

Abstract
Biomarker discovery produces lists of candidate markers whose presence and level must be subsequently verified in serum or plasma. Verification represents a paradigm shift from unbiased discovery approaches to targeted, hypothesis-driven methods and relies upon specific, quantitative assays optimized for the selective detection of target proteins. Many protein biomarkers of clinical currency are present at or below the nanogram/milliliter range in plasma and have been inaccessible to date by MS-based methods. Using multiple reaction monitoring coupled with stable isotope dilution mass spectrometry, we describe here the development of quantitative, multiplexed assays for six proteins in plasma that achieve limits of quantitation in the 1–10 ng/ml range with percent coefficients of variation from 3 to 15% without immunoaffinity enrichment of either proteins or peptides. Sample processing methods with sufficient throughput, recovery, and reproducibility to enable robust detection and quantitation of candidate biomarker proteins were developed and optimized by addition of exogenous proteins to immunoaffinity depleted plasma from a healthy donor. Quantitative multiple reaction monitoring assays were designed and optimized for signature peptides derived from the test proteins. Based upon calibration curves using known concentrations of spiked protein in plasma, we determined that each target protein had at least one signature peptide with a limit of quantitation in the 1–10 ng/ml range and linearity typically over 2 orders of magnitude in the measurement range of interest. Limits of detection were frequently in the high picogram/milliliter range. These levels of assay performance represent up to a 1000-fold improvement compared with direct analysis of proteins in plasma by MS and were achieved by simple, robust sample processing involving abundant protein depletion and minimal fractionation by strong cation exchange chromatography at the peptide level prior to LC-multiple reaction monitoring/MS. The methods presented here provide a solid basis for developing quantitative MS-based assays of low level proteins in blood.
Proteomics methods based on MS have emerged as the preferred strategy for discovery of diagnostic, prognostic, and therapeutic protein biomarkers. Most biomarker discovery studies use unbiased, “identity-based” approaches that rely on high performance mass spectrometers and extensive sample processing (1–3). Semiquantitative comparisons of protein relative abundance between disease and control patient samples are used to identify proteins that are differentially expressed (4–9) and, thus, to populate lists of potential biomarkers. De novo proteomics discovery experiments often result in tens to hundreds of candidate biomarkers that must be subsequently verified in plasma (3). However, despite the large numbers of putative biomarkers, only a small number of them are passed through the development and validation process into clinical practice, and their rate of introduction is declining (10).
Currently clinical validation of novel biomarkers relies primarily on immunoassays because of their specificity for the target analyte, sensitivity (picogram/milliliter), and high throughput. However, the development of a reliable immunoassay for quantitation of one target protein is expensive, has a long development time, and is dependent upon the generation of high quality protein antibodies. These reagent limitations along with the limited ability to multiplex immunoassays make it necessary to use alternative targeted quantitative assays to provide the critical bridge between candidate discovery and validation.
Multiple reaction monitoring (MRM)1 coupled with stable isotope dilution (SID)-MS using a triple quadrupole mass spectrometer is a powerful method for quantitative measurement of target proteins. It has long been a principal tool for quantification of small molecules in clinical chemistry (11–13). MS-based quantitative assays have the necessary characteristics required for verification studies, namely high specificity, sensitivity, multiplexing capability, and precision (coefficient of variation (CV), <10%). In MRM mode, two stages of mass filtering are used. In the first stage, an ion of interest (the precursor) is preselected and induced to fragment by collisional excitation with a neutral gas in a pressurized collision cell. In the second stage, a small number of sequence-specific fragment ions derived from the precursor (transition ions) are then mass analyzed. This targeted MS analysis using MRM enhances the lower detection limit for peptides by up to 100-fold (as compared with unbiased MS analysis) by allowing rapid and continuous monitoring of the specific ions of interest. MRM methods, in principal, provide both absolute structural specificity for the analyte and relative or absolute measurement of analyte concentration when stable, isotopically labeled standards are added to a sample in known quantities. With MRM/SID-MS, a moderate number of candidate proteins (30–100) can be simultaneously targeted and measured in a statistically relevant number of patient samples required for verification.
MRM LC-MS/MS in electrospray mode in combination with SID has recently been shown to be well suited for direct quantification of proteins in plasma (14–21). In these studies, quantification of proteins is accomplished by selecting “signature” peptides derived by enzymatic digestion (usually trypsin) of the target protein released during sample digestion. These signature peptides are used as quantitative, stoichiometric surrogates of the protein itself and, therefore, are chosen to be unique in sequence (i.e. not present in other proteins in the genome). When a synthetic, stable isotope-labeled version is used as an internal standard, protein concentration can be measured by comparing the signals from the exogenous labeled and endogenous unlabeled species. Selection of signature peptides takes into account their observed and/or predicted LC retention, molecular weight, and predicted or observed charge state with preference for moderately hydrophobic peptides likely to produce doubly or triply charged ions in the detectable mass range of the mass spectrometer. While priority is generally given to peptides detected in unbiased discovery experiments, peptides can also be selected from in silico analysis of the protein sequence. Thus, MS-based assays can be developed for candidate biomarkers resulting from proteomics and genomics experiments as well as domain and/or literature knowledge.
Although MRM/SID-MS provides a powerful method for candidate biomarker verification, its sensitivity for unambiguous detection and quantitation of proteins is constrained by sample complexity. Studies published to date indicate that it is difficult without very extensive fractionation to detect and quantify proteins at concentrations lower than 1 μg/ml in plasma (21, 22). In this study, we have systematically explored and optimized sample processing and analysis methods to define conditions with sufficient throughput, recovery, and reproducibility to enable robust detection and quantitation of candidate biomarker proteins at the low nanogram/milliliter levels in plasma without immunoaffinity enrichment at either the protein or peptide level. Such a study provides the foundation for a possible hybrid approach that includes anti-peptide antibody enrichment and moderate fractionation to extend the limit of quantitation (LOQ) of MRM assays to cover the full known protein dynamic range of plasma.
EXPERIMENTAL PROCEDURES
Reagents and Chemicals
Bovine aprotinin, bovine myelin basic protein (MBP), murine leptin, horse heart myoglobin, and horseradish peroxidase (HRP) were purchased from Sigma. Human prostate-specific antigen (PSA) was purchased from Scripps Laboratories (San Diego, CA). All other reagents (including water) were of HPLC or proteomics grade.
Plasma Depletion
All experiments were performed on 100μl aliquots of immunoaffinity depleted human plasma from a healthy female donor. Two separate immunoaffinity protocols were followed. In the first protocol, seven high abundance plasma proteins (albumin, IgG, IgA, transferrin, fibrinogen, α1-antitrypsin, and haptoglobin) were depleted using a Multiple Affinity Removal System (MARS Hu7) affinity column from Agilent Technologies (Hu-7, 4.6 × 100 mm) on an Agilent 1100 analytical LC system (Agilent Technologies, Palo Alto, CA). Depletion was performed using buffers provided with the kit and according to manufacturer’s instructions. In the second protocol, 12 high abundance plasma proteins (above plus α2-macroglobulin, IgM, α1-acid glycoprotein, apolipoprotein A-I, and apolipoprotein A-II) were depleted using a ProteomeLab IgY-12 high capacity LC10 column (12.7 × 79 mm; Beckman Coulter, Fullerton, CA) on an AKTA fast performance liquid chromatography system (FPLC; GE Healthcare/Amersham Biosciences). Depletion was performed using buffers provided with the kit and according to manufacturer’s instructions. Depleted sample from both protocols was concentrated using Vivaspin 15R concentrators (5000 molecular weight cutoff; Vivascience, Hannover, Germany). Protein concentration of depleted, concentrated plasma was determined by Coomassie Plus Bradford assay (Pierce) and estimated to be 7.5 and 2.3 mg/ml for MARS Hu7 and IgY-12 depleted plasma, respectively.
Preparation of Protein Mixtures and Digestions
Stocks of 1 mg/ml of the proteins to be spiked into nondepleted and depleted female plasma were made in water, submitted for amino acid analysis (AAA), and aliquoted into substocks. After AAA, protein stock concentrations were adjusted to reflect AAA value. Nonspiked and spiked plasma (100 μl aliquots) was denatured with 6 M urea, 10 mM Tris, pH 8.0, reduced with 20 mM dithiothreitol at 37 °C for 30 min, and alkylated with 50 mM iodoacetamide at room temperature in the dark for 30 min. Urea concentration was diluted 10-fold with water prior to overnight digestion at 37 °C with trypsin (sequencing grade modified; Promega, Madison, WI) using a 1:50 (w/w) enzyme to substrate ratio. Digests were terminated with formic acid to a final concentration of 1% and desalted using Oasis HLB 1 ml (30 mg) reverse phase cartridges (Waters, Milford, MA) according to the following procedure: wash cartridge with 3 × 500 μl of 80% acetonitrile in 0.1% formic acid, equilibrate with 4 × 500 μl of 0.1% formic acid, load total volume of digest, wash with 4 × 500 of μl 0.1% formic acid, and elute with 2 × 500 μl of 80% acetonitrile in 0.1% formic acid. Eluates were frozen at −80 °C and dried to dryness via vacuum centrifugation.
Strong Cation Exchange Chromatography (SCX)
Digested plasma samples (~230 μg of total protein) were reconstituted in 40 μl of 5 mM potassium phosphate in 25% acetonitrile, pH 3.0 and fractionated by strong cation exchange chromatography on a BioBasic 1 × 250-mm column (ThermoFisher). Separations were performed on an Agilent 1100 capillary LC system (Agilent Technologies) at a flow rate of 50 μl/min and with mobile phases that consisted of 5 mM potassium phosphate in 25% acetonitrile, pH 3.0 (A) and 500 mM potassium chloride in 5 mM potassium phosphate, 25% acetonitrile, pH 3.0 (B). After loading 40 μl of sample onto the column, the mobile phase was held at 4% B for 15 min. Peptides were then separated with a linear gradient of 4–20% B in 20 min, 20–40% B in 10 min, and 40–100% B in 10 min. Fractions were collected every minute, and acetonitrile was removed from collected fractions by vacuum centrifugation. Pools of three to four fractions each were generated according to the SCX elution profile of signature peptides injected separately (Supplemental Fig. 13). Pooled fractions were desalted using Oasis HLB 1ml (10 mg) reverse phase cartridges, and eluates were dried to dryness via vacuum centrifugation. For peptides that elute early by SCX (e.g. fractions 1–2), the flow-through from the SCX separation was also desalted and analyzed by LC-MRM/MS.
Labeled Peptide Internal Standards
Stable isotope-labeled amino acids, [13C6]leucine (98.4 atom % isotopic enrichment) and [13C5]valine (98 atom % isotopic enrichment), were purchased from Cambridge Isotope Laboratories (Andover, MA). Labeled peptide internal standards derived from the target proteins were synthesized with a single, uniformly labeled [13C6]leucine or [13C5]valine using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry (Massachusetts Institute of Technology, Cambridge, MA). Unlabeled 12C forms of each peptide were synthesized by GL Biochem (Japan). Peptide selection was based upon experimental observation using commercially available protein standards. Briefly the proteins were enzymatically digested with trypsin and analyzed by nano-LC-MS/MS on an LTQ linear ion trap mass spectrometer (Thermo, San Jose, CA) with data-dependent acquisition. Database matching of collected MS/MS spectra was performed using Spectrum Mill software (Agilent Technologies). One to three peptide standards per protein were chosen based upon the following criteria: high relative abundance, MS/MS spectral quality, and homology to the corresponding endogenous forms of the plasma proteins. Exclusion criteria included large hydrophobic or small hydrophilic peptides; flanking, tryptic ends with dibasic amino acids (KK, RR, or KR) at the N or C terminus; and identity to corresponding endogenous plasma proteins. Methionine-and cysteine-containing peptide standards were not excluded in these experiments. Individual synthetic peptide stocks of 2 nmol/μl based upon weight were made in 30% acetonitrile, 0.1% formic acid and submitted for AAA analysis. Substocks were diluted to 1 nmol/μl with 30% acetonitrile, 0.1% formic acid to reflect AAA value.
LC-MRM/MS
Nonfractionated plasma digests were reconstituted in 100 μl of 5% formic acid, 3% acetonitrile and diluted 1:10 (for MARS hu7 depleted plasma) and 1:3 (for IgY-12 depleted plasma) with 0.1% formic acid containing 13C-labeled peptides at an equimolar concentration of 5 fmol/μl. Pooled SCX fractions were reconstituted in 25 μl of 5% formic acid, 3% acetonitrile, and an aliquot of 9 μl from each SCX pool was mixed with 1 μl of 0.1% formic acid containing 13C-labeled peptides at an equimolar concentration of 50 fmol/μl. Final concentration of internal standards in SCX pools was 5 fmol/μl. All samples were analyzed by nano-LC-MRM/MS on a 4000 Q Trap hybrid triple quadrupole/linear ion trap mass spectrometer (Applied Biosystems, Framingham, MA). Chromatography was performed using an Agilent 1100 nanoflow LC system (Agilent Technologies) with Solvent A (0.1% formic acid) and Solvent B (90% acetonitrile in 0.1% formic acid). Plasma digests (1-μl injections) were eluted at 200 nl/min with PicoFrit columns (75-μm inner diameter, 10-μm tip opening, New Objective, Woburn, MA) slurry-packed in house with 10–12 cm of ReproSil-Pur C18-AQ 3-μm reverse phase resin (Dr. Maisch GmbH) and with a gradient of 3–20% Solvent B in 3 min, 20–55% Solvent B in 35 min, and 55–90% Solvent B in 3 min. Data acquisition was performed with an ion spray voltage of 2200 V, curtain gas of 20 p.s.i., nebulizer gas of 10 p.s.i., and an interface heater temperature of 150 °C. Collision energy, declustering potential, and collision cell exit potential were optimized for maximum transmission and sensitivity of each MRM transition with infusion of each peptide standard and the Quantification Optimization feature provided in Analyst. Identical declustering potential, collision energy, and collision exit potential values were used for each 12C/13C pair. A dwell time of 10 ms was used for MRM analysis of nonfractionated plasma digests, whereas a dwell time of 25–75 ms was used for SCX-fractionated plasma digests. In all experiments, cycle times did not exceed 1 s and a minimum of five to six data points were collected per peak. Three MRM transitions per peptide were monitored and acquired at unit resolution both in the first and third quadrupoles (Q1 and Q3) to maximize specificity. All LC-MRM/MS experiments were performed in triplicate.
Data analysis was performed using Multiquant development software from Applied Biosystems. The relative ratios of the three transitions selected and optimized for the final MRM assay were predefined in the absence of plasma (i.e. in buffer) for each peptide using the 13C-internal standards (Supplemental Figs. 1A–9A). Comparison of the expected relative ratios with those obtained for the analyte and internal standard peptides in plasma matrix was performed by manual inspection for each 12C/13C-peptide pair. Deviation from the expected relative ratios in any of the three MRM channels was considered to be the result of co-eluting peptide(s) with a transition that falls within the mass width of Q1 and Q3 (i.e. matrix interference). The most abundant transition for each pair was used for quantification unless interference in this channel was observed. For those cases, the second most abundant transition was used. Signal to noise ratio (S/N) was calculated by dividing peak intensity at the apex by 300 and 150 counts/s for nonfractionated and fractionated samples, respectively. These noise levels are conservative estimates based upon visual inspection of preceding regions and postregions around the analyte chromatographic peak. We also evaluated the noise level in the identical transition channels in the absence of spiked proteins. The noise levels defined by this method were equal or slightly lower than those obtained by the method described above with one peptide derived from PSA as an exception.
LC-MS/MS with Full Scan MS/MS Acquisition
Full scan MS/MS spectra were recorded on the doubly charged ion at m/z 636.7 in nonfractionated and SCX-fractionated plasma samples using the 4000 Q Trap with enhanced product ion acquisition. Enhanced product ion spectra were collected in a targeted fashion whereby only m/z 636.7 was transmitted through Q1 throughout the LC gradient. Q1 resolution was set to “low” (1.5 amu full-width half-height), and the scan rate on Q3 was 4000 amu/s.
MS/MS spectra were searched against the NCBI nonredundant database (NCBInr, March 23, 2006 www.ncbi.nlm.nih.gov) using Spectrum Mill MS Proteomics Workbench (Agilent Technologies). MS/MS spectra were extracted and filtered using Spectrum Mill Extractor with sequence tag length >1. Search parameters included carbamidom-ethylation of cysteines; trypsin digestion with two missed cleavages; variable modifications including oxidized methionines, pyroglutamic acid, and carbamylation of lysines; 50% minimum matched peak intensity; precursor mass tolerance of ±2.5 Da; product ion mass tolerance of ±0.7 Da; and scoring settings for electrospray Q Trap.
RESULTS
Design of LOQ Studies for Spiked Proteins
The overall goal of our study was to test the effects of simple plasma processing methods, alone and in combination, on the LOQ, limit of detection (LOD), and assay CV for signature peptides of candidate biomarker proteins in blood. Here we define LOQ and LOD as the concentration at which the S/N of the analyte is equal to 10 and 3, respectively, with noise levels determined as described under “Experimental Procedures.” High priority was placed on defining processing methods that enabled reasonable assay throughput (tens of patient samples/day) with high assay reproducibility and specificity.
Fig. 1 illustrates the sample processing strategy developed. The six target proteins used in all LOQ studies are shown in Table I. Five of the proteins are non-human and therefore not found in blood. For the human target protein PSA, we obtained ELISA results for free and total PSA in nondepleted and depleted female plasma. Results confirmed that both forms of PSA were below the detection limits of the immunoassay (≤0.01 ng/ml for both species). All LOQ studies were performed in female plasma from a healthy donor. Depletion of 7 versus 12 high abundance plasma proteins was compared to determine whether depletion of the additional five proteins from the background matrix had a significant effect on the LOQ in the final MRM assay. In addition to the evaluation of depletion strategies, the effect of fractionation of plasma prior to MS on LOQ was also examined. Following reduction, carbamidomethylation, and enzymatic digestion of spiked, depleted plasma, two separate processing paths were followed (Fig. 1). In path 1, digested plasma was diluted to yield ~0.75 μg of total protein onto the nano-LC column, and the 13C-internal standards were added just prior to direct analysis by LC-MRM/MS. In path 2, peptides were separated by SCX, and the 13C-internal standards were added to the corresponding SCX fraction prior to analysis by LC-MRM/MS. Recovery from immunoaffinity depletion and subsequent concentration steps were evaluated by adding the target proteins to plasma prior to depletion, whereas recovery from SCX was evaluated by adding 12C-synthetic peptides prior to fractionation.
Fig. 1. Experimental flow diagram for LOQ studies.
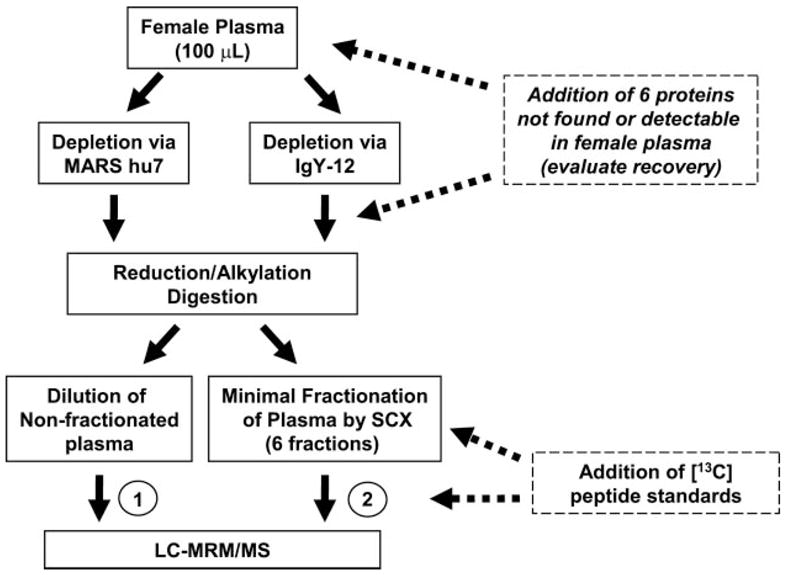
Female plasma from a healthy donor was immunoaffinity depleted of 7 and 12 high abundance proteins via commercial affinity columns. Proteins listed in Table I were added to depleted plasma at 0, 2.5, 5, 10, 25, 50, 100, 250, and 500 ng/ml prior to reduction, alkylation, and digestion. Two separate processing paths were followed after proteolytic digestion. In path 1 (left), digested plasma was diluted to yield ≤1 μg of total protein on column, and internal standards were added just prior to LC-MRM/MS. In path 2 (right), peptides were separated by SCX, and internal standards were added to corresponding SCX pooled fractions prior to LC-MRM/MS. All MRM analyses were performed in triplicate.
Table I. Target proteins and their signature peptides.
Both [12C] and [13C] versions of monitor peptides were synthesized. Uniformly labeled amino acids are indicated in boldface type.
| Protein | Source | MW (kDa) | Signature Peptide | MH+(mono) | Z (Q1) | MRM Transitionsb |
|
|---|---|---|---|---|---|---|---|
| Q1 | Q3 | ||||||
| Aprotinin | bovine lung | 6.4 | AGLCamcQTFVYGGCamcRa | 1488.7 | 2 | 744.8 | 858.3, 959.4, 1087.5 |
| AGLCamcQTF[13C5]VYGGCamcRa | 1493.7 | 2 | 747.3 | 863.4, 964.4, 1092.5 | |||
|
| |||||||
| Leptin | mouse | 10 | INDISHTQSVSAK | 1399.7 | 3 | 467.2 | 543.2, 586.8, 643.8 |
| INDISHTQS[13C5]VSAK | 1404.7 | 3 | 468.9 | 543.2, 589.3, 646.3 | |||
|
| |||||||
| Myoglobin | Horse heart | 17 | LFTGHPETLEK | 1271.7 | 3 | 424.6 | 506.2, 579.7, 716.3 |
| LFTGHPET[13C6]LEK | 1277.7 | 3 | 426.6 | 509.2, 582.8, 722.4 | |||
|
| |||||||
| Myelin Basic Protein | bovine | 18 | HGFLPR | 726.4 | 2 | 363.7 | 385.2, 532.3, 589.3 |
| HGF[13C6]LPR | 732.4 | 2 | 366.7 | 391.2, 538.3, 595.3 | |||
| YLASASTMDHAR | 1322.6 | 3 | 441.5 | 488.2, 523.7, 817.3 | |||
| Y[13C6]LASASTMDHAR | 1328.6 | 3 | 443.5 | 488.2, 523.7, 817.3 | |||
|
| |||||||
| Prostate Specific Antigen | human | 30 | IVGGWECamcEka | 1077.5 | 2 | 539.3 | 808.3, 865.3, 964.4 |
| I[13C5]VGGWECamcEKa | 1082.5 | 2 | 541.7 | 808.3, 865.3, 969.4 | |||
| LSEPAELTDAVK | 1272.7 | 2 | 636.7 | 775.4, 846.4, 943.4 | |||
| LSEPAE[13C6]LTDAVK | 1278.7 | 2 | 639.7 | 781.3, 852.5, 949.4 | |||
|
| |||||||
| Peroxidase | Horse Radish | 48 | DTIVNELR | 959.5 | 2 | 480.3 | 531.2, 630.3, 743.4 |
| DTIVNE[13C6]LR | 965.5 | 2 | 483.3 | 537.3, 636.3, 749.4 | |||
| SSDLVALSGGHTFGK | 1475.7 | 3 | 492.6 | 703.3, 790.3, 974.5 | |||
| SSDLVA[13C6]LSGGHTFGK | 1481.8 | 3 | 494.6 | 703.3, 790.3, 980.5 | |||
Cysteines are synthesized as carbamidomethyl cysteines.
All three transitions were monitored. However, only the transition in bold was used to determine LOQ.
Peptides derived from human PSA and the five non-human proteins were selected as peptide internal standards based upon experimental observations in an LC-MS/MS analysis of each protein digested separately with trypsin. Our goal in peptide selection was to design MRM assays for two to three peptides per protein due to the fact that different peptides from the same protein can vary widely in their MS response and recovery from sample processing and because of the high potential for interference in plasma. We prejudiced our selection for moderately hydrophobic peptides that exhibit good chromatographic peak shape on reverse phase chromatography and for peptides with good ionization efficiency (as indicated by relative abundance) and high MS/MS spectral quality. For those proteins with human homologs to our spiked ones, we chose unique sequences to minimize inaccurate quantitative measurements due to the presence of endogenous forms of our signature peptides. Initially 13 13C-labeled peptides representing six proteins were synthesized. However, four of these peptides were eliminated due to early elution on reverse phase resin and/or poor chromatographic peak shape. These peptides included a third peptide derived from PSA and HRP and a second peptide derived from leptin and myoglobin. Ionization efficiency, fragmentation patterns, LC behavior, and SCX elution for all peptides were studied and optimized for the final MRM assay.
MRM Assay Configuration and Optimization
The main limitation to unbiased proteomics biomarker discovery and the targeted, candidate-based verification approaches described here is the extremely large concentration range and complexity of the proteins in the biofluids analyzed. Triple quadrupole mass spectrometers, the instruments of choice for targeted analysis, are generally considered to be low resolving instruments with resolution of only ~1000–3000. Therefore, the potential for analyte-on-analyte interference and chemical noise is greater on these instruments when the analysis is performed in a complex background such as plasma. For example, two co-eluting peptides differing in mass by up to 2 Da will have overlapping isotopic distributions, which in a targeted, MRM analysis will likely result in ion contributions from both species residing in the Q1 mass transmission window at unit resolution. Another potential source of interference is in-source fragmentation of abundant peptides where the fragment ions rather than the precursors are the source of interference. Interference at the product ion level (Q3) is caused by coincidence of a primary or secondary fragment of the precursor that has the same or nearly the same mass as the analyte transition of interest. The extent to which interference will be a significant problem depends upon the detection level one is trying to achieve for the true analyte and the abundance of the peptides giving rise to the interferences. Therefore, it is critical for MRM assay development on these instruments to select transition ions that maximize specificity and potentially minimize interferences from co-eluting species that fall within the mass windows of the analyzers. Because it is not yet possible to reliably predict the likelihood of having an interference, we approach this experimentally by selecting a minimum of three transitions per peptide to increase specificity and selectivity of the assay in plasma (Table I). In general, transitions were chosen based upon relative abundance and m/z greater than the precursor m/z in the full scan MS/MS spectrum recorded on the 4000 Q Trap mass spectrometer. MRM assays for each signature peptide were optimized for collision energy to maximize transmission and sensitivity of the transitions being monitored. The final MRM method included 60 optimized MRMs for the six test proteins.
Following construction of the final MRM method, LC retention, MS detectability, and linearity were evaluated by titration curves in 0.1% formic acid (no biological matrix) using heavy labeled synthetic peptides only. Most peptides showed good linearity over the 0.1–500 fmol/μl concentration range. However, as expected, equimolar peptides showed different MS responses (Supplemental Fig. 10). Although only the 13C-peptides were used, we monitored the m/z channel for the all-12C-analyte to determine the percentage of the fully unincorporated 13C for the heavy amino acids leucine and valine. Using peak area ratios for the 12C and 13C transitions at the high end of the titration curves, we determined that there was ≤1% residual for each 13C-peptide standard (Supplemental Figs. 11, B–F, and 12, B–F). These results were consistent with full scan MS spectra recorded with high resolution on the heavy labeled peptides, LFTGHPET[13 C6]LEK (myoglobin) and I[13 C5]VGGWECamcEK (PSA) on the Orbitrap. Supplemental Figs. 11A and 12A show the distribution of 13C incorporation for [13C6]leucine and [13C5]valine, respectively. Given that the triple quadrupole instrument is operating at unit resolution, both the 13C0 and the 13C1 species will be detected in the analyte channels as noted above. The percentages of unincorporated 13C were used to determine the amounts of internal standard to add for quantitative measurements to minimize the contribution of residual in the 12C channels. The level of the isotopic impurity in the internal standard (which cannot be completely avoided) effectively limits the maximum useable ratio of analyte to internal standard.
Effect of Abundant Protein Depletion on LOQ
Previous studies in our laboratory with PSA spiked into depleted plasma showed improved LOQ for quantitative measurements of PSA (data not shown) as compared with reported studies for LOQ of proteins in nondepleted plasma (21, 22). Therefore, we evaluated the effect of depletion of plasma on LOQ in LC-MRM/MS using two different columns, the MARS hu7 (Agilent) and IgY-12 (Beckman Coulter) for depletion of 7 and 12 high abundance proteins, respectively. Calibration curves focused in the low nanogram/milliliter range were generated by adding each test protein to MARS-hu7 and IgY-12 depleted plasma at 0, 2.5, 5, 10, 25, 50, 100, 250, and 500 ng/ml. Each concentration point was digested in triplicate representing three biological replicates, and each LC-MRM/MS was performed in triplicate (technical replicates). The data are summarized in Table II and discussed in detail for several of the proteins below.
Table II. Results of quantitative measurements for target proteins spiked into IgY-12 and MARS hu7 depleted plasma.
Three biological replicates were used to generate each concentration point, and each MRM experiment was performed in triplicate for n = 9. LOQ for each depletion strategy is indicated in bold.

Value listed is average of 9 MRM experiments with 0.1 ml of starting plasma. Calculated concentration (ng/ml) × area ratio × 5 pmol/mL IS × protein MW pg/pmol × 1 ng/1000 pg × dilution factor where the dilution was 3 and 10 for IgY-12 and MARS hu7 depleted plasma, respectively.
Value listed is the average of 9 MRM experiments. Fmol peptide detected on column with 1 μl injection × area ratio × 5 fmol IS.
Average S/N for 9 MRM experiments. S/N calculation is as described in text.
Fig. 2 shows the calibration curve for the INDISHTQSVSAK peptide derived from leptin in both backgrounds. The area ratio of light peptide (analyte) to heavy peptide (internal standard) was determined from the extracted ion chromatogram (XIC) of the most abundant transition for the pair, 467.2/643.8 and 468.9/646.3, respectively. These curves in both depleted plasma matrices demonstrate linearity of about 2 orders of magnitude for the concentration range tested and are representative of linearity of the other target proteins with the exception of MBP. The relative ratios of the three transitions monitored for the INDISHTQSVSAK peptide in buffer only are shown in Fig. 3, A and D, and provide the ideal ratios of the transition ions in the absence of interference. Fig. 3, B and E, are the corresponding data for this peptide obtained by spiking 25 ng/ml leptin protein into IgY-12 depleted plasma and processed as shown in Fig. 1 (path 1). The relative ratios of the transitions agree closely with those observed in buffer indicating that no interference from the matrix is present in these channels. The most abundant transition, 467.2/643.8, was used to quantify leptin in this sample and across all concentration points. The S/N for this transition in 25 ng/ml spiked plasma was 16 (Fig. 3B, blue), indicating that the LOQ for leptin in IgY depleted plasma is 25 ng/ml (Table II). The precision/accuracy of this measurement across three biological replicates at this concentration was 12% CV.
Fig. 2. Calibration curve for quantifying mouse leptin in MARS hu7 and IgY-12 depleted plasma.
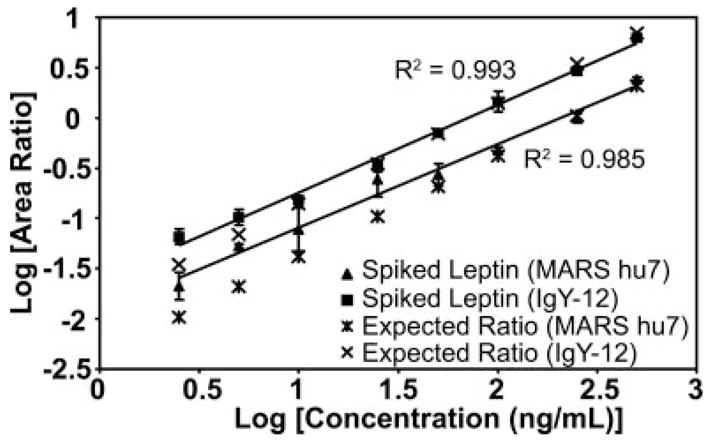
The area ratio of light peptide to heavy peptide was determined from the extracted ion chromatograms of the 467.2/643.8 and 468.9/646.3 transitions, respectively, and plotted versus protein concentration. Three biological replicates for each concentration point were generated in MARS hu7 (closed triangle) and IgY-12 (closed square) depleted plasma and analyzed in triplicate by MRM (n = 9). Error bars indicate S.D. of the measurements. Raw data are shown in Fig. 3.
Fig. 3. Extracted ion chromatograms (A–C) and MRM spectra (D–F) of transitions monitored for mouse leptin in buffer (A and D), IgY-12 (B and E), and MARS hu7 (C and F) depleted plasma.
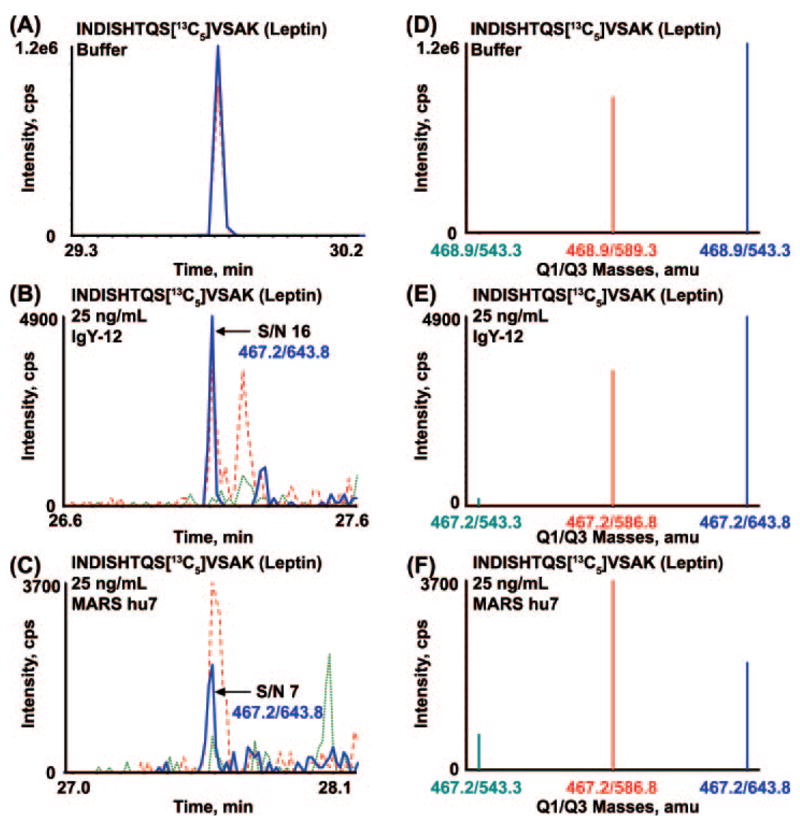
Overlay of XICs and the corresponding MRM spectra for the three transitions monitored for INDISH-TQSVSAK peptide of leptin in 0.1% formic acid (A and D) and the same peptide produced by digestion of leptin protein that had been spiked at 25 ng/ml into IgY-12 depleted plasma (B and E) and into MARS hu7 depleted plasma (C and F). XICs and ions in the MRM/MS spectra are color-coordinated. cps, counts/s.
In contrast, the relative ratios of the monitored transitions for the leptin peptide in MARS hu7 depleted plasma do not correlate with the expected ratios in buffer alone. Fig. 3, C and F, show the overlay of the XICs and the MRM spectrum, respectively, recorded on the three transitions for the INDISH-TQSVSAK analyte peptide in MARS hu7 depleted plasma spiked with 25 ng/ml leptin. The ratio of the 467.2/586.8 transition (Fig. 3, C and F, red) to the 467.2/643.8 transition (Fig. 3, C and F, blue) is ~1.0:0.6 compared with ~0.7:1.0 in buffer alone, indicating the presence of a significant interference from the plasma matrix in this channel. The S/N of the 467.2/643.8 transition (Fig. 3C, blue) is 7, which is approaching the limit of detection for this peptide in MARS hu7 depleted plasma. The LOQ for leptin protein spiked into MARS hu7 depleted plasma was 50 ng/ml with a CV of 26% and was based on an average S/N of 10 (Table II).
A similar analysis of the signal to noise ratios for the remaining signature peptides derived from the other target proteins showed similar improvements in S/N, LOQ, and CV using IgY-12 depleted plasma versus the MARS depleted plasma (Table II). With the exception of MBP, the LOQs for these target proteins were 25 ng/ml in IgY-12 depleted plasma and ≥50 ng/ml in MARS-Hu7 depleted plasma. The percent CVs of the quantitative measurements at the specified LOQ for all test proteins (Table II, bold) were <10% for five of eight measurements in IgY-12 depleted plasma and four of eight measurements in MARS hu7 depleted plasma. No LOQ for PSA based upon the LSEPAELTDAVK peptide was reported due to the significant amount of interference observed in the most abundant MRM channel for this analyte in depleted plasma with no PSA added (see below for further discussion). The amount of total protein loaded onto the nano-LC column in all experiments was normalized for both depletion strategies by dilution of MARS-Hu7 and IgY-12 depleted plasma by 1:10 and 1:3, respectively. These dilution factors were defined by previous experiments that showed that injection of ≤1 μg of total protein onto nano-LC columns is optimum for reproducible chromatographic peak shape and robust quantitative measurements (data not shown). In effect, the removal of five additional proteins with IgY-12 versus MARS hu7 reduces the total protein concentration, allowing more of the remaining sample (including the analytes of interest) to be injected and analyzed. Based upon these results, all subsequent experiments were performed in IgY-12 depleted plasma.
Evaluation of Digestion Efficiency
Since all quantitative measurements for our target proteins are reliant upon recovery of signature peptides from proteolytic digestion, we wanted to determine and better understand the effect that digestion may have on LOQ. To test digestion efficiency, we generated calibration curves for all six target proteins with 12C synthetic forms of our signature peptides. To directly compare the results of the 12C-peptide curves with those generated with intact protein, we added the light signature peptides to IgY-12 depleted and digested plasma at protein concentrations equivalent to those that were used previously. Supplemental Fig. 1C shows the comparison of the spiked light peptide and intact protein curves for the AGLCamcQTFVYG-GCamcR peptide derived from aprotinin. Both curves show excellent correlation with the expected ratio and indeed have virtually the same line shape and behavior across the concentration range. These results were typical of the rest of the signature peptides with the exception of MBP (see below). Therefore, we conclude that our digestion efficiency and reproducibility are high and do no contribute significantly to measurement variability in our studies.
Use of Multiple Peptides to Determine LOQ in Plasma
It is well known that equimolar peptides derived from the same protein can have varying MS detectability due to different ionization efficiencies. Reliable programs for predicting de novo which peptides from a protein will give the best MS response have yet to be developed. For these reasons, multiple peptides per protein are selected for MRM assay development with the assumption that at least one will be a useful surrogate for the target protein in the final MRM assay. For the determination of LOQ for HRP and MBP, we selected two peptides from each protein. However, each of these signature peptides yielded a different LOQ for its respective protein in IgY-12 depleted plasma (Table II). To determine the cause of these differences, we again compared calibration curves generated at equivalent concentrations with 12C-synthetic peptides and intact protein already described above. Supplemental Figs. 8C and 9C show the protein and 12C-synthetic peptide calibration curves for peptides DTIVNELR and SS-DLVALSGGHTFGK derived from HRP in nonfractionated, IgY-12 depleted plasma. Curves for the identical signature peptide behaved similarly regardless of the source of analyte peptide (i.e. spiked intact HRP followed by digestion or spiked 12C-synthetic peptide into digested, depleted plasma). However, each signature peptide behaved differently relative to the expected ratio (Supplemental Figs. 8C and 9C). Curves generated for spiked intact protein added to depleted plasma prior to digestion (closed triangle) and 12C-synthetic peptides spiked into digested, depleted plasma (closed square) for the SSDLVALSGGHTFGK sequence fall close to the expected ratios (Supplemental Fig. 9C), whereas the same curves generated for the DTIVNELR peptide (Supplemental Fig. 8C) are lower than the expected ratios, indicating that there is an issue with this peptide that is unrelated to proteolytic digestion, e.g. solubility. Nevertheless DTIVNELR is detected with greater efficiency than SSDLVALSGGHTFGK.
Figs. 4 and 5 show two different views (left versus right) of the three transitions monitored for the DTIVNELR peptide and the SSDLVALSGGHTFGK peptide of HRP, respectively. The relative ratios of the three transitions being monitored for each peptide were determined without plasma background (Figs. 4, A and D, and 5, A and D) and compared with those signals obtained in IgY-12 depleted plasma with 25 ng/ml (Figs. 4, B and C, and 5, B and C) and 100 ng/ml spiked HRP (Figs. 4, C and F, and 5, C and F). For each peptide, the relative ratios of the transitions monitored with each concentration point of added HRP agree with that observed without plasma, indicating that no interference from the matrix is present. Quantification of each analyte peptide derived from HRP was based upon the most abundant transition for each signature peptide (480.3/630.3 for DTIVNELR and 492.6/790.4 for SSDLVALS-GGHTFGK), and the S/Nfor these same transitions was calculated for each concentration point shown (Fig., 4B and C, blue; Fig. 5, B and C, blue). The lower LOQ for the DTIVNELR peptide in nonfractionated, depleted plasma as compared with that for SSDLVALSGGHTFGK of HRP is due to the about 3-fold better S/N for the former.
Fig. 4. Extracted ion chromatograms (A–C) and MRM spectra (D–F) of transitions monitored for DTIVN-ELR derived from HRP in nonfractionated, depleted plasma.
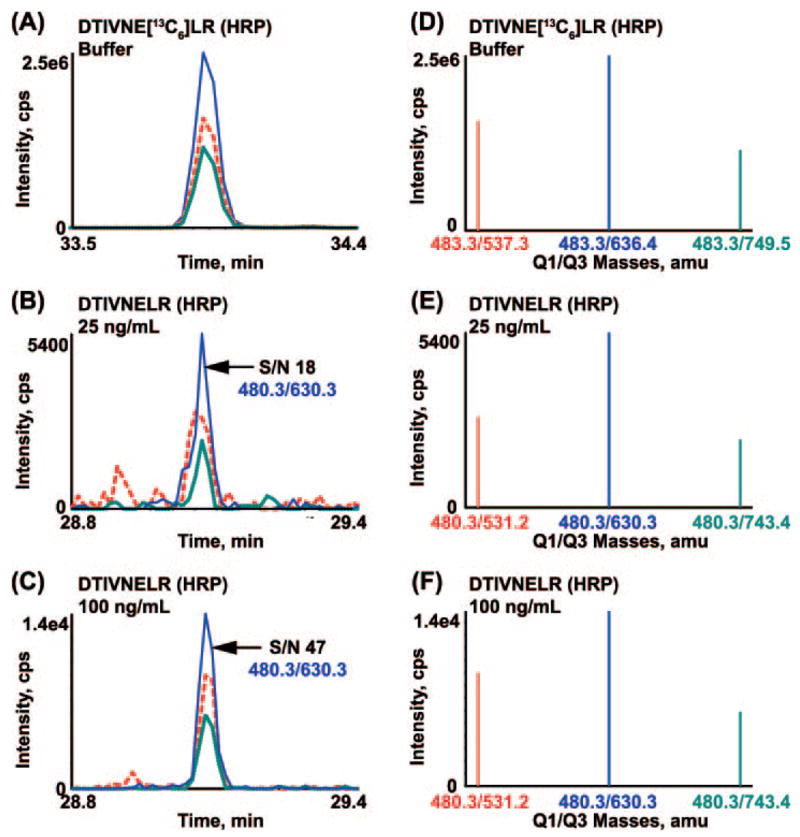
Overlay of XICs and the corresponding MRM spectra for the three transitions monitored for DTIVNELR peptide of HRP in 0.1% formic acid (A and D) and the same peptide produced by digestion of HRP protein that had been spiked at 25 (B and E) or 100 ng/ml (C and F) into IgY-12 depleted plasma. XICs and ions in the MRM/MS spectra are color-coordinated. cps, counts/s.
Fig. 5. Extracted ion chromatograms (A–C) and MRM spectra (D–F) of transitions monitored for SS-DLVALSGGHTFGK derived from HRP in nonfractionated, depleted plasma.
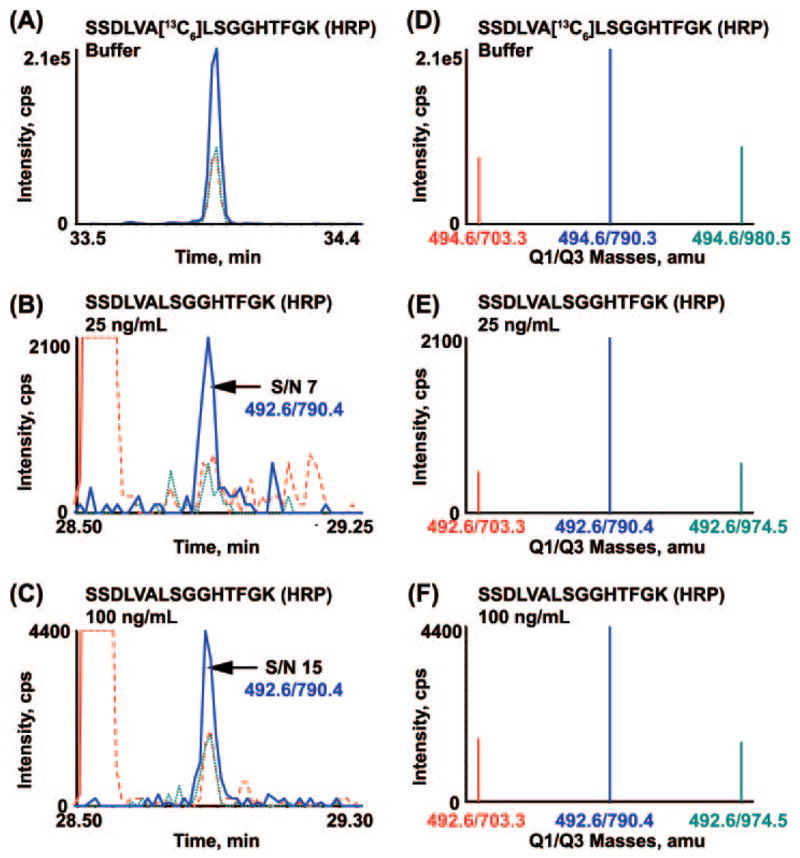
Overlay of XICs and the corresponding MRM spectra for the three transitions monitored for SSDLVALSGGHTFGK peptide of HRP in 0.1% formic acid (A and D) and the same peptide produced by digestion of HRP protein that had been spiked at 25 (B and E) or 100 ng/ml (C and F) into IgY-12 depleted plasma. XICs and ions in the MRM/MS spectra are color-coordinated. cps, counts/s.
Supplemental Figs. 4C and 5C show the protein and 12C-synthetic peptide curves for peptides HGFLPR and YLASAS-TMDHAR derived from MBP in nonfractionated, IgY-12 depleted plasma, respectively. Both curves generated by the addition of light synthetic peptides fall above the expected peak area ratios for the range of concentrations analyzed, indicating that these signature peptides respond well in the MRM assay. However, calibration curves generated from the spike of intact protein prior to digestion are lower than the expected ratio, indicating that a problem exists either in the original protein stock concentration or in the digestion efficiency of this protein. Analysis by one-dimensional SDS-PAGE showed the purity of this protein to be ~50% (data not shown), which is likely the reason for the higher LOQ obtained for MBP.
Effect of SCX Fractionation on LOQ
Identity-based biomarker discovery has become reliant upon multidimensional fractionation at the protein and/or peptide level to improve detection of proteins of lower abundance. However, this strategy has had a negative impact on overall sample throughput with small numbers of patient samples as input and large numbers of fractions per patient to analyze. Extensive fractionation of digested plasma to improve limits of quantitation and detection for verification studies is not a practical solution because the number of cases and controls required for analysis to progress a candidate biomarker into preclinical validation is in the tens to a few hundred. To take advantage of the perceived benefit that fractionation can have on increased detection and at the same time maintain reasonable throughput, we devised a strategy for limited SCX fractionation of depleted and digested plasma that relies on the generation of SCX pools (n < 10) containing multiple signature peptides per pool.
To test our hypothesis, we separated the 0, 2.5, 5, 10, 25, 50, and 100 ng/ml spiked tryptic digests of plasma by strong cation exchange chromatography. To directly compare the effect of fractionation on LOQ with our nonfractionated results, we used the identical samples for SCX that were used to generate the calibration curves in nonfractionated plasma. Supplemental Fig. 13 shows the overlay of the elution profiles for the SCX separation of the 10 ng/ml spiked plasma digest with the separation of the signature peptides in the absence of plasma. Retention time shifts for the signature peptides were routinely monitored by injection of the standards pre- and post-limited SCX fractionation of the digested plasma samples and showed minimal change. Blank injections were performed prior to injection of each plasma sample to eliminate any 13C-peptide carryover. Six pools of SCX fractions were generated to encompass eluting peptides in a single pool if possible and to increase analysis throughput (Supplemental Fig. 13).
Analysis of all of the signature peptides for the target proteins shows that fractionation improves S/N of the analyte transitions by 10- to 40-fold, resulting in LOQ for all test proteins at or below 2.5 ng/ml in plasma (Table III). For example, the calibration curves for the LFTGHPETLEK peptide derived from myoglobin in nonfractionated and fractionated plasma are shown in Supplemental Fig. 3D. In SCX-fractionated plasma, calculated concentrations based upon the area ratio of analyte to IS correlate well with the expected concentrations across the range tested. In contrast, the curve generated in nonfractionated plasma has a different shape and slope with respect to the curve generated in SCX-fractionated plasma and indeed deviates significantly from the expected concentrations at the low end of the curve. This latter observation is most likely due to the presence of interferences at the lower levels of analyte. With SCX fractionation, the LOQ for the LFTGHPETLEK peptide improves to 2.5 ng/ml with 9% CV as opposed to 25 ng/ml with 16% CV in depleted, non-fractionated plasma.
Table III. Results of quantitative measurements for target proteins in SCX fractionated, depleted plasma.
Two replicates from experiments in non-fractionated plasma were pooled and separated via SCX. All MRM experiments were performed in triplicate. LOQ for each processing strategy is indicated in bold.
| SCX Fractionated
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Protein | Signature Peptide | Q1/Q3 | Target ng/mL | 0 | 2.5 | 5 | 10 | 25 | 50 | 100 |
| Aprotinin | A GLCQTFVYGGCR | 744.8/959.4 | Calculated ng/mLa | - | 1.95 | 2.45 | 5.41 | 11.24 | 26.04 | 24.18 |
| % CV | - | 14.2 | 12.9 | 2.0 | 10.1 | 13.1 | 19.1 | |||
| Peptide Amount (fmol)b | - | 1.20 | 1.50 | 3.32 | 6.91 | 16.00 | 14.85 | |||
| S/Nc | <3 | 15 | 45 | 68 | 438 | 57 | 308 | |||
|
| ||||||||||
| Leptin | INDISHTQSVSAK | 467.2/643.8 | Calculated ng/mLa | - | 1.62 | 3.56 | 5.03 | 10.82 | 20.36 | 40.90 |
| % CV | - | 9.8 | 3.9 | 5.6 | 25 | 25.7 | 4.2 | |||
| Peptide Amount (fmol)b | - | 0.41 | 0.89 | 1.26 | 2.70 | 5.09 | 10.23 | |||
| S/Nc | < 3 | 131 | 174 | 184 | 474 | 743 | 1880 | |||
|
| ||||||||||
| Myo | LFTGHPETLEK | 424.6/579.7 | Calculated ng/mLa | - | 2.81 | 5.21 | 6.56 | 15.69 | 24.31 | 40.85 |
| % CV | - | 8.6 | 6.8 | 2.8 | 1.9 | 14.5 | 6.2 | |||
| Peptide Amount (fmol)b | - | 0.66 | 1.23 | 4.55 | 3.70 | 5.74 | 9.64 | |||
| S/Nc | <3 | 16 | 32 | 98 | 154 | 97 | 475 | |||
|
| ||||||||||
| MBP | H GFLPR | 363.7/589.3 | Calculated ng/mLa | - | 0.93 | 1.42 | 2.30 | 6.09 | 11.49 | 18.48 |
| % CV | - | 12.9 | 9.2 | 0.9 | 6.2 | 5 | 17.1 | |||
| Peptide Amount (fmol)b | - | 0.20 | 0.31 | 0.50 | 1.33 | 2.51 | 4.04 | |||
| S/Nc | < 3 | 27 | 17 | 29 | 78 | 197 | 353 | |||
|
| ||||||||||
| MBP | YLASASTMDHAR | 441.5/523.7 | Calculated ng/mLa | - | 0.62 | 1.02 | 1.64 | 3.21 | 7.49 | 13.67 |
| % CV | - | 10.2 | 5.7 | 27.1 | 26 | 2.3 | 32.4 | |||
| Peptide Amount (fmol)b | - | 0.14 | 0.22 | 0.36 | 0.70 | 1.63 | 2.98 | |||
| S/Nc | < 3 | 10 | 18 | 26 | 219 | 158 | 284 | |||
|
| ||||||||||
| PSA | I VGGWECEK | 539.3/865.3 | Calculated ng/mLa | 0.39 | 2.20 | 4.95 | 7.25 | 15.93 | 30.50 | 55.78 |
| %CV | 11.2 | 2.8 | 4 | 3.1 | 9.4 | 3.3 | 9.8 | |||
| Peptide Amount (fmol)b | 0.05 | 0.31 | 0.69 | 1.01 | 2.22 | 4.25 | 7.76 | |||
| S/Nc | 5 | 24 | 67 | 89 | 234 | 260 | 289 | |||
|
| ||||||||||
| PSA | LSEPAELTDAVK | 636.8/943.4 | Calculated ng/mLa | - | 9.58d | 12.66d | 23.74d | 50.511d | 48.2 d 8 | 115.68d |
| % CV | - | 37.0 | 8.7 | 11.1 | 5.7 | 6.6 | 10.7 | |||
| Peptide Amount (fmol)b | - | 1.33 | 1.76 | 3.30 | 7.03 | 20.64 | 16.10 | |||
| S/Nc | < 3 | 15 | 36 | 51 | 187 | 71 | 42 | |||
|
| ||||||||||
| HRP | D TIVNELR | 480.3/531.2 | Calculated ng/mLa | - | 0.94 | 1.69 | 2.50 | 6.30 | 15.02 | 18.18 |
| % CV | - | 6.6 | 5.6 | 10 | 19.9 | 4.5 | 10 | |||
| Peptide Amount (fmol)b | - | 0.10 | 0.17 | 0.26 | 0.65 | 1.55 | 1.87 | |||
| S/Nc | <3 | 14 | 21 | 48 | 76 | 36 | 15 | |||
|
| ||||||||||
| HRP | SSDLVALSGGHTFGK | 492.6/790.3 | Calculated ng/mLa | - | 2.54 | 6.38 | 11.70 | 21.79 | 44.31 | 55.52 |
| % CV | - | 2.6 | 11.8 | 16.2 | 6.5 | 2.2 | 4.7 | |||
| Peptide Amount (fmol)b | - | 0.26 | 0.66 | 1.21 | 2.25 | 4.57 | 5.72 | |||
| S/Nc | < 3 | 15 | 16 | 70 | 93 | 67 | 139 | |||
Value listed is average of 3 MRM experiments. Calculated concentration (ng/ml) = (area ratio × 5 pmol/mL IS × MW pg/pmol × 1 ng/1000 pg × 0.025 mL)/0.100 ml where 0.025 ml = SCX reconstitution volume and 0.100 ml is the starting volume of IgY-12 depleted plasma.
Fmol peptide detected on column with 1 μl injection = area ratio × 5 fmol IS.
Average S/N of 3 MRM experiments for SCX fractionated plasma. S/N calculation is as described in text.
The LSEPAELTDAVK peptide derived from PSA eluted into 2 separate SCX fractions. Interference in the 636.7/943.4 transition in the 2nd fraction precluded its use for quantification, and another transition was used. Value listed was determined from the 636.7/943.4 and 636.7/775.4 transitions for SCX fraction 1 and SCX fraction 2, respectively.
With SCX fractionation, the amount of peptide detected on column is as low as 100–200 amol for some peptides. The signature peptide, INDISHTQSVSAK from leptin, is particularly well behaved. Fig. 6 shows the XICs of the three transitions monitored for the leptin peptide in nonfractionated (Fig. 6, A–C) and SCX-fractionated (Fig. 6, D–F) depleted plasma. Fig. 6, A and B, show no significant signal in the MRM channels at the retention time where the 13C-internal standard peptide elutes (Fig. 6, A and B, insets) for the 0 and 2.5 ng/ml additions of leptin, respectively, in nonfractionated depleted plasma. In Fig. 6C (inset), the relative ratios of the transitions and the retention time correlate with that of the internal standard and show an S/N of 16 for the most abundant transition, 467.2/643.8. In the SCX-fractionated, depleted plasma, an S/N of 127 was observed for the identical transition in the 2.5 ng/ml spike of leptin (Fig. 6E) with no appreciable signal in any of the MRM channels with no leptin added (Fig. 6D). Therefore, LOQ for leptin in depleted, SCX-fractionated plasma can be extrapolated to the picogram/milliliter range.
Fig. 6. Extracted ion chromatograms of MRM transitions monitored for the INDISHTQSVSAK peptide derived from mouse leptin in nonfractionated (A–C) and SCX-fractionated (D–F), depleted plasma.

Each XIC displays an overlay of three MRM transitions being monitored for the INDISH-TQSVSAK peptide (467.2/543.3 (green), 467.2/586.8 (red), and 467.2/643.8 (blue). Each inset XIC displays the MRM transitions for the internal standard peptide, indicating retention time for the analyte peptide. The 467.2/643.8 transition (blue) was used for quantification. Overlay of MRM transitions for leptin spiked into depleted, nonfractionated plasma at 0 (A), 2.5 (B), and 25 ng/ml (C) is shown. Overlay of MRM transitions for leptin spiked into depleted plasma followed by SCX fractionation at 0 (D), 2.5 (E), and 25 ng/ml (F) is shown. The S/N at 2.5 and 25 ng/ml was 127 and 447, respectively. cps, counts/s.
Analysis of Interferences from Co-eluting Peptides: A Case Study of PSA
Despite the high specificity and selectivity of the MRM assay, peptides and other small molecules in the biological matrix can produce interferences in the m/z channels monitored, resulting in inaccurate quantitative measurements and overestimation of LOQ and LOD. For example, we have noted significant interference with the signature peptide LSEPAELTDAVK derived from PSA. The measured levels of free and total PSA in the female plasma we used were both below 0.01 ng/ml, so these experiments were carried out by addition of PSA protein. The calibration curves for this PSA-derived peptide provided the first indication of plasma interference (Fig. 7). The area ratios obtained by measuring LSE-PAELTDAVK from either exogenous PSA protein (red triangles) or spiked 12C-synthetic peptide (red squares) relative to 13C-synthetic peptide in nonfractionated plasma were both well above the expected area ratio even at high protein or peptide concentration. The other signature peptide from PSA, IVGGWECamcEK, tracks much closer to the expected area ratio, indicating little to no interference. Consistent with these observations are the high percent relative errors (>400%) calculated for the LSEPAELTDAVK peptide in non-fractionated plasma with respect to the target concentration across all concentrations of spiked protein (Table II). To determine whether the stock protein concentration for PSA was in error, we obtained ELISA results for two additions of PSA to IgY-12 depleted female plasma, 10 and 25 ng/ml, which were consistent with our target concentrations (11.82 and 27.80 ng/ml, respectively).
Fig. 7. Protein and 12C-synthetic peptide calibration curves for quantifying human PSA in nonfractionated, depleted plasma.
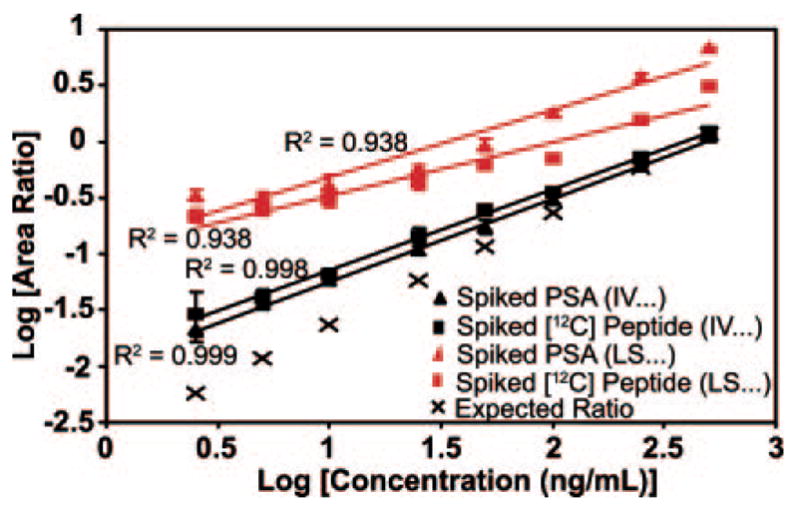
Calibration curves were generated with intact PSA added to depleted plasma prior to digestion and with 12C-synthetic peptides added to digested, depleted plasma. Two peptides, IVGGWECamcEK and LSE-PAELTDAVK, were used to quantify PSA by using the area ratio of light peptide to heavy peptide from the XICs of the 539.3/865.4 and 636.7/943.4 transitions, respectively. The concentration of 12C-peptide added was equivalent to the concentration of intact protein added. Three biological replicates for each concentration point were generated with intact PSA, whereas the calibration curves with 12C-synthetic peptides were generated once. Each point for all curves was analyzed in triplicate by MRM. Error bars indicate S.D. of the measurements.
The full scan MS/MS spectrum of the authentic PSA peptide (Fig. 8A) is dominated by the y9 ion formed by gas phase cleavage N-terminal to proline. Consequently the m/z 636.7/943.5 transition is an obvious one to monitor by MRM. We also monitored two additional transitions for this peptide (Table I and Supplemental Fig. 7A). To corroborate the presence of an interference in one or more of these channels, we recorded the full scan MS/MS spectrum of m/z 636.7, the (M ± 2H)2+ of the LSEPAELTDAVK peptide in female plasma with and without the addition of exogenous PSA protein on the 4000 Q Trap (Fig. 8, B and C, respectively). A co-eluting peptide with a precursor mass residing within the mass window of Q1 is observed in the MS/MS spectrum obtained in the absence of added PSA. This interference yields a product ion at 943.4, coinciding with the most abundant transition for the authentic PSA peptide, LSEPAELTDAVK (Fig. 8B). A co-mixture of the sequence ions for the PSA peptide and the interfering peptide is observed in the MS/MS spectrum obtained from plasma samples spiked with 100 ng/ml PSA (Fig. 8B). Clearly the presence of a strong interference in this channel would lead to a significant overestimation of the level of PSA and the LOQ for its detection in plasma at levels at or below 100 ng/ml. Without monitoring the relative ratios of multiple transitions for this peptide, it would have been difficult to identify the presence of an interfering peptide. Knowing that interference was present at levels much greater than the desired LOQ range (low nanograms/milliliter), we constructed an assay using another PSA peptide, IVGGWECamcEK (Table I). This peptide does not suffer significant interference from plasma in the m/z transitions monitored, and we were able to develop an assay for PSA with an LOQ of ~25 ng/ml in nonfractionated plasma (Table II).
Fig. 8. Full scan MS/MS spectra recorded on the doubly charged ion at m/z 636.7.
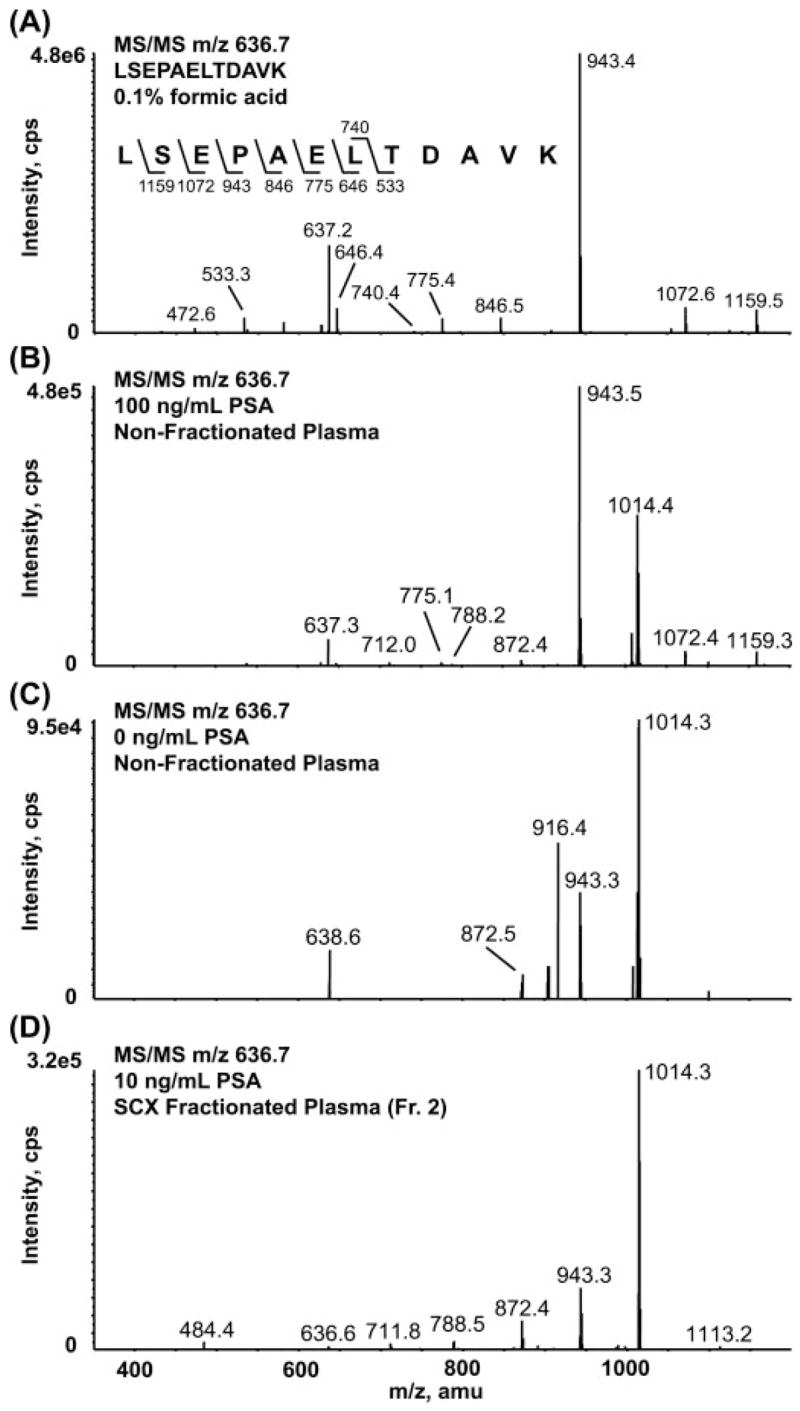
A, 12C-synthetic peptide LSEPAELTDAVK in 0.1% formic acid. B, 100 ng/ml spike of PSA in nonfractionated, depleted plasma. C, 0 ng/ml sample where no analyte protein or peptide was added. D, 10 ng/ml spike of PSA in SCX-fractionated, depleted plasma. The sequence-specific fragment ion at m/z 943.4 (y9) for the LSEPAELTDAVK peptide derived from PSA is observed in the 0 ng/ml sample (C), representing an interfering component in this channel from the matrix that limits the usefulness of this peptide for quantifying PSA in nonfractionated plasma. Fr., fraction; cps, counts/s.
Interestingly the interference of the LSEPAELTDAVK peptide persists in SCX-fractionated plasma where the interfering peptide is purified together with a fraction of the authentic PSA peptide. To obtain the identity of the interfering peptide, the MS/MS spectrum of 636.7 was recorded in the SCX fraction in which the interfering peptide co-purifies with the PSA peptide (Fig. 8D). Spectra were searched against the NCBInr database using Spectrum Mill MS Proteomics Workbench but did not produce a match. De novo interpretation of the MS/MS spectrum yielded the partial sequence [159]VAA-CamcNLPIVR where 159 is the residual mass that must be present at the N terminus to account for the mass of the peptide. This sequence matches a tryptic peptide from α1 microglobulin (TVAACamcNLPIVR) with a modification on the N terminus. The isotopic distribution of the precursor ion as well as the y8 fragment ion from the interpreted sequence overlaps with the precursor ion and the y9 fragment ion from the PSA peptide. Therefore, the interference observed in the 636.7/943.4 channel during MRM analysis is a 13C isotope of the interpreted sequence. Synthetic peptides corresponding to the α1-microglobulin sequence as well as permutations of the N terminus are being synthesized to confirm the identity of the co-eluting peptide.
Recovery of Protein after Depletion and SCX Fractionation
To evaluate loss of protein at the immunoaffinity depletion and subsequent concentration step, we added five of the six target proteins to nondepleted plasma prior to any processing at concentrations ranging from 5000 to 2.5 ng/ml (Fig. 1). Aprotinin was eliminated from this analysis because of its low molecular weight with respect to the 5000 molecular weight cutoff filter that was used for the concentration step. An aliquot of the nondepleted plasma spiked with proteins at 5000 ng/ml was reserved for LC-MRM/MS following tryptic digestion, and the remaining undigested samples were depleted via IgY-12, concentrated, and digested as described under “Experimental Procedures.” Percent recoveries were determined by LC-MRM/MS of the signature peptides pre-and postdepletion (without SCX fractionation) for those spike levels at which the signature peptides could be detected (Supplemental Table 1). To determine protein losses at the lowest protein spike levels, calibration curves were generated for the five proteins added prior to depletion of the plasma, and these samples were then depleted, fractionated by SCX, and analyzed by LC-MRM/MS as described. As anticipated, percent protein recoveries varied depending upon the specific protein. Three of the five proteins exhibited recoveries of 80–100%; PSA was recovered at ~30%. Importantly protein recoveries at the lowest spike levels were similar to those observed at 5000 ng/ml, indicating that the reproducibility of recovery is high and consistent across concentrations. MBP was not detected. By concentrating a stock solution of MBP and analyzing the retentate by gel electrophoresis, we have determined that this protein is being lost at the membrane filtration step (data not shown).
We also evaluated peptide recovery at the SCX fractionation and desalting steps separately from protein depletion by spiking eight of the well retained 12C-peptides into 100 μl of depleted and digested plasma at an equimolar amount of 50 fmol. A small aliquot was reserved for LC-MRM/MS, and the remaining sample was separated by SCX followed by desalting as described under “Experimental Procedures.” 13C-Internal standards were added to all samples at 5 fmol/μl just prior to MRM analysis (performed in triplicate). Percent recoveries ranged from >80 to 100% for the eight analyte peptides tested (data not shown).
DISCUSSION
Sensitivity for detection and quantitation of any given target peptide or protein in blood or other biologically complex fluid or tissue is largely governed by interference from real chemical signal (as opposed to electronic noise) produced by ionization of peptide, small molecule, and inorganic constituents of the sample. Plasma or serum represent “worst case” scenarios with respect to complexity given the enormous number of proteins present (likely in the many hundreds of thousands taking into account posttranslationally modified forms and splice variants), and the 1011–1012-fold span of protein abundance (10). In the absence of biological background (e.g. in buffer alone) we have shown that peptides may be quantified in the 50–100-amol range, corresponding to LOQs for protein in the low to mid-picogram/milliliter range depending on molecular weight. In sharp contrast, direct analysis of plasma by MRM LC-MS/MS can only achieve detection and quantitation limits in the low microgram/milliliter range for plasma (21, 22). Instrument performance factors are responsible for some of these limitations. Contributing factors include the relatively low resolution and mass precision of even the latest generation triple quadrupole mass analyzers and an in-spectrum dynamic range of typically ≤1000. Together these factors limit analyte detection specificity and sensitivity in the face of enormous sample complexity. Noninstrumental factors such as analyte-on-analyte ion suppression may also impair analyte detection.
The overall goal of our study was to define simple plasma processing methods that would enable development of quantitative, reproducible, and multiplexed MS-based assays for routine measurement of plasma proteins in the low nanogram to sub-nanogram/milliliter range of concentrations, a range in which many current clinical biomarkers reside. One previous study by members of this laboratory demonstrated that, with a complex fractionation approach and a large starting volume of serum (1 ml), detection limits of several hundred nano-grams/milliliter for c-reactive protein by MRM methods could be achieved in plasma (19). While the methods used in that study were impractical for routine measurement and fell short of achieving the sensitivity goals described above, they did support the notion that fractionation of plasma would improve detection of lower abundance proteins. In the present study, we have used several simple and robust plasma processing methods (both individually and in combination) to systematically evaluate their benefit for improving S/N, LOD, and LOQ for proteins at low levels in plasma. At the same time, we carefully evaluated and defined some “best practices” for detection and avoidance of biological interferences in MRM quantitation of peptides in highly complex biofluids such as plasma. Attention to these critical aspects of assay development has enabled us to develop quantitative, multiplexed MS-based assays for proteins that routinely achieved limits of quantitation in the 1–10 ng/ml range without the use of antibody enrichment.
Removal of high abundance proteins by immunoaffinity depletion followed by multidimensional protein and/or peptide fractionation has proven to be critical for increasing proteome depth and detection in unbiased biomarker discovery approaches (23–26). However, throughput during this stage of the biomarker discovery pipeline is very low (often a few samples/month on a single instrument) because of the amplification of a single sample into many tens of fractions, each of which must be analyzed by a 2–3-h reverse phase LC gradient. As a bridge to preclinical validation, verification studies using MRM-SID/MS must be able to quantify a moderate number of analytes (tens to hundreds) in a relatively large number of clinically relevant samples (tens to hundreds). Therefore the methods developed for unbiased discovery are not directly translatable to the requirements of verification.
In the most careful study to date, Anderson and Hunter (22) demonstrated the multiplexing capability of the MRM assay by obtaining quantitative measurements for 47 medium to high abundance plasma proteins in a single MRM LC-MS/MS analysis of ~1-h duration. The LOQs achieved were ~1 μg/ml in nondepleted plasma. Using depleted plasma they observed improved CVs but did not observe a general improvement in LOD or LOQ for peptides. Other attempts have been made to develop quantitative assays for target proteins in plasma without immunoaffinity depletion to maximize sample throughput (21). However, these studies also fail to quantify proteins below microgram/milliliter levels in plasma.
Abundant Protein Depletion and Limited Peptide Fractionation Improves LOD and LOQ by up to 1000-fold Compared with Direct Analysis of Plasma
In the present study we have shown that abundant protein depletion combined with limited peptide fractionation by SCX improves LOD and LOQ of proteins by up to 1000-fold compared with direct analysis of proteins in plasma. For abundant protein depletion, we evaluated the performance of two commercially available immunoaffinity depletion LC columns that remove 7 and 12 proteins, respectively, from plasma. The LOQs were improved by a factor of ~2 from an average of 50 to ~25 ng/ml by removal of the additional five proteins (Table II). In addition to removing potential interferences from the very large number of highly abundant peptides that would be derived from these highly abundant proteins, depletion also has the major benefit of reducing the overall plasma protein concentration permitting larger amounts of peptide digest to be loaded onto the LC column for subsequent MS detection. In the case of the Agilent MARS hu7 column, the concentration of protein in plasma was reduced from ~55 to 7.5 mg/ml, a nearly 7-fold decrease in concentration. The Beckman Coulter IgY-12 column reduced the protein concentration to 2.3 mg/ml, a 24-fold decrease in concentration. Since the loading amounts of digest are normalized to present ~1 μg of protein digest to the capillary column, these reductions in concentration allow for concomitant increases of 7- and 24-fold in sample enriched for the analytes of interest. Of course these estimates do not take into account potential losses of nontargeted proteins that may occur upon depletion. Here we have shown that many of the test proteins used are recovered in greater than 50% yield following depletion and membrane concentration. These results are consistent with other studies in our laboratory of cardiovascular disease-related protein biomarkers where we have measured the losses of two naturally occurring low to medium abundance proteins (myeloperoxidase and C-reactive protein) pre- and postdepletion using these columns by ELISA and found that, on average, ~50% of each protein is lost upon depletion and buffer exchange (data not shown). Therefore, it is possible that the actual enrichment for nondepleted proteins is only ~3.5- and 12-fold in the two columns, respectively.
Further improvements in LOQ on the order of 10- to 40-fold were achieved by limited peptide separation by SCX following immunoaffinity depletion. LOQs for all of the target proteins were determined to be at least 2.5 ng/ml, the lowest concentration examined. The percent CVs of the assays were all less than 15% and were ~5% on average. Based upon the S/N for the leptin signature peptide, the LOQ for this protein can be extrapolated into the picogram/milliliter level in plasma, demonstrating that it is possible to quantify clinically relevant biomarkers by MRM in this concentration range without antibody enrichment.
All sample processing steps and fractionation methods result in some level of analyte loss due to irreversible adsorption, losses at buffer exchange or desalting steps, incomplete digestion, or introduction of new interferences from the separation media (e.g. salts or ampholytes). The assays we configured for proteins based on stable isotope dilution of peptides in complex biological samples do provide absolute values for the peptides measured with experimentally defined precision, but they do not provide absolute quantitation of either the peptide or the protein as the losses incurred in the sample processing steps are not routinely measured or quantified. The percent relative errors for the determined protein concentrations varied widely among the target proteins from 5 to >100% (Table II). Sample loss can also be protein- and peptide-dependent as evidenced by the range of recoveries (>50 to 100%) after SCX fractionation observed for our peptide internal standards derived from the target proteins used in this study. Addition of labeled protein standards at the start of sample processing can be used to account for all sources of sample loss. Fully labeled protein (native or in the form of concatenated peptide sequences (22, 27)) can be made but requires considerable resources for expression, purification, and characterization of the final protein product. It is important to emphasize that, although the lack of labeled protein standards prevents true absolute quantitation, the most important use of these assays for precise measurement of relative changes in peptide/protein or posttranslational modification levels across samples is not impeded.
In the current study we have demonstrated that, despite losses of protein at the depletion steps and for peptides at the SCX/desalting step, the losses are more than offset by the reduction in sample complexity and the very high sensitivity of the MS detector. Improvements in peptide ion exchange separations or isoelectric focusing may result in further incremental improvement in the assay performance demonstrated here. We are presently evaluating the use of ammonium formate as the running buffer in SCX to be able to avoid the need for desalting postseparation. In addition we are testing off-gel electrophoresis at the peptide level, hydrophilic interaction chromatography, and reverse phase at basic pH as potential substitutes for ion exchange (28–30).
The number of transitions that can be effectively monitored while maintaining good sensitivity for the analytes is governed by the dwell time used per transition (typically 10–30 ms) and the chromatographic peak width. The latter, together with the minimum acceptable number of data points across a peak, sets the upper bound on the number of transitions that can be monitored. Although fractionation has the obvious disadvantage of requiring n additional LC-MS/MS analyses (where n is the number of fractions generated), some fractionation is advantageous for the LC-MRM-MS/MS analyses. For example, each SCX fraction contains a predetermined subset of the total list of signature peptides, and therefore different populations of the total signature peptide list can be monitored in each SCX fraction enabling the total number of signature peptides monitored to reach into the many hundreds. With knowledge of the elution times for the peptides of interest, it is possible to use the vendor-supplied software on triple quadrupole MS instruments to use different lists of transitions for differing time periods of the chromatography, further increasing the total number of analytes that can be monitored.
From a practical standpoint a critical question is whether or not the approaches described could be applied in a real world verification study and completed in an acceptable amount of time. The principal bottleneck in conducting, for example, a verification study of 200 samples is data acquisition on the mass spectrometer and data analysis, not sample processing, which can be done in parallel to sample analysis and is largely automatable. Using the processing strategy described and assuming each sample is fractionated into eight SCX fractions, each of which is analyzed in triplicate by LC-MRM/MS, the analysis of 200 patient samples for up to 100 signature peptides would take on the order of 10 months. This timeline also includes peptide synthesis and assay configuration. For comparison, antibody production for these same candidate biomarkers would likely require 9–12 months, and then substantial additional time would be required to configure useful immunoassays.
Alternative Enrichment Strategies
Bioaffinity, immunoaffinity, and chemical capture methods have been proposed to provide greatly improved means to decrease complexity and enrich for subpopulations of the plasma proteome. For example, capture of glycosylated proteins or peptides has been shown to greatly reduce plasma complexity and thereby enable detection of some lower abundance proteins (31–35), although recovery may be an issue as peptides from only ~100 plasma proteins were detected in studies of serum (32, 33). Other possible complications are that the former glycosylation site peptides obtained may not be good candidates for MRM assay development and that an observed change in the level of former glycosylation site peptide can arise either as a result of a change in the level of glycosylation at particular sites or a change in the level of the protein; these different causes cannot be distinguished.
Coupling immunoaffinity capture of peptides with stable isotope dilution is a very promising strategy for construction of MRM assays for proteins in plasma (referred to as SISCAPA (36)). While the benefits of SISCAPA are yet to be fully realized, recent studies suggest that more than a 1000-fold enrichment can be achieved for proteins in plasma using this approach (37). These results suggest that SISCAPA coupled to the approaches presented here may facilitate the development of quantitative MS-based assays for proteins present at picogram/milliliter levels in blood.
Selection and Use of Signature Peptides for Assay Development
The first step in assay development is to identify the best responding peptides of unique sequence (the signature peptides). In this study, we relied upon experimental LC-MS/MS data generated on proteolytic digestions of commercially available standards. The final panel of nine MRMs achieved quantitative measurements for all six target proteins with acceptable precision in nonfractionated and fractionated depleted plasma. Different performance characteristics were observed for peptides derived from the same test protein, resulting in each signature peptide providing somewhat different LOQs depending on the overall response and residual interferences. Other published reports have also shown that multiple peptides derived from the same target protein can have varying MRM results (19). Interpretation of these quantitative measurements in clinical samples will have to rely on the relative differences and trends across multiple cases with respect to controls rather than on the absolute values measured for the peptides. The necessity of multiple signature peptides per target protein increases the likelihood that a successful and sensitive MRM assay will be developed. Computational methods for predicting peptides from proteins that are more likely to be observed in digests are being developed, but their predictive value, especially for selection of the best responding peptides for each protein in complex matrices, remains to be established (38).
Monitoring Multiple Transitions for Each Peptide Is Required for Detection and Avoidance of Interference
Sensitivity for detection and quantitation by MS is largely limited by the sample background. Human serum is a complex protein mixture with a huge dynamic range in protein concentrations. The overwhelming presence of numerous peptides derived from predominant proteins can cause significant ion suppression when pursuing an analyte of interest. Often ion signals from low abundance analytes are masked by ions of similar m/z and retention time. Given the complexity of the plasma matrix and the low resolving power of triple quadrupole instruments, it is anticipated that many interferences from co-eluting peptides will often reside within the mass selection windows of the mass analyzers.
Monitoring a single transition per peptide has been proposed as being sufficiently specific for peptide quantitation in complex mixtures (18). There are advantages to using only one transition per peptide as the instrument’s duty cycle can be concentrated on a smaller number of transitions thereby increasing either the sensitivity, the degree of multiplexing, or both. However, as we have demonstrated here for PSA, using single fragment transitions to monitor for peptides in complex proteomes can be highly misleading and result in erroneous detection of the presence and/or the level of a peptide because what is actually being detected is an interference that cannot be distinguished from the analyte of interest. To help avoid this common problem, we have shown how one can use the ratios of multiple transitions for the same peptide to give an indication of the presence of interference in specific channels and thereby enable correct quantitation using channels in which little or no interference is detected.
Based on the results presented here, quantitative assays that rely on minimal sample processing can be configured and run for low abundance plasma proteins in the 1–10 ng/ml range with precision that is acceptable (3–15%) for verification studies. Our study used readily available standards and separation methods that are commonly available in proteomics laboratories. The methods and the benchmarks presented should facilitate testing and adoption of SID-MRM LC-MS/MS by other laboratories interested in developing multiplex assays for low abundance proteins in blood. Using the approaches described here, we have recently configured quantitative assays for seven proteins implicated as potential biomarkers of cardiovascular disease in humans and are using these methods to assay these proteins at low nanogram/milliliter levels in patient samples (Ref. 39; full results to be published elsewhere). We are presently incorporating of peptide immunoaffinity enrichment into our workflow with the aim of extending the assay range of MRM-SID/MS into the low to mid-picogram/milliliter concentration range, potentially resulting in a comprehensive assay platform for verification of candidate biomarkers in plasma.
Supplementary Material
Acknowledgments
We thank Dick Cook, Rick Schiavoni, and Katie Tone from the Biopolymers Laboratory at the Massachusetts Institute of Technology for the synthesis and purification of the heavy labeled peptides and Will Beavers from Dana Farber for amino acid analysis of all protein and peptide stocks. We also thank Nadar Rifai and Gary Bradwin from Children’s Hospital Boston and Harvard Medical School for assistance in obtaining the ELISA data for PSA and Xu Shi from Massachusetts General Hospital for laboratory assistance. We also thank Michael Gillette from the Broad Institute and Massachusetts General Hospital and Robert Gerszten from Massachusetts General Hospital for helpful discussions.
Footnotes
This work was supported by grants (to S. A. C.) from the NCI, National Institutes of Health (1U24 CA126476-02), part of NCI’s Clinical Proteomic Technologies Initiative, the NHLBI, National Institutes of Health (U01-HL081341), and the Entertainment Industry Foundation.
The on-line version of this article (available at http:www.mcponline.org) contains supplemental material.
The abbreviations used are: MRM, multiple reaction monitoring; SID, stable isotope dilution; SCX, strong cation exchange chromatography; PSA, prostate-specific antigen; MBP, myelin basic protein; HRP, horseradish peroxidase; AAA, amino acid analysis; Q1, quadrupole 1; Q3, quadrupole 3; Camc, carbamidomethylcysteine; LOQ, limit of quantitation; LOD, limit of detection; S/N, signal to noise ratio; CV, coefficient of variation; XIC, extracted ion chromatogram; SIS-CAPA, stable isotope standards with capture by anti-peptide antibodies; IS, internal standard; MARS, Multiple Affinity Removal System.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Adkins JN, Varnum SM, Auberry KJ, Moore RJ, Angell NH, Smith RD, Springer DL, Pounds JG. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Mol Cell Proteomics. 2002;1:947–955. doi: 10.1074/mcp.m200066-mcp200. [DOI] [PubMed] [Google Scholar]
- 3.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;16:1929–1935. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 4.Wang W, Zhou H, Lin H, Roy S, Shaler TA, Hill LR, Norton S, Kumar P, Anderle M, Becker CH. Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal Chem. 2003;75:4818–4826. doi: 10.1021/ac026468x. [DOI] [PubMed] [Google Scholar]
- 5.MacCoss MJ, Wu CC, Liu H, Sadygov R, Yates JR., III A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal Chem. 2003;75:6912–6921. doi: 10.1021/ac034790h. [DOI] [PubMed] [Google Scholar]
- 6.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 7.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Ong S, Blagoev B, Kratchmarova I, Kristensen D, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 9.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 10.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 11.Perchalski R, Yost R, Wilder B. Structural elucidation of drug metabolites by triple-quadrupole mass spectrometry. Anal Chem. 1982;54:1466–1471. [Google Scholar]
- 12.Lee MS, Kerns EH. LC/MS applications in drug development. Mass Spectrom Rev. 1999;18:187–279. doi: 10.1002/(SICI)1098-2787(1999)18:3/4<187::AID-MAS2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 13.Tiller PR, Cunniff J, Land AP, Schwartz J, Jardine I, Wakefield M, Lopez L, Newton JF, Burton RD, Folk BM, Buhrman DL, Price P, Wu D. Drug quantitation on a benchtop liquid chromatography-tandem mass spectrometry system. J Chromatogr A. 1997;771:119–125. doi: 10.1016/s0021-9673(97)00147-7. [DOI] [PubMed] [Google Scholar]
- 14.Desiderio D, Kai M. Preparation of stable-isotope incorporated peptide internal standards for field desorption mass spectrometry quantification of peptides in biologic tissue. Biomed Mass Spectrom. 1983;10:471–479. doi: 10.1002/bms.1200100806. [DOI] [PubMed] [Google Scholar]
- 15.Desiderio D, Kai M, Tanzer F, Trimble J, Wakelyn C. Measurement of enkephalin peptides in canine brain regions, teeth, and CSF with HPLC and mass spectrometry. J Chromatogr. 1984;297:245–260. doi: 10.1016/s0021-9673(01)89046-4. [DOI] [PubMed] [Google Scholar]
- 16.Lisek CA, Bailey JE, Benson LM, Yaksh TL, Jardine I. Quantitation of endogenous substance P by on-line microcolumn liquid chromatography/continuous-flow fast atom bombardment mass spectrometry. Rapid Commun Mass Spectrom. 1989;3:43–46. doi: 10.1002/rcm.1290030211. [DOI] [PubMed] [Google Scholar]
- 17.Barr JR, Maggio VL, Patterson DG, Jr, Cooper GR, Henderson LO, Turner WE, Smith SJ, Hannon WH, Needham LL, Sampson EJ. Isotope-dilution mass spectrometric quantification of specific proteins: model application with apolipoprotein A-1. Clin Chem. 1996;42:1676–1682. [PubMed] [Google Scholar]
- 18.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuhn E, Wu J, Karl J, Liao H, Zolg W, Guild B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 2004;4:1175–1186. doi: 10.1002/pmic.200300670. [DOI] [PubMed] [Google Scholar]
- 20.Lin S, Shaler TA, Becker CH. Quantification of intermediate-abundance proteins in serum by multiple reaction monitoring mass spectrometry in a single-quadrupole ion trap. Anal Chem. 2006;78:5762–5767. doi: 10.1021/ac060613f. [DOI] [PubMed] [Google Scholar]
- 21.Barnidge D, Goodmanson M, Klee G, Muddiman D. Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-MS/MS using protein cleavage and isotope dilution MS. J Proteome Res. 2004;3:644–652. doi: 10.1021/pr049963d. [DOI] [PubMed] [Google Scholar]
- 22.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–588. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Shen Y, Jacobs JM, Camp DG, II, Fang R, Moore RJ, Smith RD, Xiao W, Davis RW, Tompkins RG. Ultra-high-efficiency strong cation exchange LC/RPLC/MS/MS for high dynamic range characterization of the human plasma proteome. Anal Chem. 2004;76:1134–1144. doi: 10.1021/ac034869m. [DOI] [PubMed] [Google Scholar]
- 24.Shen Y, Moore RJ, Zhao R, Blonder J, Auberry DL, Masselon C, Pasa-Tolic L, Hixson KK, Auberry KJ, Smith RD. High-efficiency on-line solid-phase extraction coupling to 15–150-microm-i. d column liquid chromatography for proteomic analysis. Anal Chem. 2003;75:3596–3605. doi: 10.1021/ac0300690. [DOI] [PubMed] [Google Scholar]
- 25.Tirumalai RS, Chan KC, Prieto DA, Issaq HJ, Conrads TP, Veenstra TD. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics. 2003;2:1096–1103. doi: 10.1074/mcp.M300031-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Tang H, Ali-Khan N, Echan LA, Levenkova N, Rux JJ, Speicher DW. A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics. 2005;5:1–14. doi: 10.1002/pmic.200401275. [DOI] [PubMed] [Google Scholar]
- 27.Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ. Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat Methods. 2005;2:587–589. doi: 10.1038/nmeth774. [DOI] [PubMed] [Google Scholar]
- 28.Michel PE, Reymond F, Arnaud IL, Josserand J, Girault HH, Rossier JS. Protein fractionation in a multicompartment device using Off-Gel isoelectric focusing. Electrophoresis. 2003;24:3–11. doi: 10.1002/elps.200390030. [DOI] [PubMed] [Google Scholar]
- 29.Moritz RL, Clippingdale AB, Kapp EA, Eddes JS, Ji H, Gilbert S, Connolly LM, Simpson RJ. Application of 2-D free flow electrophoresis/RP-HPLC for proteomic analysis of human plasma depleted of multi high-abundance proteins. Proteomics. 2005;5:3402–3413. doi: 10.1002/pmic.200500096. [DOI] [PubMed] [Google Scholar]
- 30.Heller M, Ye M, Michel PE, Morier P, Stadler D, Junger MA, Aebersold R, Reymond F, Rossier JS. Added value for tandem mass spectrometry shotgun proteomics data validation through isoelectric focusing of peptides. J Proteome Res. 2005;4:2273–2282. doi: 10.1021/pr050193v. [DOI] [PubMed] [Google Scholar]
- 31.Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, Isobe T. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat Biotechnol. 2003;21:667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Li X, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Yi EC, Li X, Mallick P, Kelly-Spratt KS, Masselon CD, Camp DG, II, Smith RD, Kemp CJ, Aebersold R. High throughput quantitative analysis of serum proteins using glycopeptide capture and liquid chromatography mass spectrometry. Mol Cell Proteomics. 2005;4:144–155. doi: 10.1074/mcp.M400090-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Sun B, Ranish JA, Utleg AG, White JT, Yan X, Lin B, Hood L. Shotgun glycopeptide capture approach coupled with mass spectrometry for comprehensive glycoproteomics. Mol Cell Proteomics. 2007;6:141–149. doi: 10.1074/mcp.T600046-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Stahl-Zeng J, Lange V, Ossola R, Aebersold R, Domon B. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol Cell Proteomics. 2007;6:1809–1817. doi: 10.1074/mcp.M700132-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA) J Proteome Res. 2004;3:235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 37.Whiteaker JR, Zhao L, Zhang HY, Feng L, Piening BD, Anderson L, Paulovich AG. Antibody-based enrichment of peptides on magnetic beads for mass-spectrometry-based quantification of serum biomarkers. Anal Biochem. 2007;362:44–54. doi: 10.1016/j.ab.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, Kuster B, Aebersold R. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25:125–131. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 39.Addona T, Keshishian H, Burgess M, Shi X, Saenz-Vash V, Kuhn E, Gerszten RE, Carr SA. Quantitation of protein biomarkers of cardiovascular injury in patients by targeted MS. Proceedings of the 55th ASMS Conference on Mass Spectrometry and Allied Topics; Indianapolis. June 3–7, 2007.2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.