et al., 2004) to examine effects of Ih on Ca2+ transients that may be relevant to hippocampal memory. These distal Ca2+ spikes are thought to play an important role in the induction of LTP at the PP synapses, since somatic action potentials fail to backpropagate into the distal CA1 dendrites (Golding et al., 2002; Remondes and Schuman, 2003). Importantly, the peak Ca2+ level associated with the distal spikes can exceed 10 μM, a concentration capable of inducing LTP (Malenka et al., 1988; Yang et al., 1999). Thus, the augmentation in distal Ca2+ transients observed upon HCN1 deletion provides a potential mechanism for the enhancement in LTP at the PP synapses in the HCN1 KO mice. As both our study and a previous study of the role of HCN1 in LTP (Nolan et al., 2004) were performed in the presence of blockers of inhibitory synaptic transmission, it will be of interest in the future to explore the role of HCN1 when inhibition is intact.
Although the regulation of dendritic Ca2+ spikes that we have analyzed is due to genetic or pharmacological manipulations of Ih, HCN channels are regulated by a number of modulatory processes that are of physiological relevance. Thus, Ih is directly facilitated by the second messenger cAMP (DiFrancesco and Tortora, 1991) and the membrane lipid PI(4,5)P2 (Pian et al., 2006; Zolles et al., 2006). Enhancement of cAMP levels by dopamine depresses dendritic excitability in entorhinal cortex neurons through a mechanism that depends on an enhancement in Ih (Rosenkranz and Johnston, 2006). Moreover, both physiological (Fan et al., 2005) and pathophysiological (Shah et al., 2004) levels of intrinsic neural activity have been found to alter levels of Ih and HCN1 expression that are associated with long-lasting changes in dendritic integration and excitability. Conversely, a reduction in Ih, for example due to a reduction in resting levels of cAMP, may augment the distal Ca2+ events, potentially leading to an enhancement of LTP, as seen in the HCN1 KO mice. Thus, we suggest that the effect of Ih to constrain dendritic Ca2+ spikes may represent a physiologically important means for dynamically regulating dendritic integration and synaptic plasticity in a wide variety of pyramidal neurons in the central nervous system.
EXPERIMENTAL PROCEDURES
Tissue Preparation
Horizontal brain slices were prepared from P30–P50 HCN1 KO mice (−/−) or wild-type (+/+) littermates (Nolan et al., ■ ■ ■). Mice were deeply anesthetized with isoflurane and their brains rapidly removed and placed in cold (2°C–3°C) modified ACSF containing (in mM) NaCl (10), NaH2PO4 (1.25), KCl (2.5), NaHCO3 (25), glucose (25), CaCl2 (0.5), MgCl2 (7), sucrose (190), and Na-pyruvate (2), equilibrated with 95%/5% O2/CO2. The hemisected brain was glued to an agar block (angled 10° in the ventromedial direction for optimal preservation of SLM perforant path inputs) and cut submerged in cold ACSF into 300 μm sections with a Vibratome 1000. Slices were transferred to standard ACSF at 35°C for 30–60 min and then kept at room temperature (21°C–22°C). Experiments were performed 1.5–7 hr after slice preparation.
Electrophysiology Recordings and Solutions
The standard ACSF had the following composition (mM): NaCl (125), NaH2PO4 (1.25), KCl (2.5), NaHCO3 (25), glucose (25), CaCl2 (2), and MgCl2 (1). In all experiments, inhibitory transmission was blocked by the GABAA and GABAB receptor antagonists gabazine (1 μM) and CGP-55845 (2 μM), respectively. Whole-cell recordings were obtained from CA1 pyramidal cells in submerged slices at 33°C–35°C. Neurons were visually identified using DIC optics and contrast enhancement with a digital camera (Hamamatsu). Patch pipettes (2.5–5 MΩ) were filled with intracellular solution containing (mM) KMeSO4 (130), KCl (10), HEPES (10), NaCl (4), MgATP (4), Na2GTP (0.3), phosphocreatine (10), and calcium-insensitive (Alexa Fluor 594, 25 μM) and calcium-sensitive (Oregon Green BAPTA-5N, 500 μM) dyes. Series resistance was less than 20 MΩ, and capacitance was fully compensated throughout the experiment.
Fluorescent indicators (Alexa 594 cadaverine, Fluo-4 cadaverine, Oregon-Green BAPTA-5N) were purchased from Molecular Probes (Invitrogen, Carlsbad, CA), diluted into 100× stock solutions using standard intracellular solution, aliquoted, and frozen (−20°C). Pharmacological antagonists were added to the bath solution by dilution from stock solutions (500- to 1000-fold concentrated). All drugs were obtained from either Sigma or Tocris-Cookson and used at the following concentrations (μM): CPA (30), D-APV (50), gabazine (1), CGP-55845 (2), CNQX (10), w-conotoxin GVIA (1), mibefradil (20), and nimodipine (20).
Two-Photon Ca2+ Imaging
Two-photon imaging was performed using a BioRad Radiance 2100 MP (Zeiss, Jena, Germany), powered by a MaiTai Ti:sapphire pulsed laser (Spectra-Physics, Fremont, CA) tuned to 800 nm. Red (Alexa 594 cadaverine; Ca2+ insensitive) and green (Oregon Green BAPTA-5N or Fluo-4 cadaverine; Ca2+ sensitive) epifluorescent signals were collected through a 60× 1.1 NA objective (Olympus, Center Valley, PA) and measured by custom external GaAsP detectors (Multiphoton Peripherals Inc., Ithaca, NY). Optical signals were digitized through the Radiance system using Lasersharp 2000 software (Zeiss) and analyzed offline in Igor Pro (Wavemetrics, Lake Oswego, OR). The Ca2+ signal (S) was defined as the ratio of the calcium-dependent green fluorescence versus the calcium-independent red fluorescence (G/R), using low-pass filtered averages of three to five individual line-scans, normalized to the prestimulus baseline (see Supplemental Methods for details).
Electrophysiological Data Acquisition and Analysis
Recordings were obtained using a two-channel Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA). Data were digitized on a Windows PC using an ITC-18 A/D board (Instrutech Instruments, Port Washington, NY) controlled by either PULSE acquisition software (Heka Instruments) or custom routines written in Igor Pro (Wavemetrics, Eugene, OR). All current-clamp data were acquired at 20 kHz and low-pass filtered at 4 kHz using the Multiclamp 700B Bessel filter. Analysis was performed using custom routines written in Igor Pro. Statistical tests were performed using Excel (Microsoft, Redmond, WA) and SigmaStat (Systat Software, Inc., San Jose, CA).
Computational Modeling
Nonlinear, synaptically evoked regenerative events at distal dendrites were simulated in NEURON (Carnevale and Hines, ■ ■ ■ ; available at http://www.neuron.yale.edu/neuron) using a single-compartment model (dendrite of 25 μm length and 1 μm diameter) with the following active channel conductances (in pS/μm2): gNa (45), gK,DR (4), gK,A (640), gH (20), gLEAK (10), gCa,T (60), and gCa,N (30). A specific capacitance of 2 μF/cm2 was used to account for spine surface area. The Ih conductance value was chosen to yield a membrane potential shift of ~6 mV upon removal of 80% of the conductance, matching our experimental observations. Synaptic current (ten synapses, 350 pS conductance each) was simulated using custom models of AMPA and NMDA receptor kinetics (Supplemental Data).
The predicted fluorescence signal was simulated by implementing a model of Ca2+ binding and extrusion assuming first-order enzyme kinetics in Matlab 7 (Mathworks, Natick, MA). The model included (concentrations in μM) calbindin (40), extrusion via membrane pumps (240), a very low-affinity endogenous buffer (10,000), and OGB-5N (500). The model was constrained by varying densities of buffers and transporters to yield a Ca2+ fluorescence signal whose amplitude and time course matched our experimental observations. Further details of simulations with references are given in Supplemental Data.
Supplementary Material
Supplemental Data
The Supplemental Data for this article can be found online at http://www.neuron.org/cgi/content/full/56/6/■■■/DC1/.
Acknowledgments
This work was partially supported by HHMI (S.A.S.), a grant from NIH (NS R01-36658 to S.A.S.), NYSTAR, the NIH MSTP (D.T.), and an NIH training grant (J.T.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Kao JP, Tang CM, Thompson SM. Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron. 2004;44:351–364. doi: 10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Chen C. ZD7288 inhibits postsynaptic glutamate receptor-mediated responses at hippocampal perforant path-granule cell synapses. Eur J Neurosci. 2004;19:643–649. doi: 10.1111/j.0953-816x.2003.03174.x. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc Natl Acad Sci USA. 2002;99:9538–9543. doi: 10.1073/pnas.142213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie BR, Eliot LS, Miyakawa H, Johnston D. Different Ca2+ channels in somata and dendrites of hippocampal pyramidal neurons mediate spike-induced Ca2+ influx. J Neurophysiol. 1995;73:2553–2557. doi: 10.1152/jn.1995.73.6.2553. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- Eller P, Berjukov S, Wanner S, Huber I, Hering S, Knaus HG, Toth G, Kimball SD, Striessnig J. High affinity interaction of mibefradil with voltage-gated calcium and sodium channels. Br J Pharmacol. 2000;130:669–677. doi: 10.1038/sj.bjp.0703352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h) Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Felix R, Sandoval A, Sanchez D, Gomora JC, De la Vega-Beltran JL, Trevino CL, Darszon A. ZD7288 inhibits low-threshold Ca(2+) channel activity and regulates sperm function. Biochem Biophys Res Commun. 2003;311:187–192. doi: 10.1016/j.bbrc.2003.09.197. [DOI] [PubMed] [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J Physiol. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S, Migliore M, Magee JC. On the initiation and propagation of dendritic spikes in CA1 pyramidal neurons. J Neurosci. 2004;24:11046–11056. doi: 10.1523/JNEUROSCI.2520-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Spruston N. Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron. 1998;21:1189–1200. doi: 10.1016/s0896-6273(00)80635-2. [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Golding NL, Mickus TJ, Katz Y, Kath WL, Spruston N. Factors mediating powerful voltage attenuation along CA1 pyramidal neuron dendrites. J Physiol. 2005;568:69–82. doi: 10.1113/jphysiol.2005.086793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarsky T, Roxin A, Kath WL, Spruston N. Conditional dendritic spike propagation following distal synaptic activation of hippocampal CA1 pyramidal neurons. Nat Neurosci. 2005;8:1667–1676. doi: 10.1038/nn1599. [DOI] [PubMed] [Google Scholar]
- Kole MH, Hallermann S, Stuart GJ. Single Ih channels in pyramidal neuron dendrites: properties, distribution, and impact on action potential output. J Neurosci. 2006;26:1677–1687. doi: 10.1523/JNEUROSCI.3664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Notomi T, Tamas G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Post-synaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, Turner RW. Ca(V)3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci. 2006;24:2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- Mills LR, Niesen CE, So AP, Carlen PL, Spigelman I, Jones OT. N-type Ca2+ channels are located on somata, dendrites, and a subpopulation of dendritic spines on live hippocampal pyramidal neurons. J Neurosci. 1994;14:6815–6824. doi: 10.1523/JNEUROSCI.14-11-06815.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O’Carroll C. Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:773–786. doi: 10.1098/rstb.2002.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DA, Trana R, Katz Y, Kath WL, Spruston N, Geinisman Y. Distance-dependent differences in synapse number and AMPA receptor expression in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:431–442. doi: 10.1016/j.neuron.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Lisman JE. Pathway-specific properties of AMPA and NMDA-mediated transmission in CA1 hippocampal pyramidal cells. J Neurosci. 2002;22:1199–1207. doi: 10.1523/JNEUROSCI.22-04-01199.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pian P, Bucchi A, Robinson RB, Siegelbaum SA. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J Gen Physiol. 2006;128:593–604. doi: 10.1085/jgp.200609648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- Randall AD, Tsien RW. Contrasting biophysical and pharmacological properties of T-type and R-type calcium channels. Neuropharmacology. 1997;36:879–893. doi: 10.1016/s0028-3908(97)00086-5. [DOI] [PubMed] [Google Scholar]
- Remondes M, Schuman EM. Molecular mechanisms contributing to long-lasting synaptic plasticity at the temporoam-monic-CA1 synapse. Learn Mem. 2003;10:247–252. doi: 10.1101/lm.59103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes KJ, Carroll KI, Sung MA, Doliveira LC, Monaghan MM, Burke SL, Strassle BW, Buchwalder L, Menegola M, Cao J, et al. KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. J Neurosci. 2004;24:7903–7915. doi: 10.1523/JNEUROSCI.0776-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Johnston D. Dopaminergic regulation of neuronal excitability through modulation of Ih in layer V entorhinal cortex. J Neurosci. 2006;26:3229–3244. doi: 10.1523/JNEUROSCI.4333-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol. 1997;505:605–616. doi: 10.1111/j.1469-7793.1997.605ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, et al. alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Wei DS, Mei YA, Bagal A, Kao JP, Thompson SM, Tang CM. Compartmentalized and binary behavior of terminal dendrites in hippocampal pyramidal neurons. Science. 2001;293:2272–2275. doi: 10.1126/science.1061198. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Zolles G, Klocker N, Wenzel D, Weisser-Thomas J, Fleischmann BK, Roeper J, Fakler B. Pacemaking by HCN channels requires interaction with phosphoinositides. Neuron. 2006;52:1027–1036. doi: 10.1016/j.neuron.2006.12.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data
The Supplemental Data for this article can be found online at http://www.neuron.org/cgi/content/full/56/6/■■■/DC1/.