

Abstract
The hypoxic microenvironment of solid tumors is associated with malignant progression and it renders tumors more resistant to cancer therapies. Endothelial cell damage may occur following hypoxic conditions and lead to dysfunction; however endothelial cells in tumors survive hypoxic conditions providing nutrients and oxygen to facilitate tumor growth. In this study, we investigated the effects of tumor-conditioned medium on hypoxia-induced changes in endothelial cell growth, migration and survival. Tumor conditioned medium collected from human U87 glioblastoma cells were applied to endothelial cultures in normoxia or hypoxia conditions. Hypoxia caused a reduction in clonogenic cell survival response and an increase of the sub-G1 phase of the cell cycle in endothelial cells. Cell migration was measured by spheroid and wound-induced migration assays and hypoxia compared with normoxia significantly increased the number of migrating endothelial cells. Nuclear staining with Hoechst 33258 and caspase-9 and -3 activation in endothelial cells show that hypoxia induced apoptosis involves caspase-dependent mechanism. Exposure to hypoxia caused an increase in gene expression of VEGF and VEGFR2 and activities of MMP-2 and MMP-9. Furthermore, hypoxia induced an increase capillary-like structure formation in endothelial cells seeded into Matrigel. Tumor conditioned medium enhanced survival and rescued endothelial cells from apoptosis induced by hypoxia. These molecular changes in endothelial cells could, in part, contribute to the angiogenic response that occurs during hypoxic-induced angiogenesis in glial tumors.
Keywords: Hypoxia, invasion, angiogenesis, glioblastoma, endothelial cells
Introduction
Malignant gliomas are the most common primary brain tumors in adults characterized by massive and diffuse infiltration of the surrounding normal brain tissue (1,2). Dismal overall prognosis remains for patients with malignant glioma due to a local recurrence despite surgery, radiotherapy and chemotherapy (3,4) and these facts stress the need for more effective novel therapeutic strategies. Angiogenesis is a fundamental process by which new blood vessels are formed from pre-existing ones. Induction of angiogenesis is an essential prerequisite for tumor growth and spread and precedes malignant tumor formation (5). Glioblastomas are one of the most vascularized human cancers, and the formation of tumor-specific vessels occurs early at the onset of tumor growth (6). Hypoxic states occur during tumorigenesis when deregulated cellular proliferation allows neoplastic tissue to outgrow the available oxygen supply. The hypoxic microenvironment of glioblastoma tumors is associated with malignant progression and is an adverse prognostic indicator for tumor treatment (7–9). Tumor vascularization is often poorly organized and marginally functional due to tumor structural abnormalities, inducing hypoxic conditions and nutritional shortages in tumor tissues (6,10,11). Tumor cell survival is thus dependent on the stimulation of angiogenesis and metabolic adaptation to hypoxia.
Vascular endothelial cells (ECs) can undergo apoptosis in response to a number of pathophysiological stimuli including hypoxia (12). The endothelial cells supporting tumor vascular structures survive the hypoxic microenvironment. The underlying regulatory mechanisms by which endothelial cells overcome these conditions are not completely understood. Cancer cells release factors that may protect endothelial cellular injury and death from hypoxia. This study was undertaken to investigate the role of U87 glioblastoma tumor cell conditioned medium on endothelial cell growth, migration and survival under hypoxic conditions. We investigated whether tumor cells influence the expression of endothelial cell genes by growing human microvascular endothelial cells (HMECs) in conditioned medium from U87 human glioblastoma cells.
Materials and Methods
Cell Culture
The human glioblastoma U87 cell line was obtained from American Type Culture Collection (Manassas, VA) and were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 cg/ml) and maintained at 37°C in a 95% air/5% CO2 humidified incubator. Human microvascular endothelial cells (HMECs) were maintained as described earlier (13). For hypoxic experiments, unless stated otherwise, cells were placed in a humidified chamber maintained at 1% O2, 5% CO2 and balance in N2 for the indicated times. Cells cultured in a standard incubator (normoxia; air with 5% CO2) for the same period of time served as controls.
Tumor conditioned medium was prepared from the U87 cell culture grown to near confluency. After being washed twice with serum free medium, cells were incubated in serum free MCDB medium at 37°C in a humidified 5% carbon dioxide atmosphere for 24 h. The supernatant was harvested, centrifuged at 2000g at 4°C for 10 min and supplemented with EGF and hydrocortisone prior to use.
Cell proliferation assay
Cells were plated at a density of 1x105 cells/well in microtiter plates and cultured under normoxic or hypoxic conditions for indicated time periods as mentioned. Then 20 μl of 5 mg/ml MTT in PBS was added to each well. After 4h of incubation at 37fC, 100 μl of DMSO was added to each well to dissolve the formazan crystals. Absorbance values at 550 nm were measured with a microplate reader.
Colony-Formation Assay
Endothelial cells grown under normal or hypoxic conditions for 24 or 48 h were plated on 100-mm dishes at 500 cells per dish. Medium was subsequently changed every 3 days. After 10 days, the resulting colonies were fixed in methanol, stained with 0.5% crystal violet (dissolved in 20% methanol) for 10 min and counted as a measure of clonogenicity
DNA Cell Cycle Analysis
Cells were grown under normal or hypoxic conditions, harvested, washed twice with PBS, and fixed in 3.7% paraformaldehyde in PBS for 10 min at room temperature. Cells were pelleted, washed once with PBS, and resuspended in a peopidium iodide (PI) solution (50 μg/ml PI, Sigma; 0.1 mg/ml RNase A in PBS, pH 7.4) for 30 min in the dark. Flow cytometry analysis was performed using FASCalibur flow cytometry system (BD Biosciences, San Jose, CA) as described previously (14). Forward light scatter characteristics were used to exclude the cell debris from the analysis. The sub-G1 population was calculated as an estimate of the apoptotic cell population. The percentages of cells within the G1, S, and G2/M phases of the cell cycle were determined by analysis with the program CellQuest.
Cell migration from spheroids
Migration from spheroids was assayed as described previously (15). Multicellular spheroids were placed in 96-well microplates, one in each and were cultured for 24 h, after which the spheroids were fixed and stained with crystal violet and cellular migration from the spheroids was assessed under light microscopy.
Wound-induced migration assay
Migration through a wound introduced in a cell monolayer was assayed as described elsewhere (15). Briefly, subconfluent monolayers of cells were wounded by scraping with a plastic pipette tip and the distance that the advancing cells had moved into the cell-free (wound) area was measured after 24 h by staining with Crystal violet..
Hoechst 33342 staining
Endothelial cells were cultured under normal or hypoxic conditions in the presence or absence of U87 conditioned medium for 24 h. Then, cells were washed with PBS and fixed with 10% neutral buffered saline. After fixation, cells were stained with 1 μg/ml of Hoechst 332258 (Molecular Probes, Eugene, OR) for 10 min at 37 °C in the dark. Cells were washed twice with PBS and were analyzed under a fluorescent microscope (Olympu, Melville, NY). Apoptotic cells were identified by nuclear condensation, formation of membrane blebs and apoptotic bodies. The mean percentage of apoptotic cells was estimated by counting three random fields in duplicate wells per group.
SDS-PAGE and Western blot analysis
Cells were extracted in a buffer solution containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 1mM sodium fluoride, 1mM PMSF, 10 cg/ml aprotinin on ice for 20 min. Samples were subjected to SDS-PAGE and separated proteins were transferred onto membrane followed by blocking of membrane with 5% nonfat milk powder (w/v) in tris- buffered saline (10 mM Tris, 100 mM NaCl, 0.1% Tween 20) for 1 hour at room temperature or overnight at 4°C. Membranes were probed for apoptotic molecules, caspase-9 and caspase-3 using their specific primary antibodies (Cell signaling, Beverly, MA) followed by appropriate secondary antibody and enhanced chemiluminescence visualization. Membranes were stripped and reprobed with GAPDH antibody (Novus Biologicals, Littleton, CO) as a protein loading control.
RNA extraction and reverse transcription–polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA thus obtained was further purified by digesting with DNase for 20 min at 37°C and then reverse-transcribed using the cDNA cycle kit (Invitrogen, Carlsbad, CA) with random primers. RT products were then amplified using IQ Sybr green Supermix (Bio-Rad, Hercules, CA), according to the protocol provided. Real-time PCR was performed on an iCycler (Bio-Rad) with activation at 95°C for 5 min followed by 40 cycles of 30 sec 95°C denaturing, 30 sec 55°C annealing and 45 sec 68°C elongation. The forward (F) and reverse (R) primers used were: 5′-AGCCTTGCCTTGCTGCTCTA-3′ (F) and 5′-GTGCTGGCCTTGGTGAGG -3′ (R) for VEGF165, 5′-CTGGCATGGTCTTCTGTGAAGCA-3′ (F) and 5′-AATACCAGTGGATGTGATGCGG-3′ (R) for VEGFR-2 and 5′-ACCTCATGAAGATCCTCACCGAGCG-3′ (F); 5′-TCTACAATGAGCTGCGTGTGGCTCC-3′ (R) for β-actin. Threshold cycles were normalized according to the level of β-actin expression in different treatments.
Zymographic assays
Conditioned medium was resolved under nonreducing conditions on 10% SDS-PAGE gels embedded with gelatin (16). Gels were rinsed three times in 2.5% Triton X-100 for 30 min at room temp and then incubated in 50 mM Tris-HCl, 10mM CaCl2 buffer pH 7.6 overnight at 37°C. Gels were stained with Amido Black and areas of lysis were visualized as transparent bands. Bands of lysis representing gelatinase activity were then visualized against a dark background.
In vitro angiogenesis assay
Matrigel (BD Biosciences, Bedford, MA) diluted 1:2 in cold DMEM medium was plated into flat-bottomed 96 well tissue culture plates and allowed to gel for 20 min at 37°C before cells were added. After a 24 h incubation period, images were taken with a phase contrast microscope and capillary tubes were defined as cellular extension linking cell masses or branch points. Experiments were performed in triplicate.
Statistical analysis
Statistical significance of the experimental results was determined by the Student’s t-test. For all analyses p < 0.05 was accepted as a significant probability level.
Results
Conditioned medium of U87glioma cells increases survival of hypoxic HMECs
Our aim was to investigate whether exposure to hypoxia imparts an anti-proliferative effect against HMECs. Cells exposed to hypoxia (1% O2) for 24 and 48 h proliferate at a significantly slower rate than HMECs in normoxic conditions (control). Cell numbers in cultures of HMECs exposed to 0.1% O2 hypoxic conditions decreased furthermore (Fig 1A). We used U87 conditioned medium to evaluate the effect of mediators produced by glioma cells on HMECs proliferation. HMECs grown under hypoxic conditions (0.1% O2) in presence of U87 conditioned medium failed to show reduction in cell numbers (Fig 1b). We also assayed clonogenic survival of HMECs exposed to hypoxic conditions and found that hypoxia decreased survival of HMECs (Fig 2A) and U87 conditioned medium significantly prevented reduction in cell numbers in cultures of HMECs exposed to hypoxia (Fig 2B).
Figure 1.
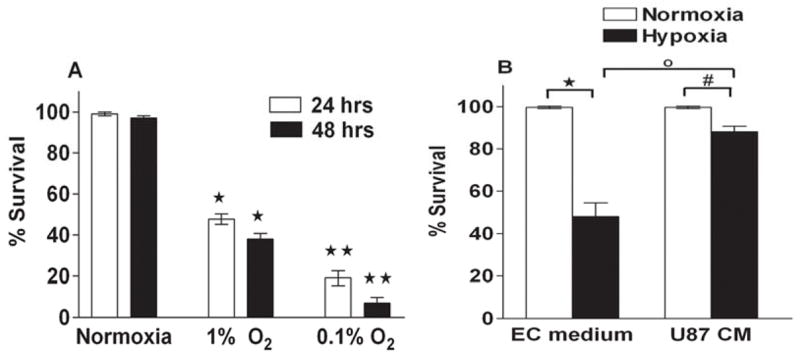
Effect of U87 conditioned medium on the proliferation of endothelial cells grown under normoxic or hypoxic conditions. A. HMECs were cultured to 90% confluence, subjected to normoxia or hypoxia conditions for 24 h and 48 h. *p < 0.05; **p < 0.01. versus normoxic control at the same time point.
B. HMECs were grown under normoxic or hypoxic (1% O2) conditions in presence of EC medium or U87 conditioned medium for 24 h. Viable cells were scored using metabolic- dye based MTT assay. Mean ± SD of three independent experiments; each assayed in duplicate. *p < 0.05 versus hypoxic (in EC medium); #p < 0.051, Not significant versus hypoxic (in U87 Conditioned medium); °p < 0.05 EC medium versus U87 conditioned medium.
Figure 2.
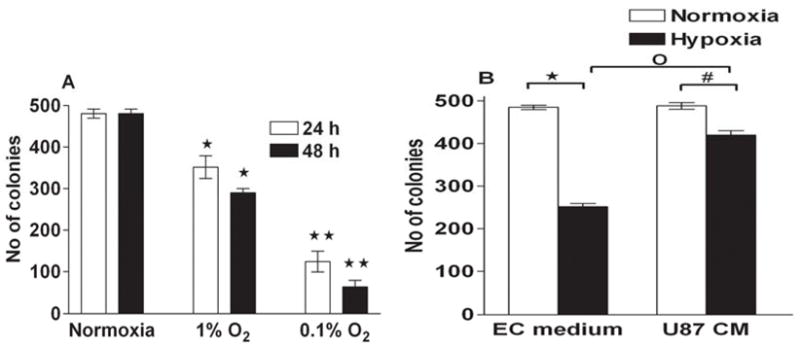
Clonogenic assay. A. HMECs were cultured under normoxic or hypoxic (0.1% or 1 %) conditions for 24 or 48 h. *p < 0.05; **p < 0.01. versus normoxic control at the same time point. B. HMECs were grown under normoxic or hypoxic conditions in presence of EC or U87 conditioned medium for 24 h. Then the cells were plated into 100 mm cell culture dishes with a total of 500 cells/dish and allowed to grow at normoxic conditions. After incubation at 37 °C in 5% CO2 for 10 days, cells were stained with crystal violet and colonies containing > 50 cells were counted under light microscopy. *p < 0.05 versus hypoxic (in EC medium); #p < 0.05 versus hypoxic (in U87 conditioned medium); °p < 0.05 EC medium versus U87 conditioned medium.
Glioma-conditioned medium prevents hypoxia- induced apoptosis of endothelial cells
Hypoxia induces cell cycle arrest and apoptosis in HDECs. Since hypoxia induces alterations in cell cycle and apoptosis in certain cell types (17), an analysis of cell cycle and apoptosis was performed in hypoxic HMECs. To determine whether the reduction of cell numbers might result from hypoxia-induced apoptosis, two different types of apoptosis assays were performed to assess hypoxia-induced apoptosis in HMECs. We exposed HMECs to hypoxic conditions for 24 h and then analyzed the cells by fluorescence microscopy following Hoechst 33258 staining. . Under normoxic conditions, few apoptotic cells were observed. In contrast, in cells grown under hypoxic conditions significant morphological changes and chromosomal condensation, which is indicative of apoptotic cell death occurred. Within 24 h of hypoxic exposure, HMECs clearly exhibited significant morphological changes and chromosomal condensation, which is indicative of apoptotic cell death (Fig 3). However, there was a marked reduction in dead or apoptotic cells in cultures exposed to U87 conditioned medium compared to endothelial cell medium under hypoxic conditions (Fig 3). To further analyze the hypoxic effect, FACS analysis was carried out on HMECs exposed for 24 hours with hypoxia. Quantification of sub-G1 phase cells was considered an apoptosis marker. Presented in Fig 4 are representative DNA histograms obtained by flow cytometry that describe the cell cycle distributions of HMECs exposed to hypoxic treatment for 24h. Exposure of HMECs to hypoxia for 24 h resulted in increased apoptosis, as evidenced by an increase in the sub-G1 fraction of cells at 24 h after hypoxic exposure (Fig 4). Treatment of HMECs with U87 conditioned medium during the exposure to hypoxia resulted in a significant reduction in the magnitude of the sub-G1 population consistent with U87 conditioned medium mediated antiapoptopic effect There was no major change under normoxic conditions in U87conditioned medium in the cell cycle distribution of apoptotic sub-G1 population of HMECs compared with control cells at 24 h (Fig 4). By using both apoptosis detection methods (i.e., Hoechst 33258 and sub-G1), we determined that hypoxia exposure significantly enhanced apoptosis incidence over control values. Exposure of HMECs to U87 conditioned medium significantly protected against hypoxia-induced apoptosis, regardless of which of the two apoptosis detection methods were used.
Figure 3.
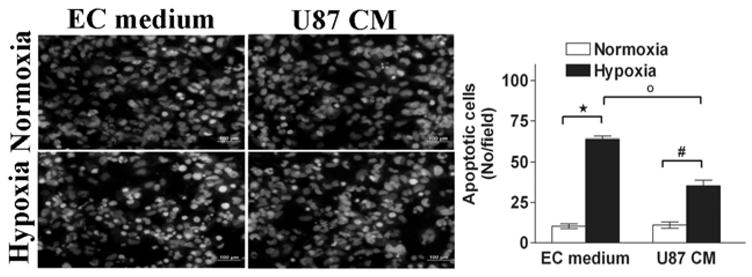
Cell apoptosis following hypoxia culture conditions. HMECs were treated with EC medium or U87 conditioned medium under normoxic or hypoxic culture conditions for 24 h. Cell apoptosis was assessed by fluorescence microscopy using the chromatin stain Hoechst 33258 as described in the Materials and Methods. The apoptotic index was assessed by counting three random fields (in duplicate wells) per group. *p < 0.05 versus hypoxic (in EC medium); #p < 0.05 versus hypoxic (in U87 conditioned medium); °p < 0.05 EC medium versus U87 conditioned medium.
Figure 4.

Flow cytometry. HMECs were grown in EC medium or U87 tumor conditioned medium for 24 h under normoxic and hypoxic (1% O2) and then the cells were harvested, washed twice in PBS, centrifuged, resuspended in staining solution containing propidium iodide and incubated for 30 min. Fluorescence that emitted from the propidium iodide–DNA complex was quantitated after excitation of the fluorescent dye by flow cytometry.
Role of glioma conditioned medium on caspase activation in hypoxia HMECs
Caspase-9 activation is a characteristic indicator of apoptosis. Based on the above results showing induction of apoptosis, we determined the effect of hypoxia on the activation of caspase-9, a cysteine protease activated upon stimulation of the mitochondria-mediated apoptotic death signal following 24 h of exposure. Cleavage of caspases is directly related to their activation status. Hypoxic exposure of HMECs for 24 h caused an increase in cleaved caspase-9 (Fig 5). In addition, we measured the activation of caspase-3, a key enzyme in the process of cell apoptosis activity. As shown in Fig 5, 24 hours of hypoxia caused an increase of cascapse-3 activation, whereas exposure to hypoxia in presence of U87 conditioned medium resulted in decrease of proteolytic cleavage of proforms of both caspase-9 and caspase-3. Equal protein loading was confirmed by probing the same membrane with GAPDH antibody. Taken together, these results show that U87 conditioned medium reduces activation of caspase-9 and caspase-3 in hypoxic endothelial cells.
Figure 5.
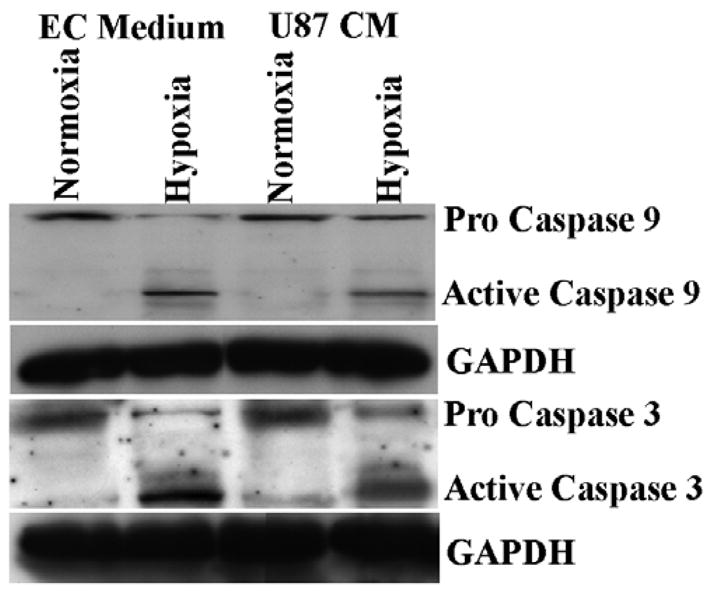
Western blot analysis for caspase-3 and –9 levels in HMECs. Caspase-3 and –9 levels in HMECs following incubation in normoxic or hypoxic culture conditions in presence of EC or U87 conditioned medium for 24 h. All bands were normalized to GAPDH expression.
Glioma conditioned medium increases VEGF and VEGFR-2 expression in hypoxic HMECs
To clarify angiogenic factors derived from HMECs, we studied the gene expression of VEGF and VEGFR-2 in HMECs under normoxic and hypoxic conditions using real time RT–PCR. Increased expression of VEGF and VEGFR-2 transcripts were detected in HMECs under hypoxic conditions compared with normoxia. Levels of these gene transcripts were significantly elevated in U87 conditioned medium compared with EC conditioned medium under normoxic as well as hypoxic conditions (Fig 6).
Figure 6.
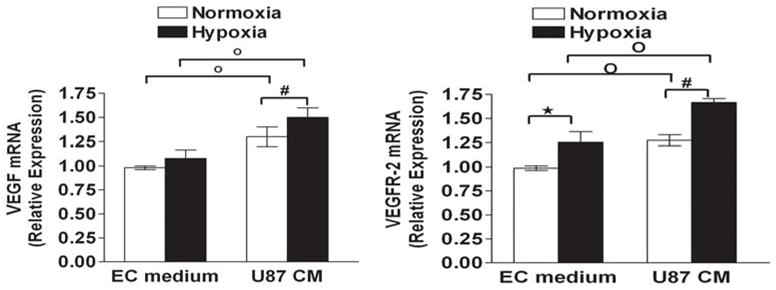
Effect of U87 conditioned medium on A)VEGF (Left) and B)VEGFR-2 (Right) levels of HMECs. Cells were cultivated under the normoxia or hypoxia condition for the 24 h in presence of EC medium or U87 conditioned medium, total RNA was isolated and the expression level of mRNA was analyzed by quantitative RT-PCR. The relative expression of VEGF and VEGFR-2 was normalized by comparison of its value with that of β-actin. . Data are expressed as the mean ± SD and are representative of two separate experiments. Each experiment was carried out in duplicate. *p < 0.05 versus hypoxic (in EC medium); #p < 0.05 versus hypoxic (in U87 conditioned medium); °p < 0.05 EC medium versus U87 conditioned medium.
Conditioned medium from glioblastoma cells modulates MMP-2 and MMP-9 activities in HMECs
Zymography was used to evaluate MMP-2 and MMP-9 secreted by HMECs after exposure to hypoxia. The HMECs were found to constitutively express latent MMP-2, whereas MMP-9 activity was relatively low. Hypoxia for 24 hours led to a significant increase in the level of MMP-2 and MMP-9 activities. U87 conditioned medium enhanced the activity of MMP-2 secreted under hypoxic conditions compared with EC medium; the increase in MMP-9 activity is slight although significant (Fig 7).
Figure 7.

Hypoxia modulates the gelatinolytic activities in endothelial cells. HMECs cultured for 24h were exposed to hypoxia or normoxia with EC medium or U87 conditioned medium and the activities of MMP-2 and MMP-9 in culture supernatants were assessed using gelatin zymography. HT1080 conditioned medium was used a reference for MMP-2 and MMP-9.
Glioma-conditioned medium increases hypoxia-induced migration of HMECs
To determine the effect of hypoxia on cell motility, we analyzed the migration of HMECs using the spheroid migration assay. Single multicellular spheroids of HMECs were cultured for 24 h under normoxic or hypoxic conditions and cellular migration from the spheroids was assessed under light microscopy. Compared with controls, HMECs showed significantly higher numbers of migrated cells under hypoxic cells (Fig. 8). U87 conditioned medium also enhanced migration under normoxic conditions; furthermore, a significant increase was observed under hypoxic conditions. In a further set of experiments, we investigated cell motility in a wound-induced migration assay, in which a subconfluent cell monolayer was disrupted and cells allowed to migrate into the cell-free area. HMECs cells under hypoxic conditions as well as in presence of U87 conditioned medium showed higher numbers of migrating cells (Fig. 9). Hypoxia (24 h) compared with normoxia significantly enhanced the number of migrating HMECs and U87 conditioned medium altered migration of hypoxic HMECs further in both migration assays.
Figure 8.

Spheroid migration assay. Multicellular HMEC spheroids were prepared by seeding 2X103 cells on Micro-well mini-trays and cultured for 2 days. Spheroids were placed in 96-well plate, one in each well and cultured for 24 h in presence of HMEC medium or U87 conditioned medium under normoxic or hypoxic conditions, after which the spheroids were fixed and stained with Crystal Violet, and cellular migration from the spheroids was assessed under light microscopy. *p < 0.05 versus hypoxic (in EC medium); #p < 0.05 versus hypoxic (in U87 conditioned medium); °p < 0.05 EC medium versus U87 conditioned medium.
Figure 9.

Monolayer wound-induced migration assay. A line was scratched with a plastic pipette tip in HMEC cultures, washed twice with PBS, and the medium was replaced with EC medium or U87 conditioned medium. After 24 h exposure of normoxia or hypoxia, cells that had migrated to the wounded areas were counted under a microscope for quantification of cell migration. Migration was calculated as the average number of cells observed in five random high power wounded fields/per well in duplicate wells. *p < 0.05 versus hypoxic (in EC medium); #p < 0.05 versus hypoxic (in U87 conditioned medium); °p < 0.05 EC medium versus U87 conditioned medium.
Glioma-conditioned medium enhances HMEC tube formation
HMECs seeded on Matrigel and maintained develop tube-like structures. We checked whether hypoxia-mediated tube formation is modulated by U87 conditioned medium. HMECs seeded on Matrigel and cultured in the EC medium or U87 conditioned medium developed tube-like structures. Under hypoxic conditions, the tube-like structures were more extensive in cells cultured with U87 conditioned medium compared with HMEC medium (Figure 10). Taken together, these results suggest that tumor- conditioned medium stimulates endothelial cell tube formation, through released growth factors.
Figure 10. Capillary-like structure formation.

Matrigel diluted 1:2 in cold DMEM medium was plated into flat-bottomed 96 well tissue culture plates and allowed to gel for 20 min at 37°C before HMECs were added. HMECs were grown in the presence of EC medium or U87 conditioned medium for 24 h under normoxic or hypoxic conditions. Cells were stained with Crystal Violet, capillary length was determined and represented using computer-assisted image analysis with the Image-Pro plus program. *p < 0.05 versus hypoxic (in EC medium); #p < 0.05 versus hypoxic (in U87 conditioned medium); °p < 0.05 EC medium versus U87 conditioned medium.
Discussion
Angiogenesis is one of the most important biological processes for glioma growth. The proliferation of HMECs, as a crucial step of angiogenesis is triggered and modulated by a variety of stimuli. Endothelial cells undergo a variety of biological responses when placed in hypoxic conditions, including activation of signaling pathways that regulate proliferation, angiogenesis and death (18). It has been recognized that glioblastoma cells are able to secrete soluble factors, which are mitogenic for endothelium. We studied the responses of HMECs to oxygen deprivation and using in vitro techniques, we compared the response of endothelial cells in presence of normal and tumor-conditioned medium. In this study, we have shown that the conditioned medium of U87 glioma cells rescued endothelial cells from apoptosis induced by hypoxia and enhanced angiogenesis in vitro. These findings suggest that glioma cells secrete soluble factors enhancing endothelial cell survival in hypoxic environment.
Studies regarding the role of endothelial cells in hypoxia have been performed in vitro. Whereas some studies indicate an enhancement of endothelial proliferation in hypoxia (19) others have shown a reduction of endothelial cell number in hypoxia (20) and an arrest in the cell cycle, respectively (21). The latter is similar to our findings (reduction of cell number and increased subG1 phase of the cell cycle). It has been shown that endothelial cell cycle arrest at quiescent G0/G1 phase of the cell cycle during hypoxia is in part mediated by HIF-1a (21). We found U87 conditioned medium blocked the decrease in proliferation and cell cycle arrest at sub-G1 phase induced by hypoxia.
Apoptosis has been suggested to be a critical determinant of angiogenesis in vitro (22, 23) and in vivo (23). Upon exposure to hypoxia, HMECs clearly exhibited significant morphological changes and chromosomal condensation, which is indicative of apoptotic cell death. Next, we explored the activation of caspases in HMECs following hypoxia. Caspase-3 has been shown to play a pivotal role as a downstream member of the protease cascade. It was found that the hypoxic treatment of HMECs enhanced activation of caspase-3. In an attempt to characterize the pathway, upstream of caspase-3, responsible for hypoxia-induced apoptosis of HMECs, the activation of caspase-9 was next investigated. These findings confirmed that caspase-3 and caspase-9 activation was involved in the apoptosis induced by hypoxia. Activation of caspase-9 and caspase-3 has been recognized as hallmarks of mitochondrial cell death in a variety of different cell types (24). Our results strongly suggest an involvement of the mitochondrial pathway during apoptosis induced by hypoxia in HMECs. Glioma cells secrete cytokines such as VEGF (25) and it has been shown that VEGF inhibits the apoptosis of endothelial cells induced by TNFα supporting its role in inhibiting endothelial cell apoptosis (26). In our studies, U87 conditioned medium resisted apoptotic cell death of HMECs induced by hypoxia in part, through blockade of caspase activation.
Since angiogenesis is essential for tumor progression, we determined migration and angiogenesis of human microvascular endothelial cells after exposure to hypoxia. In addition, we performed in vitro capillary-like structure formation assays to examine the importance of U87 conditioned medium in the angiogenic process. The results here demonstrate that exposure of HMECs to hypoxia increased cell migration and angiogenic potential. . Hypoxia is a strong inducer of angiogenesis-promoting factors that orchestrate complex interactions between ECs and the extracellular matrix, leading to neovascularization (27). Endothelial MMPs have been shown to play a pivotal role in these interactions because of their ability to degrade basement membrane components, a primary step in the process of angiogenesis (28). There is considerable accumulating evidence suggesting a role of MMP-2 and MMP-9 in angiogenesis. Enhanced activities of MMP-2 and MMP-9 in HMECs exposed to hypoxia correlated with increased capillary-like structure formation in in vitro studies. Current studies confirm that gelatinase activity is increased in several types of endothelial cells exposed to hypoxia (29,30).
During hypoxia there is activation of the transcription factor HIF-1α which induced expression of target genes with hypoxia-responsive elements (HREs) in their promoters (31) including VEGF (32). In the present study we examined the expression of proangiogenic genes, VEGF and VEGFR2 in HMECs. Whereas some studies could not detect any VEGF-release neither in normoxia nor in hypoxia and anoxia [33) others showed a clear expression of VEGF mRNA and protein in ECs induced by hypoxia (34). In our studies, we observed upregulation of VEGF and VEGFR2 transcripts in HMECs by hypoxic treatment. An in vitro model of angiogenesis was used to study hypoxia-induced effects. HMECs plated on Matrigel matrix were exposed to hypoxia and examined for capillary-like structure formation. Hypoxia stimulated the angiogenic response in vitro, whereas U87 conditioned medium exposure induced further VEGF and VEGFR2 expression and tube formation. The stimulation of angiogenesis in vitro by hypoxic conditions is mediated by an increase in the responsiveness of ECs to the different angiogenesis influencing factors
The present study shows that both hypoxia and U87 conditioned medium can influence angiogenic process in HMECs. The influence of hypoxia was significant on apoptosis of HMECs whereas U87 conditioned medium caused a consistent protection.
Acknowledgments
This work was supported by NIH Grant R01-NS-051625 (to S.M.).
References
- 1.Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Pomeroy SL, Cairncross JG. Focus on central nervous system neoplasia. Cancer Cell. 2002;1:125–128. doi: 10.1016/s1535-6108(02)00040-5. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, Levin VA, Yung WK. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999;17:2572–2578. doi: 10.1200/JCO.1999.17.8.2572. [DOI] [PubMed] [Google Scholar]
- 5.Plate KH. Mechanisms of angiogenesis in the brain. J Neuropathol Exp Neurol. 1999;58:313–320. doi: 10.1097/00005072-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Fischer I, Gagner JP, Law M, Newcomb EW, Zagzag D. Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol. 2005;15:297–310. doi: 10.1111/j.1750-3639.2005.tb00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen RL. Hypoxia in the tumorigenesis of gliomas and as a potential target for therapeutic measures. Neurosurg Focus. 2006;20:E24. doi: 10.3171/foc.2006.20.4.16. [DOI] [PubMed] [Google Scholar]
- 8.Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang WT, Nelson PT, Lustig RA, Jenkins K, Magarelli DP, Hahn SM, Collins RA, Grady MS, Koch CJ. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res. 2004;10:8177–8184. doi: 10.1158/1078-0432.CCR-04-1081. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237:10–21. doi: 10.1016/j.canlet.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 10.Soini Y, Paakko P, Lehto VP. Histopathological evaluation of apoptosis in cancer. Am J Pathol. 1998;153:1041–1053. doi: 10.1016/S0002-9440(10)65649-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leon SP, Folkerth RD, Black PM. Microvessel density is a prognostic indicator for patients with astroglial brain tumors. Cancer. 1996;77:362–372. doi: 10.1002/(SICI)1097-0142(19960115)77:2<362::AID-CNCR20>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Tan Z, Tran ND. Chemical hypoxia-ischemia induces apoptosis in cerebromicrovascular endothelial cells. Brain Res. 2000;877:134–140. doi: 10.1016/s0006-8993(00)02666-4. [DOI] [PubMed] [Google Scholar]
- 13.Jadhav U, Chigurupati S, Lakka SS, Mohanam S. Inhibition of matrix metalloproteinase-9 reduces in vitro invasion and angiogenesis in human microvascular endothelial cells. Int J Oncol. 2004;25:1407–1414. [PubMed] [Google Scholar]
- 14.Chandrasekar N, Mohanam S, Gujrati M, Olivero WC, Dinh DH, Rao JS. Downregulation of uPA inhibits migration and PI3k/Akt signaling in glioblastoma cells. Oncogene. 2003;22:392–400. doi: 10.1038/sj.onc.1206164. [DOI] [PubMed] [Google Scholar]
- 15.Mohanam S, Chandrasekar N, Yanamandra N, Khawar S, Mirza F, Dinh DH, Olivero WC, Rao JS. Modulation of invasive properties of human glioblastoma cells stably expressing amino-terminal fragment of urokinase-type plasminogen activator. Oncogene. 2002;21:7824–7830. doi: 10.1038/sj.onc.1205893. [DOI] [PubMed] [Google Scholar]
- 16.Yanamandra N, Gumidyala KV, Waldron KG, Gujrati M, Olivero WC, Dinh DH, Rao JS, Mohanam S. Blockade of cathepsin B expression in human glioblastoma cells is associated with suppression of angiogenesis. Oncogene. 2004;23:2224–2230. doi: 10.1038/sj.onc.1207338. [DOI] [PubMed] [Google Scholar]
- 17.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 18.Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 19.Nomura M, Yamagishi S, Harada S, Hayashi Y, Yamashima T, Yamashita J, Yamamoto H. Possible participation of autocrine and paracrine vascular endothelial growth factors in hypoxia-induced proliferation of endothelial cells and pericytes. J Biol Chem. 1995;270:28316–28324. doi: 10.1074/jbc.270.47.28316. [DOI] [PubMed] [Google Scholar]
- 20.Peters K, Schmidt H, Unger RE, Kamp G, Prols F, Berger BJ, Kirkpatrick CJ. Paradoxical effects of hypoxia-mimicking divalent cobalt ions in human endothelial cells in vitro. Mol Cell Biochem. 2005;270:157–166. doi: 10.1007/s11010-005-4504-z. [DOI] [PubMed] [Google Scholar]
- 21.Iida T, Mine S, Fujimoto H, Suzuki K, Minami Y, Tanaka Y. Hypoxia-inducible factor-1alpha induces cell cycle arrest of endothelial cells. Genes Cells. 2002;7:143–149. doi: 10.1046/j.1356-9597.2001.00512.x. [DOI] [PubMed] [Google Scholar]
- 22.Peters K, Troyer D, Kummer S, Kirkpatrick CJ, Rauterberg J. Apoptosis causes lumen formation during angiogenesis in vitro. Microvasc Res. 2002;64:334–338. doi: 10.1006/mvre.2002.2438. [DOI] [PubMed] [Google Scholar]
- 23.Segura I, Serrano A, De Buitrago GG, Gonzalez MA, Abad JL, Claveria C, Gomez L, Bernad A, Martinez-A C, Riese HH. Inhibition of programmed cell death impairs in vitro vascular-like structure formation and reduces in vivo angiogenesis. FASEB J. 2002;16:833–841. doi: 10.1096/fj.01-0819com. [DOI] [PubMed] [Google Scholar]
- 24.Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zuniga-Pflucker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 25.Sawa H, Murakami H, Ohshima Y, Murakami M, Yamazaki I, Tamura Y, Mima T, Satone A, Ide W, Hashimoto I, Kamada H. Histone deacetylase inhibitors such as sodium butyrate and trichostatin A inhibit vascular endothelial growth factor (VEGF) secretion from human glioblastoma cells. Brain Tumor Pathol. 2002;19:77–81. doi: 10.1007/BF02478931. [DOI] [PubMed] [Google Scholar]
- 26.Spyridopoulos I, Brogi E, Kearney M, Sullivan AB, Cetrulo C, Isner JM, Losordo DW. Vascular endothelial growth factor inhibits endothelial cell apoptosis induced by tumor necrosis factor-alpha: balance between growth and death signals. J Mol Cell Cardiol. 1997;29:1321–1330. doi: 10.1006/jmcc.1996.0365. [DOI] [PubMed] [Google Scholar]
- 27.Michiels C, Arnould T, Remacle J. Endothelial cell responses to hypoxia: initiation of a cascade of cellular interactions. Biochim Biophys Acta. 2000;1497:1–10. doi: 10.1016/s0167-4889(00)00041-0. [DOI] [PubMed] [Google Scholar]
- 28.Haas TL, Madri JA. Extracellular matrix-driven matrix metalloproteinase production in endothelial cells: implications for angiogenesis. Trends Cardiovasc Med. 1999;9:70–77. doi: 10.1016/s1050-1738(99)00014-6. [DOI] [PubMed] [Google Scholar]
- 29.Ben-Yosef Y, Lahat N, Shapiro S, Bitterman H, Miller A. Regulation of endothelial matrix metalloproteinase-2 by hypoxia/reoxygenation. Circ Res. 2002;90:784–791. doi: 10.1161/01.res.0000015588.70132.dc. [DOI] [PubMed] [Google Scholar]
- 30.Kolev K, Skopal J, Simon L, Csonka E, Machovich R, Nagy Z. Matrix metalloproteinase-9 expression in post-hypoxic human brain capillary endothelial cells: H2O2 as a trigger and NF-kappaB as a signal transducer. Thromb Haemost. 2003;90:528–537. doi: 10.1160/TH03-02-0070. [DOI] [PubMed] [Google Scholar]
- 31.Dachs GU, Patterson AV, Firth JD, Ratcliffe PJ, Townsend KM, Stratford IJ, Harris AL. Targeting gene expression to hypoxic tumor cells. Nat Med. 1997;3:515–520. doi: 10.1038/nm0597-515. [DOI] [PubMed] [Google Scholar]
- 32.Berra E, Pages G, Pouyssegur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19:139–145. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- 33.Kroon ME, Koolwijk P, van der Vecht B, van Hinsbergh VW. Urokinase receptor expression on human microvascular endothelial cells is increased by hypoxia: implications for capillary-like tube formation in a fibrin matrix. Blood. 2000;96:2775–2783. [PubMed] [Google Scholar]
- 34.Minchenko A, Bauer T, Salceda S, Caro J. Hypoxic stimulation of vascular endothelial growth factor expression in vitro and in vivo. Lab Invest. 1994;71:374–379. [PubMed] [Google Scholar]