

Abstract
Tryprostatin A is a potent inhibitor of breast cancer resistance protein, consequently a series of structure-activity studies on the cell cycle inhibitory effects of tryprostatin A analogues as potential antitumor antimitotic agents have been carried out. These analogues were assayed for their growth inhibition properties and their ability to perturb the cell cycle in tsFT210 cells. SAR studies resulted in the identification of the essential structural features required for cytotoxic activity. The absolute configuration L-Tyr-L-pro in the diketopiperazine ring along with the presence of the 6-methoxy substituent on the indole moiety of 1 was shown to be essential for dual inhibition of topoisomerase II and tubulin polymerization. Biological evaluation also indicated the presence of the 2-isoprenyl moiety on the indole scaffold of 1 was essential for potent inhibition of cell proliferation. Substitution of the indole Na-H in 1 with various alkyl or aryl groups, incorporation of various L-amino acids into the diketopiperazine ring in place of L-proline, and substitution of the 6-methoxy group in 1 with other functionality provided active analogues. The nature of the substituents present on the indole Na-H or the indole C-2 position influenced the mechanism of action of these analogues. nalogues 68 (IC50 = 10 μM) and 67 (IC50 = 19 μM) were 7-fold and 3.5-fold more potent, respectively than 1 (IC50 = 68 μM) in the inhibition of the growth of tsFT210 cells. Diastereomer-2 of tryprostatin B 8 was a potent inhibitor of the growth of three human carcinoma cell lines: H520 (IC50 = 11.9 μM), MCF7 (IC50 = 17.0 μM) and PC3 (IC50 = 11.1 μM) and was equipotent with etoposide, a clinically used anticancer agent. Isothiocyanate analogue 71 and 6-azido analogue 72 were as potent as 1 in the tsFT210 cell proliferation and may be useful tools in labeling BCRP.
Introduction
The cell cycle coordinates a variety of cellular functions involved in the accurate replication of the genome and cell division.1 These processes are tightly regulated primarily at the G1/S and G2/M phase transitions by a series of checkpoints. It has become clear that checkpoint control defects in cancer cells contribute to tumorigenesis and are a significant reason for the increased selectivity of tumors over normal cells towards chemotherapy.2,3 Cell cycle inhibitors or modulators are highly promising new therapeutic agents against human cancers.
With an increased understanding of the molecular biology of cell cycle control it has become possible to develop bioassays and screen for agents that specifically interfere with these processes. One such method was developed by Osada et al.4,5 which utilizes the synchronous culture of the murine temperature-sensitive mutant cell line, tsFT210, defective in the p34cdc2 gene. With this assay a family of 2-isoprenylated diketopiperazine indole alkaloids which effect cell cycle arrest at the G2/M phase was isolated from the fermentation broth of a marine fungal strain of Aspergillus fumigatus BM939. It was found that tryprostatin A 1 (Chart 1) and tryprostatin B 2 (Chart 1) completely inhibited cell cycle progression of tsFT210 cells in the G2/M phase at a final concentration of 50 μg/mL of 1 and 12.5 μg/mL of 2, respectively.6–8 Since these indole alkaloids were isolated only in small amounts, studies on the mechanism and SAR were not reported earlier.
Chart 1.

Tryprostatin A and B have previously been synthesized9–11 the aim of which was to study their mechanisms of action. The similarities in the structures of the tryprostatins with etoposide (chart 1) and azatoxin (Chart 1), a dual inhibitor of topoisomerase II (G2)/tubulin polymerization (M), led to the investigation of the ability of the two tryprostatins to inhibit topoisomerase II and tubulin polymerization. Biological evaluations12 of 1 and 2 indicated that both alkaloids were very weak inhibitors of topoisomerase II in the topoisomerase II assay; while only 1 had marginal activity in the tubulin polymerization assay. This latter result was in agreement with the data reported for 1 by Osada et al.13 Osada et al.13 also reported 1 inhibited cell cycle progression of rat normal fibroblast 3Y1 cells specifically in the M phase. The concentration of 1 that arrested cell cycle progression in the M phase corresponded to that which induced a marked depolymerization in situ of the microtubules containing both cytoplasmic network and spindle apparatus. Although tryprostatin B 2 arrested cell cycle progression at a lower concentration than 1, the inhibition was not due to inhibition of the M phase. It was shown that 1 inhibited microtubule assembly through a different type of mechanism than colchicine (Chart 1), vinblastine (Chart 1), or maytansine-rhizoxin.13 Tryprostatin A 1 inhibited microtubule assembly by interfering with the interaction between microtubule associated proteins (MAPs) and the C-terminal domain of tubulin. Since 1 operated by an entirely novel mechanism this may be important in cancer chemotherapy, especially in multiple drug resistance (MDR) cancers.
Microtubules are hollow cylindrical tubes found in almost all eukaryotic cell types. They play an important role in a variety of cellular functions, such as cell division, cell movement, cell shape, and transport of organelles inside the cell.14 Tubulin exists as a heterodimer of α- and β-tubulin and is the major building block of microtubules. Proteins such as the MAPs bind to and modify microtubule properties.14,15 In the absence of MAPs, α/β-tubulin heterodimers polymerize only by treatment with high concentrations of glycerol or organic acids such as glutamate.16
The discovery of numerous compounds from natural sources which display a wide structural diversity and are cytotoxic by perturbation of the dynamic instability of microtubules has attracted much attention within the last two decades.17–20 Microtubules have, therefore, become an attractive pharmacological target for anticancer drug discovery.17–20 Almost all antimitotic agents interact with the α/β-tubulin dimer, rather than microtubule-associated proteins (MAPs) or other proteins involved in microtubule functions. The Vinca alkaloids, exemplified by vinblastine (Chart 1) and vincristine (Chart 1), as well as the taxanes, such as paclitaxel (Chart 1) and the semisynthetic analogue docetaxel (Chart 1), are the most commonly used antimitotic agents in the clinical treatment of cancer.21 Colchicine is another important antimitotic agent; however, it has limited medicinal utility due to its narrow therapeutic index.
Additionally, the natural products combretastatin A-422, curacin A,23 podophyllotoxin24, epothilones A and B,25 and dolastatin26 to cite just a few, are known to be cytotoxic through binding interactions with tubulin. Another antimitotic agent, estramustine phosphate inhibits microtubule assembly by binding to both microtubule-associated protein 2 (MAP2) and tubulin27, while 5,5′-bis[8-(phenylamino)-1-naphthalenesulphonate] (bis-ANS) specifically inhibits MAP-dependent microtubule assembly by interaction with the C-terminal domain of the tubulin heterodimer.28 These compounds may lead to useful cancer therapeutic agents. Indeed, estramustine in combination with other antimicrotubule agents exhibits synergistic cytotoxicity both in vitro and in vivo.27 However, no new tubulin polymerization inhibitor of low molecular weight has reached clinical status, as yet. Clinically available compounds such as paclitaxel or vincristine face several disadvantages; principally: (i) high toxicity, (ii) development of drug resistance in patients, (iii) marginal oral bioavailability and poor solubility, and (iv) complex synthesis or isolation procedures.17–20 Therefore, a pressing need to develop simpler, more effective antitumor drugs still remains.
The development of MDR to chemotherapeutic agents remains one of the primary obstacles in cancer treatment. These arise from intrinsic or acquired mechanisms of resistance. The overexpression of energy dependent (ATP) transmembrane drug-efflux pumps, such as P-glycoprotein (MDR1), multidrug resistance protein (MRP), and breast cancer resistance protein (BCRP), have been shown to produce resistance to several commonly used chemotherapeutic agents. Breast cancer resistance protein is a 72 kDa protein which probably homodimerizes to form an active transport complex.29 BCRP was first identified in drug resistant MCF-7/adrVp cells30 and has been recently reviewed.31–33 Like other members of the ATP binding cassette family of membrane transporters, such as MDR1 and MRP1, BCRP is expressed in a variety of malignancies where it may produce resistance to chemotherapeutic agents. In addition, it has also been reported that BCRP expression may be a prognostic indicator in certain cancers and is associated with poor response to chemotherapy.34,35 Overexpression of BCRP has been reported in a number of tumor types including: adenocarcinomas (arising from the digestive tract, the endometrium, and the lung), melanoma, soft tissue sarcomas,36 hematological malignancies such as acute myeloid leukemia (AML),37 and acute lymphoblastic leukemia (ALL).38 Elevated expression of BCRP results in resistance of various cancer cell lines to antitumor drugs including: topotecan, mitoxantrone, daunorubicin, doxorubicin, and bisantrene.39 In addition flavopiridol resistance is mediated by BCRP.40
The clinical significance of BCRP along with its limited expression in normal tissues makes BCRP a viable target for inhibition to reverse MDR. Several potent and specific inhibitors of BCRP have been developed. This has potentially opened the door to clinical applications of BCRP inhibition. These inhibitors include the targeted agents: gefitinib and imatinib mesylate,41 as well as the more specific inhibitors: fumitremorgin C42, tryprostatin A43, and GF12091844. At concentrations of 10–50 μM, tryprostatin A 1 was shown to reverse a mitoxantrone-resistant phenotype and inhibited the cellular BCRP-dependent mitoxantrone accumulation in the human gastric carcinoma cell line EPG85–257RNOV and the human breast cancer cell line MCF7/AdrVp (both exhibited acquired BCRP-mediated MDR). No cytotoxicity was observed at effective concentrations.43
In the search for potent and selective antitumor agents, the synthesis of a series of analogues (3–8, Figure 1) of 1 and 2 was carried out in order to probe the importance of the stereochemistry of the diketopiperazine ring on the inhibition of topoisomerase II and/or tubulin polymerization. Tryprostatin analogues 1–8 were evaluated for their ability to inhibit topoisomerase II and/or tubulin binding protein as well as their tumor cell growth inhibitory activity.12
Figure 1.

More recently, Osada et. al.13 reported the presence of the 6-methoxy substituent on the indole moiety of 1 confered lower cytotoxicity to tryprostatin A and enhanced the specificity for inhibition of microtubule assembly. The lower cytotoxicity of 1 in comparison to 2, combined with a unique mechanism for inhibition of tubulin polymerization, as well as BCRP prompted investigation of the structure activity relationships (SAR) in 1 in order to enhance tubulin polymerization and/or BCRP inhibition. To our knowledge, no SAR of tryprostatin A has appeared in the literature, to date. The SAR studies were designed to determine the minimum structural requirements of tryprostatin A required to exhibit potent and selective cytotoxic activity, in the search for new anticancer agents. Several modifications, which maintained the same backbone were carried out as outlined in Figure 1: (A) substitution of the 6-position of the aromatic ring, (B) alkylation of the indole NH, (C) substitution of the 2-position of the indole moiety, and (D) substitution of the L-proline residue in the diketopiperazine ring with other L-amino acids.
Chemistry
The synthesis of optically active 1 and 2 have been reported.9–11 This method was also extended to the synthesis of the enantiomers (3 and 4) and diastereomers (5–8) of 1 and 2 (Scheme 1).11,12 The synthesis of 1–8 (Scheme 1) began with indoles 9a and 9b which were coupled with the anion of the Schöllkopf chiral auxiliary 11 (derived from L-valine), to afford the trans diastereomers 14 and 15 respectively, with 100% diastereoselectivity. The diastereoselectivity of the addition to the Schöllkopf chiral auxillary was found to be 100% by analysis of the 1H spectrum of the crude mixture of the respective compounds. When indoles 14 and 15 were treated with LDA at −78 °C, followed by addition of isoprenyl bromide, the 2-isoprenylpyrazine derivatives 18 and 19 were obtained, respectively. The pyrazine moiety was removed from pyrazines 18 or 19 under acidic conditions (aq HCl, THF) in 94% yield to afford the 2-isoprenyl tryptophan 22 or 23, respectively. The coupling of 2-isoprenyl tryptophan 22 or 23 with Fmoc-D-proline 25 using triethylamine as the base was followed by formation of the diketopiperazine ring. The Boc protecting group was removed from the indole N(H) function in refluxing xylene to afford 3 and 4, respectively. Similarly, coupling of 2-isoprenyl tryptophan 22 or 23 with Fmoc-L-proline 24 afforded 5 or 6 respectively. The natural products (1 and 2) were prepared from the trans transfer of chirality from D-valine (Schöllkopf) and from L-proline.9–11 The diastereomers 7 and 8 of 1 and 2 respectively, were prepared from the trans transfer of chirality from D-valine (Schöllkopf) and from D-proline as outlined in Scheme 1.9–11
Scheme 1a.

aConditions: (a) THF, n-BuLi, −78 °C; (b) LDA (1.5 eq), THF, isoprenylbromide (3 eq); (c) 2N aq HCl, THF, −78 °C to rt; (d) TEA, CHCl3; DEA, CH3CN, rt; xylene, reflux.
Analogues 37–40 were intended as tryprostatin A mimics in which the alkyl substituent was moved from the indole 2-position to the indole NH. This was expected to result in analogues that are more readily accessible than the natural products which require methods for prenylation of tryptophan at C-2. As shown in Scheme 2, the ortho-iodoaniline 26 was coupled with the internal alkyne10 27 in the presence of catalytic Pd(OAc)2 with Na2CO3 as the base to provide indole 28 in 77% yield.45,46
Scheme 2a.

aConditions: (a) Pd(OAc)2, LiCl, Na2CO3, DMF, 100 °C, 77%; (b) NaH, DMF, RX; (c) 2N aq HCl, EtOH, THF, −78 °C to rt; (d) 24, TEA, CHCl3; DEA, CH3CN, rt; xylene, reflux.
Alkylation of 28 with methyl iodide, isoprenyl bromide, benzyl bromide or allyl bromide in the presence of sodium hydride afforded the indole Na-substituted analogues 29–32 in 72–99% yields. Hydrolysis of pyrazines 29–32 with 2N aqueous HCl in THF resulted in removal of both the bislactim ether moiety and the silyl group. Tryptophans 33–36 were readily transformed into analogues 37–40 of tryprostatin A under conditions analogous to the steps in Scheme 1.
In order to synthesize the Na-substituted analogues (46 and 47) of 1, in region B in which the indole 2-position carried the isoprenyl group, an analogous strategy was employed. Thermal removal of the Boc protecting group of 16 (Scheme 3) in refluxing xylene afforded pyrazine 41. Alkylation of 41 with benzyl bromide or allyl bromide using sodium hydride afforded analogues 42 and 43 in 91% and 82% yields, respectively. These intermediates were readily transformed into analogues 46 and 47 of tryprostatin A under conditions analogous to the steps in Scheme 1, as illustrated in Scheme 3.
Scheme 3a.
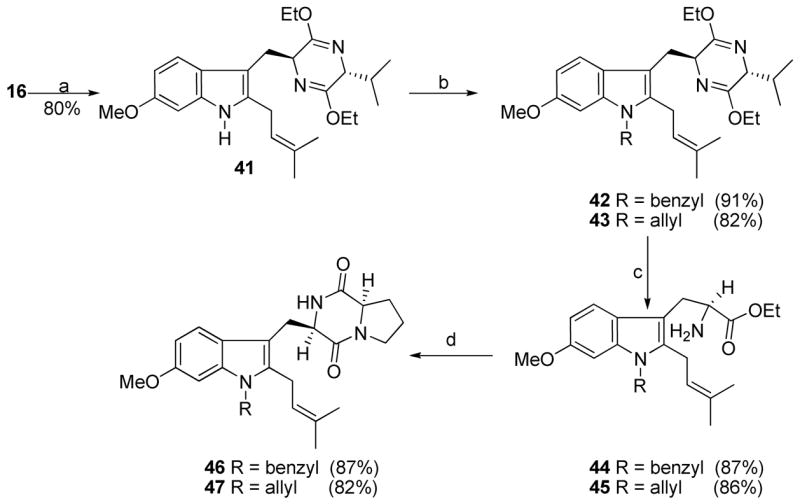
aConditions: (a) xylene, reflux, 80%; (b) NaH, DMF, RX; (c) 2N aq HCl, EtOH, THF, −78 °C to rt; (d) 24, TEA, CHCl3; DEA, CH3CN, rt; xylene, reflux.
To obtain analogues with different substitution at the indole 2-position (region C) of 1, 2-bromo-indole 48 (Scheme 4) served as a common intermediate which was easily obtained from indole 28 after sequential treatment with NBS and Boc anhydride. As shown in Scheme 4, lithium-halogen exchange, followed by treatment with benzyl bromide or allyl bromide furnished the 2-substituted analogues 49 and 50. Analogues 49 and 50 were further transformed into 2-substituted indoles 55 and 56, respectively, under standard conditions (Scheme 4). For the synthesis of 51 (Scheme 4), the zinc reagent was prepared through lithium-halogen exchange followed by treatment with zinc chloride. Negishi coupling47 of the zinc reagent with phenyl iodide using Pd(OAc)2 in the presence of trifuryl phosphine afforded the 2-phenyl indole 51 in 65% yield which was then transformed to analogue 57 under conditions outlined in Scheme 4. A variety of other conditions were attempted to prepare the 2-phenyl indole 51, however the Negishi coupling was the only method that was successful in a practical sense.
Scheme 4a.
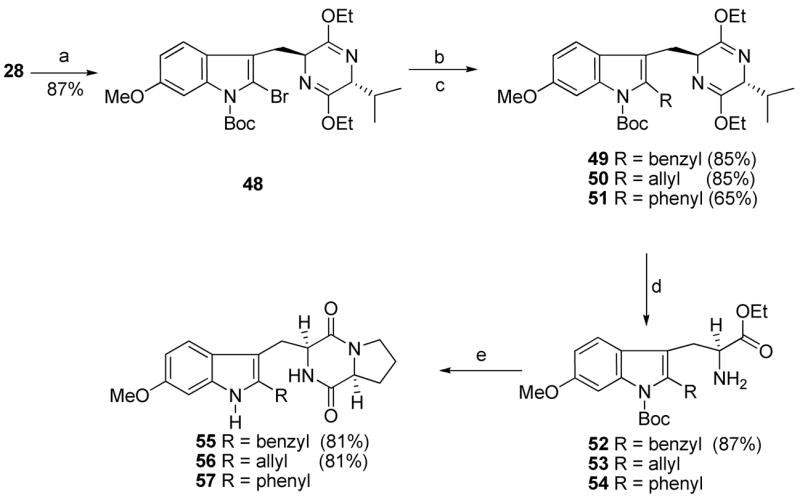
aConditions: (a) NBS, CH3CN; (Boc)2O, DMAP, CH3CN, rt, 87%; (b) n-BuLi, THF, −78 °C; RX; (c) n-BuLi, THF, −78 °C; ZnCl2; Pd(OAc)2, PhI, tri-2-furyl phosphine, rt, 65%; (d) 2N aq HCl, EtOH, THF, −78 °C to rt; (e) 24, TEA, CHCl3; DEA, CH3CN, rt; xylene, reflux.
Heck coupling48 of bromide 48 with methyl acrylate in the presence of 5% Pd(PPh3)4 and Cy2NEt afforded the coupled product 58 in 94% yield (Scheme 5). Analogue 58 (Scheme 5) was then transformed into ester 60, analogous to well-developed processes outlined previously in Scheme 1. It was well known in the literature that the 6-methoxy group activated the C-2 position of the indole nucleus towards electrophilic substitution. As illustrated in Scheme 6, indole 28 was hydrolyzed with aqueous 2N HCl to afford the 6-methoxy-L-tryptophan ethyl ester 61 in 86% yield. Ester 61 was further transformed into the Na-H analogue 62 under conditions similar to that described for 1 in Scheme 1. Analogue 62 was treated with NBS at −78 °C to afford 2-bromoindole 63 in 80% yield. The corresponding 2-chloro analogue 64 was prepared in 85% yield (from NCS) under analogous conditions to those described above for the preparation of 2-bromoindole 63.
Scheme 5a.
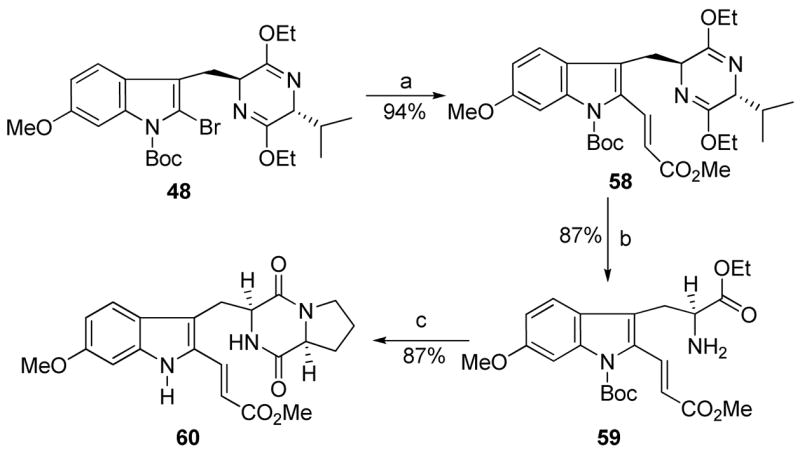
aConditions: (a) Pd(PPh3)4, methyl acrylate, Cy2NMe, toluene, 95 °C, 94%; (b) 2N aq HCl, EtOH, THF, −78 °C to rt, 80%; (c) 24, TEA, CHCl3; DEA, CH3CN, rt; xylene, reflux.
Scheme 6a.
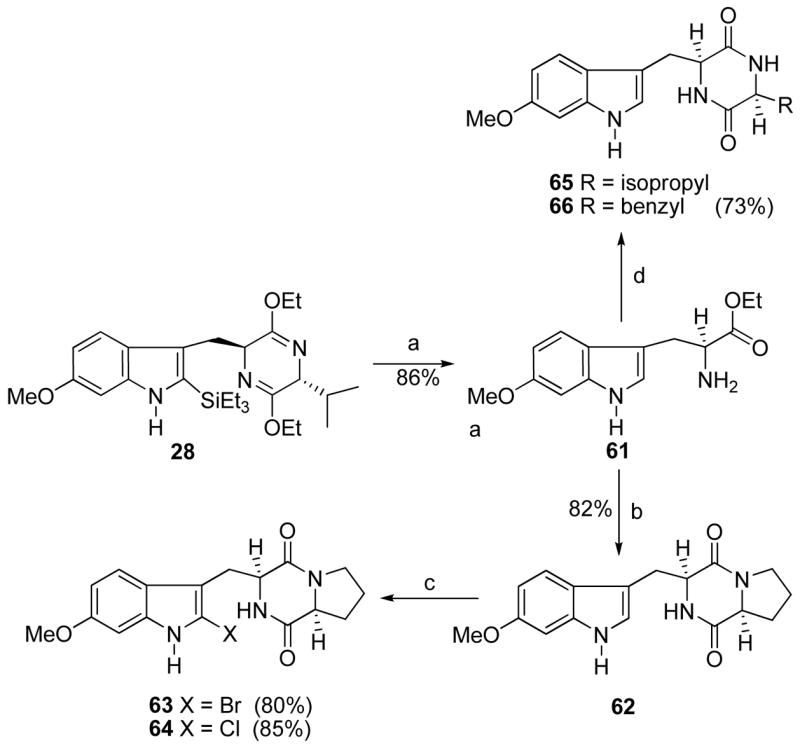
aConditions: (a) 2N aq HCl, EtOH, THF, −78 °C to rt, 86%; (b) 24, TEA, CHCl3; DEA, CH3CN, rt; xylene, reflux; (c) NBS or NCS, THF, −78 °C to rt; (d) Fmoc-L-amino-acyl chloride, TEA, CHCl3; DEA, CH3CN, rt; xylene, reflux.
Two additional amino acids, other than proline, were also incorporated into 1 (region D). As shown in Scheme 6, 6-methoxytryptophan 61 was transformed into the cyclic dipeptides 65 and 66 with L-valine and L-phenylalanine respectively, under conditions illustrated in Scheme 6. The 2-isoprenyl indole 20 was also transformed to analogues 67 and 68, as shown in Scheme 7.
Scheme 7a.
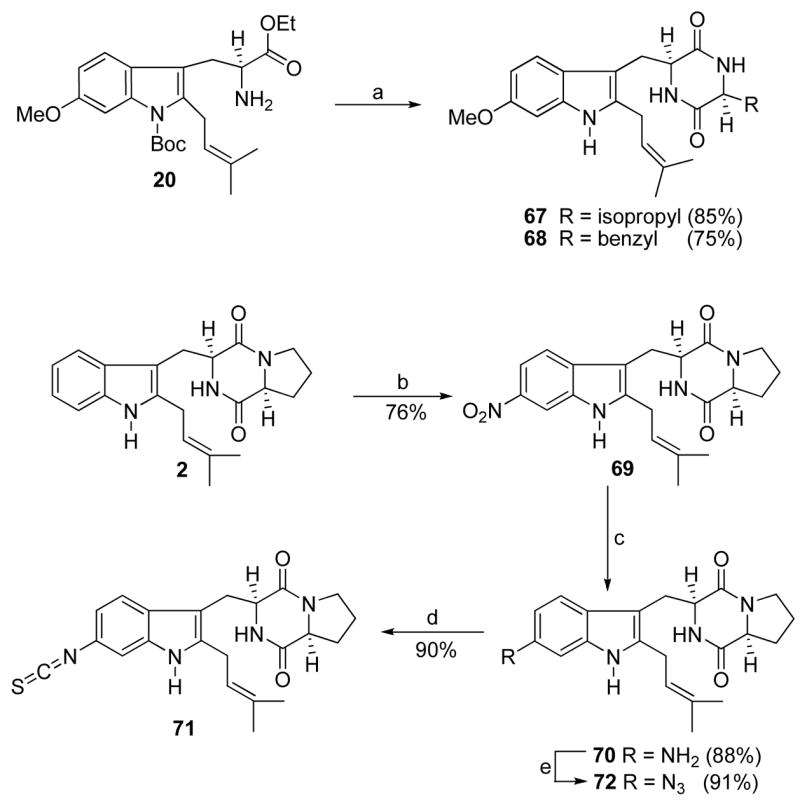
aConditions: (a) Fmoc-L-amino-acylchloride, TEA, CHCl3; DEA, CH3CN, rt; xylene, reflux; (b) NaNO2, TFA, −78 °C to −20 °C, 75%; (c) NH2NH2, FeCl3/6H2O, active C, MeOH, reflux, 91%; (d) CHCl3, ClC(S)Cl, 93%; (e) TfN3, aq CuSO4, Et3N, CH2Cl2/MeOH, 89%.
The synthesis of C-6 substituted analogues of tryprostatin A modified in region A is depicted in Scheme 7. The synthesis began with a highly regioselective process for nitration of 2. Although several methods were attempted to incorporate the nitro group at the desired 6-position with only minimal success, this was successfully carried out when 2 was treated with NaNO2 in the presence of TFA49 at low temperature to afford 69. To determine the regiochemistry, detailed NMR analysis of the 6-nitro analogue 69 was carried out. The coupling patterns of the aromatic ring protons could be employed to distinguish the 4- and 7-substituted indoles from the 5- and 6-substituted regioisomers, since one would not expect singlet protons in the spectrum of the 4- or 7-substituted indoles. The 1H NMR spectrum of 69 clearly contained one singlet (8.29 ppm) corresponding to one proton in the aromatic region, consequently, the product of this mononitration was either the 5- or 6- substituted regioisomer. In case of the 5-nitrosubstituted indole this singlet would correspond to the proton at C(4), whereas in the case of 6-nitrosubstituted indole this singlet would correspond to the proton at C(7). The 6-nitro regioisomer would be expected to exhibit a much stronger NOE signal between the indole N(H) proton and the proton at C(7) than the one between the indole N(H) proton and the proton at C(4). A strong NOE signal was observed between this proton singlet and the indole N(H) and vice versa. This further ruled out the 5-nitro regioisomer. Reduction of the nitro group in 69 (Scheme 7) with hydrazine in the presence of FeCl3•H2O and activated carbon in refluxing methanol45 furnished analogue 70 which was purified by column chromatography and stored as the hydrochloride salt. Amine 70 was stirred with thiophosgene in dry chloroform to afford the 6-isothiocyanate analogue 71 in high yield. Treatment of amine 70 with triflyl azide (TfN3) in the presence of copper sulfate afforded analogue 72 in 89% yield.50,51
Biological Evaluation and Discussion
Effects of Analogues 1–8 on Topoisomerase II
Tryprostatins 1–8 were evaluated as inhibitors of topoisomerase II in the topoisomerase II-mediated DNA relaxation assay.52,53 This assay measures the ability of the compound to inhibit the ability of topoisomerase II to relax supercoiled DNA. The inhibitory activities against topoisomerase II of compounds 1–8 were evaluated by agarose-gel electrophoresis experiments. The photopicture of 1–4’s agarose-gel electrophoresis experiment is presented in Figure 2. The agent, m-AMSA, a known inhibitor of topoisomerase II, was employed as the control (lane 3). The other controls employed were no enzyme (lane 1), enzyme (lane 2), and 1% DMSO (lane 4). The gels were analyzed qualitatively by examination of the presence of DNA bands that migrate farther down on the gel than the negative controls. Topoisomerase II-mediated relaxation of the DNA prevents the band from migrating down the gel as far as one that is still in a supercoiled form. Therefore, DNA incubated with topoisomerse II inhibitors will migrate farther on the gel than the no-enzyme or DMSO controls. Lane 1 is DNA alone, existing in two forms- supercoiled DNA and loosened DNA; lane 2 is topoisomerase II together with DNA, and supercoiled DNA was relaxed by topoisomerase II completely. As illustrated in Figure 2, 1 (lane 5) and 2 (lane 7) are both weak inhibitors of topoisomerase II; however, the potency cannot be determined from this data. The laddering is evidence of inhibition of topoisomerase II. The enantiomers of tryprostatin A 3 (lane 6) and B 4 (lane 8) were both inactive. The four diastereomers 5–8 (data not shown) were also found to be inactive as topoisomerase II inhibitors in this assay. Tryprostatin A 1 and B 2 are, therefore, weak inhibitors of topoisomerase II but their enantiomers (3 and 4) and diastereomers (5–8) are not.
Figure 2.
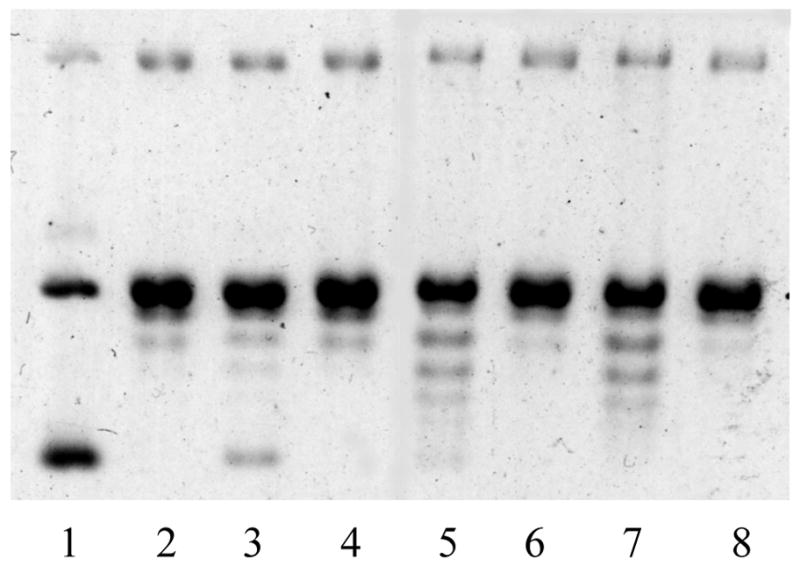
Representative agarose gel from the topoisomerase II mediated DNA relaxation assay. Data for all compounds are not shown. Lane 1: DNA only; lane 2: DNA + topoisomerase II; lane 3: DNA + topoisomerase II + m-AMSA (100 μM); lane 4: DNA + topoisomerase II + 1% DMSO; lane 5: DNA + topoisomerase II + 1 (100 μM); lane 6: DNA + topoisomerase II + 3 (100 μM); lane 7: DNA + topoisomerase II + 2 (100 μM); lane 8: DNA + topoisomerase II + 4 (100 μM).
Effects of Analogues 1–8 on Tubulin Polymerization
Tryprostatins 1–8 were also evaluated as inhibitors of tubulin polymerization.13,54 Purified tubulin, containing MAPs and GTP, was incubated at 37 °C with either DMSO (as a solvent control), colchicine (standard), or analogues 1–8 and the change in absorbance was measured at 351 nM over 10 minutes. The concentration of the standard (colchicine) and analogues (1–8) was varied for different runs to obtain a delta absorbance versus concentration curve. Illustrated in Figures 3 and 4 are the results of the tubulin polymerization assay. Colchicine (the positive control) strongly suppressed tubulin assembly (IC50 = 44.9 μM), while 1 (Figure 3) caused a moderate reduction in the rate of tubulin polymerization. Tryprostatin B 2 (Figure 3) as well as the enantiomers of tryprostatin A 3 and B 4 (Figure 4) were inactive in this assay (IC50s >700 μM). Compounds 5–8 (data not shown) were also found to be inactive in this assay (less than 2% inhibition at 60 μM: IC50s > 700 μM).
Figure 3.
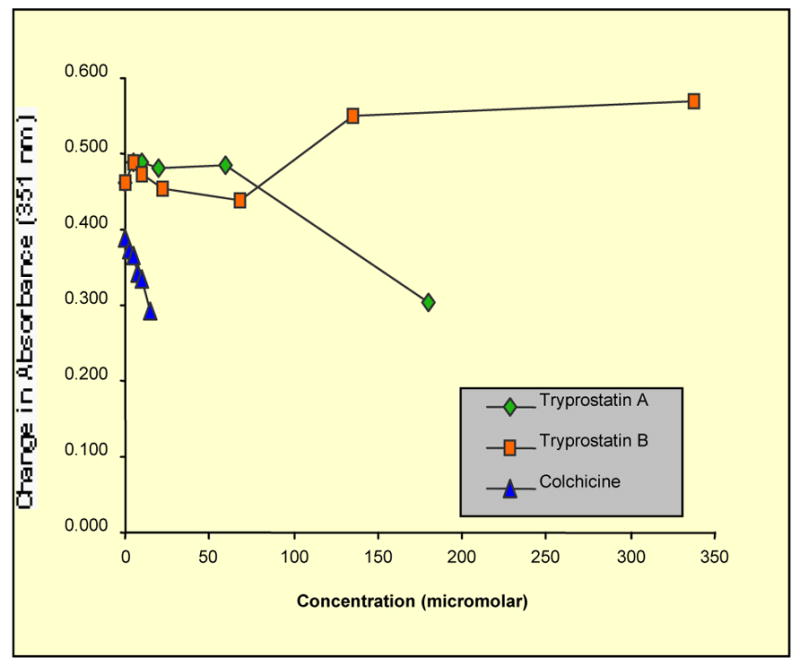
Inhibition of tubulin polymerization by tryprostatin A 1 and B 2, colchicine, a known tubulin polymerization inhibitor, was used as a control.
Figure 4.
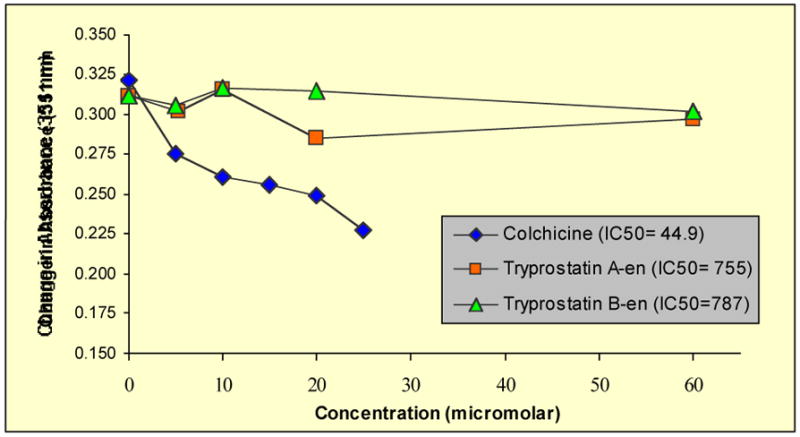
Inhibition of tubulin polymerization by analogues 3 and 4. Colchicine was employed as a control.
Osada et al.13 recently reported that tryprostatin A 1 was a novel inhibitor of MAP-dependent microtubule assembly and through disruption of the microtubule spindle, specifically inhibited cell cycle progression at the M phase. Thus biological evaluation of analogues 1–8 illustrated above indicated that 1 was a weak inhibitor of both topoisomerase II and tubulin polymerization, whereas 2 was only a weak inhibitor of topoisomerase II. The enantiomers (3 and 4) and diastereomers (5–8) were inactive in both the tubulin polymerization assay and topoisomerase II-mediated DNA relaxation assay. In terms of the stereochemistry of the amino acids present in the diketopiperazine ring, biological evaluation indicated that ligands with the absolute configuration L-Tyr-L-Pro (natural stereochemistry as in 1 and 2) were essential for inhibition of tubulin polymerization and/or topoisomerase II. Modification of the absolute configuration of the diketopiperazine ring from L-Tyr-L-Pro (1 and 2) to D-Tyr-D-Pro (3 and 4), D-Tyr-L-Pro (6 and 7), and L-Tyr-D-Pro (7 and 8) resulted in analogues that were very poor inhibitors of topoisomerase II and/or tubulin polymerization. Additionally, comparisons of analogues 1 and 2 indicated that presence of the 6-methoxy substituent in 1 resulted in analogues that are dual inhibitors of microtubule assembly and topoisomerase II.
In order to determine whether the absolute configuration of L-Tyr and/or L-Pro in the diketopiperazine ring was required to inhibit cell proliferation, analogues 1–8 were evaluated as inhibitors of three human lung (H520), breast (MCF-7), and prostrate (PC-3) cancer cell lines (Table 1).12 Analogues 1–7 were not potent inhibitors (GI50 >100 μM) of the growth of tumor cells in the three human cancer cell lines evaluated. However, the diastereomer 8 of tryprostatin B 2 exhibited potent cytotoxic activity at 100 μM against all three human cancer cell lines evaluated. It was found, in agreement with Danishefsky et al.55, that the inhibition of tryprostatin B 2 against the growth of the three human cancer cell lines evaluated occurred at higher concentrations (GI50 >100 μM) than that reported earlier6–8 for isolated tryprostatin B 2. Danishefsky et al.55 have shed some light on the apparent discrepancies in the cytotoxicity of isolated 2 versus synthetic 2. Their studies55 indicated that a DMSO solution of 2, upon standing in air, undergoes slow transformation to a mixture of products. The solutions of 2 containing detectable byproducts were considerably more cytotoxic (ca. 50-fold) than those containing apparently homogenous tryprostatin. Growth inhibitory (GI50) potency of 8 was also compared to that of etoposide against the growth of the three human lung (H520), breast (MCF-7), and prostrate (PC-3) cancer cell lines (Table 2).12 Outlined in Table 2 are the results obtained from the National Cancer Institute (NCI)56 screening of analogue 8 on the same three human cancer cell lines. The data obtained from the NCI for 8 were in complete agreement with the data obtained for 8 in the present study against all three human cancer cell lines evaluated. Analogue 8 was 3-fold more potent than etoposide in inhibition of the growth of the MCF-7 human cancer cell line. Also, analogue 8 was equipotent with etoposide against the growth of H520 and PC-3 human cancer cell lines.
Table 1.
Cell Growth Inhibition of Tryprostatins 1–8 (at 10, 100 μM) on Human Lung (H520), Breast (MCF7) and Prostate (PC-3) Cancer Cell Linesa.
| Percent Cell Survivalb |
||||||
|---|---|---|---|---|---|---|
| H520 | MCF7 | PC-3 | ||||
| Compd | 10 μM | 100 μM | 10 μM | 100 μM | 10 μM | 100 μM |
| 1 | 80.1 ± 4.1 | 79.4 ± 4.2 | >100 | 95.0 ± 4.7 | 99.2 ± 4.2 | 95.6 ± 5.0 |
| 2 | 77.6 ± 3.6 | 60.5 ± 3.5 | 88.2 ± 5.8 | 66.7 ± 5.3 | 95.5 ± 2.8 | 68.9 ± 6.6 |
| 3 | 81.7 ± 3.9 | 75.2 ± 3.5 | >100 | >100 | >100 | 83.7 ± 4.2 |
| 4 | >100 | 99.8 ± 1.6 | >100 | >100 | 95.8 ± 1.3 | 78.9 ± 2.1 |
| 5 | >100 | >100 | >100 | >100 | >100 | >100 |
| 6 | >100 | 76.5 ± 11.2 | >100 | >100 | 97.3 ± 5.9 | 68.5 ± 3.4 |
| 7 | 99.3 ± 1.8 | 98.5 ± 3.1 | >100 | 99.0 ± 4.6 | >100 | >100 |
| 8 | 88.3 ± 8.4 | 0.1 ± 0.1 | 73.6 ± 5.3 | 0.0 ± 0.1 | 59.3 ± 3.9 | 0.2 ± 0.0 |
CellTiter 96™ AQueous nonradioactive cell proliferation assay (Promega) was used to determine growth inhibition. Percent inhibition values were calculated versus control wells and were done in quadruplicate. Control wells contained 0.2% DMSO and the positive control was either etoposide or m-AMSA (20 μM, 20 μM). Values are reported ± the standard deviation of the mean.
Table 2.
Growth Inhibition (GI50) in μM of Human Cancer Cell Lines by 8 and etoposide.
| Compd | H520 | MCF-7 | PC-3 |
|---|---|---|---|
| 8a | 15.8 | 15.9 | 11.9 |
| 8 | 11.9 | 17.0 | 12.3 |
| Etoposide | 8.7 | 55.6 | 11.1 |
Data were obtained from NCI.
If one examines the structures of tryprostatins (1–7) and compares them with that of the active analogue 8, one can generate the following conclusion: the L-Tyr unit in the diketopiperazine ring was essential for potent tumor cell growth inhibition since none of the other tryprostatins (3–6), which contained the D-Tyr unit, exhibited activity. Biological evaluation of analogue 8 also indicated that the inhibition of the growth of human cancer cells by analogue 8 was not due to the inhibition of topoisomerase II or tubulin polymerization since analogue 8 was inactive against these two molecular targets. Further studies to identify the precise molecular targets are required. The presence of the 6-methoxy group on 7 compared to 8 nearly eliminated the potent tumor cell growth inhibitory activity against the three human cancer cell lines evaluated. The potent cytotoxic activity of analogue 8 against human cancer cells led to the evaluation of its activity against the growth of normal human cell lines. In preliminary studies, 8 was found to be cytotoxic to normal human cell lines; however, further studies are required in this regard.
Analogues 1–8 were selected by the NCI for evaluation in its in vitro preclinical antitumor screening program. The ability of compounds 1–8 to inhibit the growth of tumor cells was measured as GI50 values, the concentration required to inhibit the growth of tumor cells in culture by 50%, as compared to a control (Table 3). In two of the 60 tumor cell lines evaluated, tryprostatin A 1 showed GI50 values of ≤ 10−5 M. Again, tryprostatin B 2 (GI50 >40 μM) was considerably less active against the growth of the 60 tumor cell lines evaluated. In 9 of the 60 tumor cell lines evaluated, the most active analogue 8 showed GI50 values of ≤ 10−5 M. Analogue 5, the diastereomer of tryprostatin A, 1 was more active than 1 in the inhibition of the growth of tumor cells in most of the tumor cell lines evaluated. Analogues 3 and 7 were both considerably less active than 1 in inhibition of the growth of the tumor cells in the NCI screening program. However, analogue 4, the enantiomer of tryprostatin B 2, as well as both of the diastereomers 6 and 8 were more active than 2 in inhibition of the growth of tumor cells in most of the tumor cell lines evaluated. It is noteworthy that compounds 4, 5, 6 and 8 were not general cell toxins but showed selectivity both within a type of tumor cell line and across different tumor cell lines, with inhibitory values, which in some instances, differed by 100-fold.
Table 3.
Cytotoxicity Evaluation (GI50, μM) of Compounds 1–8 Against Selected Tumor Cell Lines56
| Cell line | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Leukemia | ||||||||
| CCRF-CEM | >100 | >100 | >25 | 25.1 | 11.9 | 22.2 | 99.4 | 3.22 |
| HL-60 (TB) | 11.3 | >100 | >25 | 29.0 | 35.0 | 55.7 | >100 | 22.4 |
| K-562 | 2.73 | 56.1 | >25 | 25.8 | 17.7 | 31.8 | >100 | 20.1 |
| MOLT-4 | ND | >100 | >25 | 21.0 | 12.2 | 34.9 | >100 | 5.96 |
| RPMI-8226 | 37.1 | >100 | >25 | 12.9 | 7.76 | 18.6 | 92.0 | 5.54 |
| SR | 5.68 | 50.6 | >25 | 12.1 | 11.3 | 25.4 | 76.9 | 9.46 |
| Non-small Cell Lung Cancer | ||||||||
| HOP-92 | 21.2 | 43.1 | 23.1 | ND | ND | 2.94 | 16.8 | 1.70 |
| EKVX | >100 | >100 | >25 | 39.4 | >50 | 20.7 | >100 | 6.90 |
| Colon Cancer | ||||||||
| COLO 205 | >100 | >100 | >25 | 21.2 | 12.4 | 39.3 | >100 | 17.7 |
| HT-29 | ND | ND | ND | 37.5 | 16.1 | ND | 40.3 | 5.25 |
| Melanoma | ||||||||
| LOX IMVI | >100 | >100 | >25 | >50 | 33.5 | 17.8 | >100 | 9.23 |
| Ovarian | ||||||||
| OVCAR-3 | >100 | >100 | >25 | 38.2 | >50 | 28.7 | 85.5 | 9.69 |
| IGROV1 | 90.5 | >100 | >25 | 16.0 | >50 | 32.3 | 50.0 | 11.5 |
| Prostrate Cancer | ||||||||
| PC-3 | 94.0 | >100 | >25 | 21.2 | 21.7 | 24.8 | >100 | 11.9 |
| DU-145 | >100 | >100 | >25 | >50 | 40.0 | 59.5 | >100 | 14.0 |
| Breast Cancer | ||||||||
| MDA-MB-231/ATCC | >100 | >100 | >25 | 11.9 | 13.0 | 49.6 | >100 | 14.9 |
| BT-549 | 79.7 | 58.2 | >25 | 7.21 | 9.77 | 26.9 | >100 | 13.2 |
| MCF-7 | >100 | >100 | >25 | 40.0 | 25.5 | 25.9 | >100 | 15.9 |
| Renal Cancer | ||||||||
| UO-31 | >100 | >100 | >25 | 12.6 | 27.3 | 45.7 | >100 | 14.7 |
Structure-Activity Relationships of Tryprostatin A Analogues
Because 1 was a potent inhibitor of BCRP, the tryprostatin A-related analogues (37–40, 46, 47, 55, 56, 60, and 62–72) were evaluated in vitro for the ability to disrupt the cell cycle and to inhibit tsFT210 cell proliferation.4,13,58 The inhibitory potency (IC50) values are listed in Table 4 and compared with tryprostatin A 1
Table 4.
Effect of tryprostatin A-related analogues on cell cycle progression and tsFT210 cells proliferation.
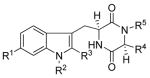 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | R4 R5 | IC50 (μM)a | Effect on cell cycle arrestb | |
| 1 | OMe | H | isoprenyl | -(CH2)- | 68 | M phase at 30 μM | |
| 37 | OMe | Me | H | -(CH2)- | > 100 | no effectc | |
| 38 | OMe | isoprenyl | H | -(CH2)- | > 100 | no effect | |
| 39 | OMe | benzyl | H | -(CH2)- | 46 | G1 phase at 100 μM | |
| 40 | OMe | allyl | H | -(CH2)- | 62 | G1 phase at 100 μM | |
| 46 | OMe | benzyl | isoprenyl | -(CH2)- | 55 | no effect | |
| 47 | OMe | allyl | isoprenyl | -(CH2)- | 75 | no effect | |
| 55 | OMe | H | Benzyl | -(CH2)- | > 100 | no effect | |
| 56 | OMe | H | Allyl | -(CH2)- | 60 | G1, G2/M phase at 100 μM | |
| 60 | OMe | H | -(CH2)- | > 100 | no effect | ||
| 62 | OMe | H | H | -(CH2)- | > 100 | no effect | |
| 63 | OMe | H | Br | -(CH2)- | 96 | no effect | |
| 64 | OMe | H | Cl | -(CH2)- | > 100 | no effect | |
| 65 | OMe | H | H | isopropyl H | > 100 | no effect | |
| 66 | OMe | H | H | benzyl H | 100 | M phase at 100 μM | |
| 67 | OMe | H | isoprenyl | isopropyl H | 19 | G1, G2/M phase at 100 μM | |
| 68 | OMe | H | isoprenyl | benzyl H | 10 | no effect | |
| 69 | NO2 | H | isoprenyl | -(CH2)- | > 100 | no effect | |
| 70 | NH2 | H | isoprenyl | -(CH2)- | > 100 | no effect | |
| 71 | NCS | H | isoprenyl | -(CH2)- | 50 | G1, G2/M phase at 100 μM | |
| 72 | N3 | H | isoprenyl | -(CH2)- | 60 | G1, G2/M phase at 100 μM | |
Exponentially growing tsFT210 cells were treated with test compounds at 32 °C for 48 h. Cell viability was measured using the color reagent, WST-8™.
Exponentially growing tsFT210 cells were treated with test compounds at 32 °C for 18 h. Then, flow cytometric analysis and nuclei staining were carried out, as described in the Experimental Section.
No effect even at 100 μM.
A 30 μM concentration of 1 arrested cell cycle progression in the M-phase, as previously reported.8,13 Many of these analogues were found to have similar activity as tryprostatin A against tsFT210 cell proliferation. Analogue 38, which closely resembled tryprostatin A, was inactive. Substitution of the 2-isoprenyl moiety in 38 with a smaller methyl substituent (37) also resulted in an inactive analogue. Replacement of the Na-isoprenyl group in analogue 38 with an allyl group (40) resulted in an analogue that was equipotent to 1 in the inhibition of cell proliferation. Similarly, replacement of the Na-isoprenyl group in 38 with a Na-benzyl group 39 also resulted in an analogue that was equipotent to 1 in inhibiting cell proliferation. However, analogues 39 and 40 inhibited cell cycle progression at the G1 phase. The biological data of analogues 39 and 40 indicated substitution of the indole N(H) with a benzyl moiety or allyl moiety was highly conducive for inhibition of cell proliferation and caused cell cycle arrest in the G1 phase. Tryprostatin A 1 analogues in which the indole NH was substituted with a benzyl moiety 46 or allyl moiety 47 also afforded active analogues that were equipotent with 1 in inhibition of the growth of tsFT210 cells. However, this inhibition was not cell cycle dependent. Removal of the 2-isoprenyl group in 1 afforded analogue 62 which was inactive. Similarly, removal of the 2-isoprenyl group in 1 and substitution of it with a bromine atom 63 or chlorine atom 64 also resulted in inactive analogues. Comparison of the analogues 62–64 with the activity of the active analogue 1 indicated the lipophilic 2-isoprenyl group in 1 played an important role in the inhibition of cell proliferation. The lipophilic 2-isoprenyl moiety may play an important role in the interaction with the molecular target and/or may increase the lipophilicity of the molecule thereby facilitating passive diffusion into the cells. Substitution of the 2-isoprenyl group of 1 with a 2-benzyl group 55 or 2-methyl acrylate moiety 60 afforded inactive analogues. However, a 2-allyl substituted analogue 56 of 1 was found to be equipotent to 1 in the inhibition of cell proliferation. Analogue 56 also arrested cell cycle progression at the G1, G2/M phase. Substitution of the L-proline residue in the diketopiperazine ring of 1 with an L-valine residue (67) afforded a 3.5-fold more potent inhibitor of the growth of tsFT210 cells than 1. Similarly replacement of the L-proline residue in 1 with an L-phenyl alanine residue (68) resulted in an analogue that was 7-fold more potent than 1 in the inhibition of the growth of tsFT210 cells, but this inhibition was not cell cycle dependent. The biological data of analogues 67 and 68 indicated substitution of the L-proline residue in the diketopiperazine ring of 1 with other L-amino acids was highly conducive for inhibition of cell proliferation. Removal of the 2-isoprenyl group from analogue 67 afforded 65 which slightly inhibited (IC50 >100 μM) cell proliferation. Again, removal of the 2-isoprenyl moiety from analogue 68 afforded analogue 66 which was 10-fold less potent than 68 in the cell proliferation assay again indicating the importance of the 2-isoprenyl moiety in the inhibition of cell proliferation. Analogue 66 also arrested cell cycle progression in the M-phase at 100 μM. Replacement of the 6-methoxy group in 1 with a nitro group (69) or amino group (70) resulted in analogues that were poor (IC50 > 100 μM) inhibitors of the growth of tsFT210 cells. However, substitution of the 6-methoxy group in 1 with an isothiocyanate group 71 or azide group 72 resulted in analogues that were equipotent with 1 in the inhibition of the growth of tsFT210 cells. Both compounds 71 and 72 inhibited the cell cycle progression of tsFT210 cells at the G1, G2/M phase.
Turner and Sullivan et al.57 have recently shown that tryprostatin A 1 is a specific and potent inhibitor of BCRP (breast cancer resistance protein), which further indicates the potential of analogues of tryprostatin A 1 synthesized in the present study in the potential inhibition of BCRP. Some of the analogues are currently being evaluated as inhibitors of BCRP and will be a topic of future communication. The isothiocyanate analog 71 and the 6-azido analog 72 may be excellent irreversible inhibitors in studies of BCRP.
Conclusion
In summary, the first structure-activity investigation into the cell cycle inhibitory effects of the tryprostatins A 1 analogues has been carried out. The SAR of tryprostatin A 1 suggests that the search for a potent and selective antitumor agent, in the tryprostatin series, still looks promising. Studies on elucidation of the mechanism of action of the tryprostatins indicate that tryprostatin A 1 is a weak inhibitor of topoisomerase II and tubulin polymerization, whereas tryprostatin B 2 is only a weak inhibitor of topoisomerase II. The absolute configuration of L-Tyr-L-pro in the diketopiperazine ring of the tryprostatins was shown to be essential for inhibition of tubulin polymerization and/or topoisomerase II. The 6-methoxy substituent in 1 was shown to promote inhibition of both topoisomerase II and tublin polymerization in in vitro assays. Biological evaluation indicated that the presence of the 2-isoprenyl moiety on the indole scaffold in 1 was essential for inhibition of cell proliferation. Removal of the 2-isoprenyl group in 1 and substitution of the indole NH with a benzyl group or allyl group also afforded analogues that inhibited cell proliferation. The 6-methoxy substituent in 1 could be replaced with various groups to afford active analogues. Various L-amino acids other than L-proline could be incorporated into the diketopiperazine ring of 1 to afford active analogues. The nature of the substituent present on the indole NH or the C-2 position influenced the mechanisms of action of the analogue and highlights the versatility of the tryprostatin skeleton as a template for drug discovery. Analogue 8 was more potent than etoposide (a clinically used anticancer drug) against the three human cancer cell lines evaluated. However, preliminary biological evaluation against normal cells indicated that it was toxic which may limit its potential use in this regard. More work is required to define this. In the NCI preclinical screening program analogue 5, the diastereomer of tryprostatin A 1, was more active than 1 in the inhibition of growth of tumor cells in most of the tumor cell lines evaluated. Similarly, analogue 4, the enantiomer of tryprostatin B 2 as well as both the diastereomers 6 and 8, were more active than 2 in inhibition of the growth of tumor cells in most of the tumor cell lines evaluated.
Experimental Section
All melting points were determined using a Thomas-Hoover capillary melting point apparatus or an Electrothermal model IA8100 digital melting point apparatus and are uncorrected. Reagents and starting materials were obtained from commercial suppliers and used without further purification unless otherwise indicated. Unless specified otherwise, solvents were freshly distilled prior to use: tetrahydrofuran (THF), benzene, toluene, dioxane, and diethyl ether were distilled under nitrogen from sodium metal utilizing benzophenone as an indicator; MeOH and EtOH were distillated over Mg metal and I2; dichloromethane was dried over MgSO4 and then distilled over P2O5; riethylamine was dried over KOH and then distilled over KOH. Flash column chromatography was carried out on silica gel purchased from E. M. Laboratories (grade 60). HPLC grade solvents were used for all chromatography. Analytical thin-layer chromatography (TLC) was conducted on precoated plates: silica gel 60 F-254, 0.25 mm thickness, manufactured by E. Merck & Co., Germany. Indoles were visualized with Dragendorf's reagent or a saturated solution of ceric ammonium sulfate in 50% H2SO4. Ketones or aldehydes were visualized with an aq solution of 2,4-dinitrophenylhydrazine in 30% H2SO4. The 1H NMR spectra were recorded on a Bruker 300-MHz or 500-MHz multiple-probe instrument. Infrared spectra were recorded on a Nicolet Dx FTIR DX V5.07 spectrometer or a Perkin Elmer 1600 Series FT-IR spectrometer. Low resolution mass spectral data (EI/CI) were obtained on a Hewlett-Packard 5985B gas chromatography-mass spectrometer. High resolution mass spectral data were taken on a VG autospectrometer (Double Focusing High Resolution GC/Mass Spectrometer, UK). Optical rotations were measured on a JASCO DIP-370 polarimeter. Microanalyses were performed on a CE Elantech EA1110 elemental analyzer.
(3S,6R)-3-[1-t-Butyloxycarbonyl-6-methoxy)-3-indoyl]methyl-3,6-dihydro-6-isoprophyl-2,5-diethoxypyrazine (12)
To a solution of 10 (3.41 g, 16.1 mmol) in dry THF (60 mL) under nitrogen, n-BuLi (2.5 M, 7.08 mL, 17.7 mmol) was added dropwise at −78 °C. The solution which resulted was stirred at −78 °C for 30 min and treated slowly with a solution of crude 3-bromomethylindole 9a (4.79 g, 14.1 mmol) in THF (30 mL). The mixture which resulted was stirred at −78 °C for 20 h, and then allowed to slowly warm to rt. The solution was concentrated under reduced pressure and diluted with a saturated aq solution of NaHCO3. The aq layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine (30 mL) and dried (K2CO3). After removal of solvent under reduced pressure, the residue was purified by flash chromatography (silica gel, hexane/EtOAc, 10:1) to afford 12 as an oil (6.04 g, 91%): [α]27D = +24.7° (c = 0.9, CHCl3); IR νmax (NaCl) 2970, 1730, 1690 cm−1; 1H NMR (250 MHz, CDCl3) δ 0.63 (d, 3H, J = 6.8 Hz), 0.92 (d, 3H, J = 6.9 Hz), 1.21 (t, 3H, J = 7.1 Hz), 1.29 (t, 3H, J = 7.1 Hz), 1.62 (s, 9H), 2.15 (m, 1H), 3.13 (d, 2H, J = 4.8 Hz), 3.53 (t, 1H, J = 3.4 Hz), 3.84 (s, 3H), 3.94–4.16 (m, 4H), 4.25 (dd, 1H, J = 3.8 Hz), 6.80 (dd, 1H, J = 2.2 and 8.6 Hz), 7.21 (s, 1H), 7.42 (d, 1H, J = 8.6 Hz), 7.67 (br, s, 1H). 13C NMR (62.90 MHz, CDCl3) δ 14.43, 16.68, 19.04, 28.23, 29.37, 31.72, 55.58, 56.13, 60.42, 60.51, 60.67, 82.93, 99.03, 111.49, 116.74, 120.10, 122.83, 125.28, 136.10, 149.81, 157.67, 162.29, 163.49. EIMS m/e (relative intensity) 471 (M+, 47), 261 (21), 212 (100), 169 (67), 141 (20), 57 (51). Anal. Calcd for (C26H37N3O5) C, H, N. This material was used directly in a later step.
(3S,6R)-3-(1-t-Butyloxycarbonyl-3-indoyl)methyl-3,6-dihydro-6-isopropyl-2,5-diethoxypyrazine (13)
Indole 13 (6.7 g) was prepared in 92% yield from 9b (5.1 g, 16.4 mmol) and 10 (3.7 g, 17.6 mmol) analogous to the procedure described for the synthesis of 12. 13: 1H NMR (300 MHz, CDCl3) δ 0.66 (d, 3H, J = 6.8, Hz), 0.95 (d, 3H, J = 6.9 Hz), 1.23 (t, 3H, J = 7.1 Hz), 1.32 (t, 3H, J = 7.1 Hz), 1.64 (s, 9H), 2.18 (m,1H), 3.19 (m, 2H), 3.56 (t, 1H, J = 3.4 Hz), 3.96–4.21 (m, 4H), 4.29 (dd, 1H, J = 4.3 Hz), 7.15–7.29 (m, 2H), 7.36 (s, 1H), 7.59 (d, 1H, J = 7.5 Hz), 8.08 (d, 1H, J = 7.6 Hz). 13C NMR (62.90 MHz, CDCl3) δ 14.40, 16.66, 19.01, 28.21, 29.26, 31.60, 31.73, 56.05, 60.56, 60.64, 83.11, 114.89, 116.73, 119.60, 121.97, 123.96, 124.19, 131.42, 135.13, 149.72, 162.25, 163.50. CIMS m/e (relative intensity) 442 (M+, +1). HRMS Calcd for C25H35N3O4 m/z = 441.2628, found m/z = 441.2536. This material was used directly in a later step.
(3R,6S)-3-[(1-t-Butyloxycarbonyl-6-methoxy)-3-indoyl)]methyl-3,6-dihydro-6-isopropyl-2,5-diethoxypyrazine (14)
Indole 14 (5.5 g) was prepared in 92% yield from 9a (4.4 g, 12.7 mmol) and 11 (3.06 g,14.5 mmol), analogous to the procedure described for the synthesis of 12. 14: [α]27D = −25.1° (c = 0.8, CHCl3); IR νmax (NaCl) 2970, 1730, 1690 cm−1; 1H NMR (250 MHz, CDCl3) δ 0.63 (d, 3H, J = 6.8, Hz), 0.92 (d, 3H, J = 6.9 Hz), 1.21 (t, 3H, J = 7.1 Hz), 1.29 (t, 3H, J = 7.1 Hz), 1.62 (s, 9H), 2.15 (m, 1H), 3.13 (d, 2H, J = 4.8Hz), 3.53 (t, 1H, J = 3.4 Hz), 3.83 (s, 3H), 3.94–4.16 (m, 4H), 4.25 (dd, 1H, J = 3.8 Hz), 6.80 (dd, 1H, J = 2.2 and 8.6 Hz), 7.21 (s, 1H), 7.42 (d, 1H, J = 8.6 Hz), 7.67 (br, s, 1H). 13C NMR (62.90 MHz, CDCl3) δ 14.43, 16.68, 19.04, 28.23, 29.37, 31.72, 55.58, 56.13, 60.42, 60.51, 60.67, 82.93, 99.03, 111.49, 116.74, 120.10, 122.83, 125.28, 136.10, 149.81, 157.67, 162.29, 163.49. CIMS m/e (relative intensity) 472 (M+, +1). HRMS Calcd for C26H37N3O5 m/z = 471.2733, found m/z = 471.2739. This material was used directly in a later step.
(3R,6S)-3-(1-t-Butyloxycarbonyl-3-indoyl)methyl-3,6-dihydro-6-isopropyl-2,5-diethoxypyrazine (15)
Indole 15 (6.6 g) was prepared in 91% yield from 9b and 11, analogous to the procedure described for the synthesis of 12. 15: 1H NMR (300 MHz, CDCl3) δ 0.66 (d, 3H, J = 6.8, Hz), 0.95 (d, 3H, J = 6.9 Hz), 1.23 (t, 3H, J = 7.1 Hz), 1.32 (t, 3H, J = 7.1 Hz), 1.64 (s, 9H), 2.18 (m, 1H), 3.19 (m, 2H), 3.56 (t, 1H, J = 3.4 Hz), 3.96–4.21 (m, 4H), 4.29 (dd, 1H, J = 4.3 Hz), 7.15–7.29 (m, 2H), 7.36 (s, 1H), 7.59 (d, 1H, J = 7.5 Hz), 8.08 (d, 1H, J = 7.6 Hz). 13C NMR (62.90 MHz, CDCl3) δ 14.40, 16.66, 19.01, 28.21, 29.26, 31.60, 31.73, 56.05, 60.56, 60.64, 83.11, 114.89, 116.73, 119.60, 121.97, 123.96, 124.19, 131.42, 135.13, 149.72, 162.25, 163.50. CIMS m/e (relative intensity) 442 (M+, +1). HRMS Calcd for C25H35N3O4 m/z = 441.2628, found m/z = 441.2634. This material was used directly in a later step.
(3S,6R)-3-[(1-t-Butyloxycarbonyl-2-isoprenyl-6-methoxy)-3-indoyl]-methyl-3,6-dihydro-6-isopropyl-2,5-diethoxypyrazine (16)
To a solution of pyrazine 12 (1.94 g, 4.11 mmol) in dry THF (30 mL) at −78 °C under nitrogen, a solution of lithium diisopropylamide (LDA, 1.5 M in THF, 4.2 mL, 6.17 mmol) was added dropwise. The mixture which resulted was stirred at −78 °C for 60 min. The dry (HBr free) 4-bromo-2-methylbutene (1.03 g, 6.91 mmol) was then added dropwise at −78 °C. The mixture was stirred at −78 °C for 1 h and allowed to warm to rt overnight. The solvent was removed under reduced pressure. The residue was taken up in CH2Cl2 and washed with a 5% aq solution of NaHCO3. The aq layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried (K2CO3). After removal of the solvent under reduced pressure, the residue was separated by flash chromatography (silica gel, hexane/EtOAc, 15:1) to provide 16 (1.89 g, 85%) as an oil: IR νmax (NaCl) 2970, 1730, 1690 cm−1, 1H NMR (250 MHz, CDCl3) δ 0.61 (d, 3H, J = 6.8 Hz), 0.94 (d, 3H, J = 6.8 Hz), 1.18 (t, 3H, J = 7.1 Hz), 1.31 (t, 3H, J = 7.1 Hz), 1.61 (s, 3H), 1.63 (s, 9H), 1.70 (s, 3H), 2.19 (m, 1H), 2.88 (dd, 1H, J = 7.4 and 14.2 Hz), 3.23 (dd, 1H, J = 3.9 and 14.3 Hz), 3.56 (t, 1H, J = 3.4 Hz), 3.69 (d, 2H, J = 6.0 Hz), 3.83 (s, 3H), 3.94–4.22 (m, 5H), 5.16 (t, 1H, J = 5.8 Hz), 6.78 (dd, 1H, J = 2.3 and 8.5 Hz), 7.37 (d, 1H, J = 8.6 Hz), 7.65 (d, 1H, J = 2.3 Hz). EIMS m/e (relative intensity) 539 (M+, 65), 439 (11), 328 (16), 272 (58), 228 (100), 212 (55), 169 (31), 141 (16), 57 (48); Anal. Calcd for (C31H45N3O5) C, H, N. This material was used directly in a later step.
(3S,6R)-3-[(1-t-Butyloxycarbonyl-2-isoprenyl)-3-indoyl]-methyl-3,6-dihydro-6-isopropyl-2,5-diethoxypyrazine (17)
Indole 17 (3.4 g) was prepared in 82% yield from 13 under the conditions described above for the preparation of 16. 17: 1H NMR (300 MHz, CDCl3) δ 0.59 (d, 3 H, J = 6.8 Hz), 0.91 (d, 3 H, J = 6.9 Hz), 1.15 (t, 3 H, J = 7.1 Hz), 1.28 (t, 3 H, J = 7.1 Hz), 1.60 (s, 9 H), 1.62 (s, 3 H), 1.68 (s, 3 H), 2.09 (m, 1 H), 2.92 (m, 1H), 3.23 (m, 1 H), 3.56 (t, 1 H, J = 3.4 Hz), 3.71 (d, 2 H, J = 6.1 Hz), 3.98–4.20 (m, 5 H), 5.14 (t, 1 H, J = 6.0 Hz), 7.10–7.15 (m, 2 H), 7.49 (dd, 1H, J = 2.1 and 6.9 Hz), 7.99 (dd, 1H, J = 1.3 and 7.2 Hz). 13C NMR (62.90 MHz, CDCl3) δ 14.34, 14.36, 16.53, 18.13, 19.05, 25.60, 26.10, 28.09, 29.33, 31.41, 56.86, 60.36, 60.40, 60.69, 83.29, 114.98, 115.27, 119.04, 121.80, 122.40, 123.15, 130.54, 131.47, 135.94, 137.83, 150.47, 162.78, 163.23. CIMS m/e (relative intensity) 510 (M+ +1). Anal. Calcd for (C30H43N3O4•0.25H2O) C, H, N. This material was used directly in a later step.
(S)-1-t-Butyloxycarbonyl-2-isoprenyl-6-methoxytryptophan ethyl ester (20)
To a solution of 2-prenylpyrazine 16 (1.27 g, 2.36 mmol) in THF (30 mL) at 0 °C was added an aq solution of 2 N HCl (10 mL). The reaction mixture was allowed to warm to rt and stirred for 1.5 h. A cold aq solution of 15% NH4OH was added. The solution was concentrated under vacuum and diluted with CH2Cl2. The aq layer was extracted with CH2Cl2. The combined organic layers were dried (K2CO3) and the solvent was removed under vacuum. The residue was separated by flash chromatography (silica gel, EtOAc) to provide 20 (0.95 g, 94%) as an oil: [α]27D = +15.2° (c = 0.92, CHCl3); IR νmax (NaCl) 2975, 1730, 1615 cm−1; 1H NMR (250 MHz, CDCl3) δ 1.22 (t, 3H, J = 7.1 Hz), 1.46–1.55 (m, 2H), 1.63 (s, 9H), 1.66 (s, 3H), 1.71 (s, 3H), 2.82 (dd, 1H, J = 8.8 and 14.2 Hz), 3.12 (dd, 1H, J = 5.0 and 14.2 Hz), 3.68 (d, 2H, J = 5.1 Hz), 3.70 (m, 1H), 3.83 (s, 3H), 4.13 (qd, 2H, J = 2.1 and 7.1 Hz), 5.16 (t, 1H, J = 5.1 Hz), 6.82 (dd, 1H, J = 2.3 and 8.5 Hz), 7.33 (d, 1H, J = 8.5 Hz), 7.69 (d, 1H, J = 2.3 Hz); 13C NMR (62.90 MHz, CDCl3) δ 14.04, 18.02, 25.48, 26.03, 28.05, 30.35, 55.62, 60.85, 83.47, 100.31, 111.32, 113.98, 118.46, 122.28, 123.66, 131.70, 136.56, 136.95, 150.35, 157.41, 175.02; EIMS m/e (relative intensity) 430 (M+, 3), 272 (36), 228 (100). HRMS Calcd for C24H34N2O5 m/z = 430.2468, found m/z = 430.2481. This material was used directly in a later step.
(S)-1-t-Butyloxycarbonyl-2-isoprenyltryptophan ethyl ester (21)
Ester 21 (1.1 g) was prepared in 93% yield from 17 as described above for the preparation of 20. 21: [α]26D = +19.7° (c = 0.8, CH3OH); 1H NMR (300 MHz, CDCl3) δ 1.23 (t, 3H, J = 7.1 Hz), 1.66 (s, 9H), 1.68 (s, 3H), 1.74 (s, 3H), 1.80 (brs, 2H), 2.89 (dd, 1H, J = 8.9 and 14.2 Hz), 3.19 (dd, 1H, J = 5.0 Hz and 14.3 Hz), 3.73–3.77 (m, 3H), 4.13–4.19 (m, 2H), 5.19 (t, 1H, J = 1.4 Hz), 7.20–7.25 (m, 2H), 7.49 (dd, 1H, J = 1.0 and 7.0 Hz), 8.08 (dd, 1H, J = 1.5 Hz and 8.8 Hz); 13C NMR (62.90 MHz, CDCl3) δ 14.11, 18.15, 25.60, 25.99, 28.09, 30.27, 54.94, 61.02, 83.72, 114.04, 115.38, 118.09, 121.91, 122.43, 123.66, 129.68, 132.11, 136.00, 138.03, 150.33, 175.09. CIMS m/e (relative intensity) 401 (M+ +1). This material was used directly in a later step.
(R)-1-t-Butyloxycarbonyl-2-isoprenyl-6-methoxytryptophan ethyl ester (22)
Ester 22 (0.86 g) was prepared in 73% yield from 14 as described above for the preparation of 20. 22: [α]27D = −15.9° (c = 0.9, CHCl3); All spectroscopic data were identical to that for 20 (the enantiomer of 22) reported in the previous experiment except the optical rotation was opposite in sign. This material was used directly in a later step.
(R)-1-t-Butyloxycarbonyl-2-isoprenyltryptophan ethyl ester (23)
Ester 23 (0.77 g) was prepared in 70% yield from 15 as described above for the preparation of 20. 23: [α]27D = −19.9° (c = 0.9, CH3OH); All spectroscopic data were identical to that for 21 (the enantiomer of 23) reported in the previous experiment except the optical rotation was opposite in sign. This material was used directly in a later step.
Tryprostation A (1)
Fmoc-L-proline (126 mg, 0.374 mmol) was dissolved in thionyl chloride (1 mL). The solution which resulted was stirred overnight at rt. Excess thionyl chloride was removed under reduced pressure. The Fmoc-L-proline chloride 24 which resulted was dissolved in dry CHCl3 (1 mL). This solution was added dropwise at 0 °C to a solution of 20 (107 mg, 0.249 mmol) and triethylamine (63.0 mg, 0.623 mmol) in dry CHCl3 (6 mL). The mixture which resulted was stirred at 0 °C for 0.5 h and then at rt overnight. After removal of solvent under reduced pressure, a solution of diethylamine (DEA, 2.5 mL) in acetonitrile (2.5 mL) was added in the same flask. The reaction mixture was stirred at rt for 2 h [monitored by TLC (silica gel) until the disappearance of starting material]. Acetonitrile and excess DEA were removed under reduced pressure. Xylene (25 mL) was added into the same reaction vessel and the solution degassed. The reaction mixture was stirred at reflux for 2 d at which time examination by TLC (silica gel) indicated the disappearance of starting material. After removal of xylene under reduced pressure, the residue was subjected to flash chromatography (silica gel, CHCl3/CH3OH, 95:5) to provide tryprostatin A 1 (78 mg, 82%) as a solid: [α]27D = −65.9° (c = 0.97, CHCl3) [lit.7 [α]27D = −69.7° (c = 0.70, CHCl3)]; 1H NMR (250 MHz, CDCl3) δ 1.73 (s, 3H), 1.76 (s, 3H), 1.85–2.07 (m, 3H), 2.27–2.34 (m, 1H), 2.89 (dd, 1H, J = 11.4 and 15.0 Hz), 3.41 (d, 2H, J = 7.2 Hz), 3.53–3.72 (m, 3H), 3.81 (s, 3H), 4.05 (dd, 1H, J = 6.9 and 7.7 Hz), 4.32 (dd, 1H, J = 2.7 and 11.1 Hz), 5.28 (dd, 1H, J = 5.8 and 8.6 Hz), 5.61 (s, 1H), 6.74 (dd, 1H, J = 2.2 and 8.6 Hz), 6.81 (d, 1H, J = 2.1 Hz), 7.32 (d, 1H, J = 8.6 Hz), 7.80 (brs, 1H); 13C NMR (62.90 MHz, CDCl3) δ 17.92, 22.63, 25.07, 25.69, 25.71, 28.32, 45.38, 54.56, 55.73, 59.23, 94.87, 104.38, 109.27, 118.33, 119.98, 122.27, 135.14, 135.27, 136.25, 156.31, 165.80, 169.37. EIMS m/e (relative intensity) 381 (M+,4), 228 (100), 212 (14), 198 (9). HRMS Calcd for C22H27N3O3 m/z = 381.2052, found m/z = 381.2044. Anal. Calcd for C22H27N3O3•1/3H2O (C, H, N). The spectral data for 1 were identical to that reported by osada et al.7 in the literature.
Tryprostatin B (2)
Indole 2 (300 mg) was prepared in 81% yield under conditions described above for the preparation of tryprostatin A 1. 2: [α]26D = −70.9° (c = 0.80, CHCl3) {lit.7 [α]27D = −71.1 (c = 0.63, CHCl3)}; IR νmax (NaCl) 3303, 2971, 1678, 1661 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.76 (s, 3H), 1.79 (s, 3H), 1.75–2.03 (m, 3H), 2.32 (m, 1H), 2.96 (dd, 1H, J = 11.4 and 15.9 Hz), 3.46–3.49 (m, 2H), 3.59–3.72 (m, 3H), 4.06 (dd, 1H, J = 7.5, 8.0 Hz), 4.36 (dd, 1H, J = 3.5, 11.0 Hz), 5.31 (dd, 1H, J = 6.5, 7.0 Hz), 5.62 (brs, 1H), 7.09–7.18 (m, 2H), 7.31 (d, 1H, J = 7.7 Hz), 7.48 (d, 1H, J = 7.7 Hz), 8.00 (brs, 1H), 13C NMR (62.90 MHz, CDCl3) δ 18.37, 23.03, 25.50, 25.98, 26.13, 28.74, 45.80, 54.95, 59.66, 105.03, 111.17, 118.13, 120.07, 120.30, 122.26, 128.37, 135.82, 135.91, 136.80, 166.183, 169.74. CIMS m/e (relative intensity) 352 (M++1,100), 198 (28). Anal. Calcd for (C21H25N3O2•1/4H2O) C, H, N. The spectral data for 2 were identical to that reported by osada et al.7 in the literature.
Enantiomer of tryprostatin A (3)
Enantiomer 3 (75 mg) was prepared in 78% yield from 22 as described above for the preparation of tryprostatin A 1. The starting material used here was D-tryptophan derivative 22 and Fmoc-D-Pro-Cl 25. 3: [α]28D = 70.3° (c = 1.0, CHCl3). Anal. Calcd for (C22H27N3O3•3/5H2O) C, H, N. All spectroscopic data were identical to that reported for 1 (the enantiomer of 3) in a previous experiment except the optical rotation was opposite in sign.
Enantiomer of tryprostatin B (4)
Enantiomer 4 (80 mg) was prepared in 83% yield from 23 as described above for the preparation of tryprostatin A 1. The starting material used here was D-tryptophan derivative 23 and Fmoc-D-Pro-Cl 25. 4: [α]26D = 71.9° (c = 1.1, CHCl3). Anal. Calcd for (C21H25N3O2•3/4H2O) C, H, N. All spectroscopic data were identical to that for 2 (the enantiomer of 4) reported in the previous experiment except the optical rotation was opposite in sign. All spectroscopic data were identical to that reported for 2 (the enantiomer of 4) in a previous experiment except the optical rotation was opposite in sign.
Diastereomer-1 of tryprostatin A (5)
Indole 5 (105 mg) was prepared in 84% yield from 22 as described above for the preparation of tryprostatin A 1. The starting material used here was D-tryptophan derivative 22 and Fmoc-L-Pro-Cl 24. 5: [α]26D = −20.0° (c = 0.12, CHCl3), IR νmax (NaCl) 3269, 2971, 1673, 1650 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.18–1.43 (m, 2H), 1.60–1.71 (m, 1H), 1.74 (s, 3H), 1.78 (s, 3H), 1.99–2.08 (m, 1H), 2.67 (dd, 1H, J = 6.3, 10.7 Hz), 3.08 (dd, 1H, J = 4.5, 14.7 Hz), 3.16 (dd, 1H, J = 2.5, 9.5 Hz), 3.35 (d, 1H, J = 5.0 Hz), 3.39 (d, 2H, J = 7.7 Hz), 3.47–3.57 (m, 1H), 3.81 (s, 3H), 4.23 (dd, 1H, J = 4.3, 8.5 Hz), 5.26 (tt, 1H, J = 1.3, 6.0 Hz), 6.26 (d, 1H, J = 3.6 Hz), 6.74 (dd, 1H, J = 2.3, 8.6 Hz), 6.78 (d, 1H, J = 2.1 Hz), 7.37 (d, 1H, J = 8.6 Hz), 7.92 (brs, 1H), 13C NMR (62.90 MHz, CDCl3) δ 17.88, 21.51, 24.85, 25.74, 28.96, 29.33, 45.03, 55.62, 57.70, 58.62, 94.31, 104.20, 109.08, 118.95, 119.80, 122.70, 135.20, 135.34, 135.73, 155.97, 165.65, 169.23. EIMS m/e (relative intensity) 381 (M+, 15), 228 (100). Anal. Calcd for (C22H27N3O3•3/4H2O) C, H, N.
Diastereomer-1 of tryprostatin B (6)
Indole 6 (77 mg) was prepared in 86% yield from 23 as described above for the preparation of tryprostatin A 1. The starting material used here was D-tryptophan derivative 23 and Fmoc-L-Pro-Cl 24. 6: [α]26D = −41.9° (c = 0.45, CHCl3). Anal. Calcd for (C21H25N3O2•1/5H2O) C, H, N. All spectroscopic data were identical to that reported for 8 except the optical rotation was opposite in sign.
Diastereomer-2 of tryprostatin A (7)
Indole 7 (48 mg) was prepared in 84% yield from 20 as described above for the preparation of tryprostatin A 1. The starting material used here was L-tryptophan derivative 20 and Fmoc-D-Pro-Cl 25. 7: [α]27D = 21.0° (c = 0.32, CHCl3). Anal. Calcd for (C22H27N3O3•3/8H2O) C, H, N. All spectroscopic data were identical to that reported for 5 in a previous experiment except the optical rotation was opposite in sign.
Diastereomer-2 of tryprostatin B (8)
Diastereomer 8 (104 mg) was prepared in 79% yield from 21 as described above for the preparation of tryprostatin A 1. The starting material used here was L-tryptophan derivative 21 and Fmoc-D-Pro-Cl 25. 8: [α]26D = 42.8° (c = 0.65, CHCl3). IR νmax (NaCl) 3266, 2977, 1666 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.30–1.44 (m, 1H), 1.58–1.71 (m, 2H), 1.75 (s, 3H), 1.79 (s, 3H), 2.00–2.09 (m, 1H), 2.69 (dd, 1H, J = 6.3, 16.8 Hz), 3.08–3.19 (m, 2H), 3.37–3.43 (m, 1H), 3.44 (d, 2H, J = 6.3 Hz), 3.48–3.58 (m, 1H), 4.24 (dd, 1H, J = 4.4, 8.6 Hz), 5.30 (tt, 1H, J = 1.4, 7.3 Hz), 6.09 (d, 1H, J = 3.4 Hz), 7.07–7.13 (m, 2H), 7.07–7.13 (m, 2H), 7.23–7.28 (m, 1H), 7.51 (dd, 1H, J = 2.1, 6.8 Hz), 8.00 (brs, 1H). 13C NMR (62.90 MHz, CDCl3) δ 17.94, 21.53, 24.91, 25.76, 28.99, 29.27, 45.08, 57.74, 58.66, 104.46, 110.37, 118.27, 119.52, 119.67, 121.56, 128.33, 134.97, 135.55, 136.66, 165.59, 169.09. EIMS m/e (relative intensity) 351 (M+, 13), 198 (100). Anal. Calcd for (C21H25N3O2•1/8H2O) C, H, N.
(5R,2S)-3,6-Diethoxy-5-[6-methoxy-2-(triethylsilyl)-3-indolyl]methyl-2,5-dihydropyrazine (28)
To a three-neck flask (3 L) equipped with an overhead stir were added iodoaniline derivative 26 (150 g), Schöllkopf derivative 27 (265 g), LiCl (2.55 g), Na2CO3 (159 g), palladium (II) acetate (1.75 g) and anhydrous DMF (2 L). The mixture was then degassed with a vacuum pump three times at rt with Ar. The suspension which resulted was heated for 36 h at 100 °C under an atmosphere of Ar. At this point TLC (silica gel) indicated 26 had been consumed and the reaction mixture was cooled to rt and the DMF was removed under vacuum (aspirator). Methylene chloride (2 L) was added to the residue and the suspension which resulted was filtered to remove unwanted salts. After removal of the CH2Cl2, the crude product was purified by flash chromatography (silica gel, 2% EtOAc in hexane) to give 77% of the desired 6-methoxy substituted indole 28. IR νmax (NaCl) 3388, 2944, 1683 cm−1; 1H NMR (300 MHz, CDCl3) δ 0.67 (d, 3H, J = 6.8 Hz), 0.85–1.05 (m, 18H), 1.20 (t, 3H, J = 7.1 Hz), 1.30 (t, 3H, J = 7.1 Hz), 2.25 (m, 1H), 2.80 (dd, 1H, J = 13.5 Hz and J = 10.6 Hz), 3.46 (dd, 1H, J = 14.1 Hz and J = 3.1 Hz), 3.84 (s, 3H), 3.88 (t, 1H, J = 3.9), 4.01–4.21 (m, 5H), 6.70 (dd, 1H, J = 8.7 Hz and J = 2.2 Hz), 6.82 (d, 1H, J = 2.1 Hz), 7.60 (d, 1H, J = 8.7 Hz), 7.77 (s, br, 1H); 13C NMR (75.5 MHz, CDCl3) δ 4.1, 7.9, 14.7, 14.8, 17.1, 19.5, 32.1, 32.5, 56.0, 59.3, 60.9, 61.0, 61.1, 93.9, 109.3, 121.8, 124.4, 124.7, 130.5, 139.5, 157.0, 163.1, 164.2. MS (CI, CH4) m/e (relative intensity) 486 (M+ + 1, 100), 456 (13), 372 (51), 274 (27). HRMS Calcd for C27H43N3O3Si m/z = 485.3074, found m/z = 485.3055. This material was used directly in a later step.
Na-Methyl-(2S,5R)-3,6-diethoxy-5-[6-methoxy-2-(triethylsilyl)-3-indolyl]methyl-2,5-dihydropyrazine (29)
Sodium hydride (60% in mineral oil, 0.2 g) in several portions was added to a mixture of 28 (1.5 g, 3.08 mmol), CH3I (0.65 g, 4.55 mmol) and anhydrous DMF (20 mL) at 0 °C. After this mixture was stirred for 2 h, analysis by TLC (silica gel) indicated the absence of starting material. The reaction solution was quenched with water (1 mL) and then was neutralized with an aq solution of NH4Cl after which it was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine (2 × 30 mL) and dried (K2CO3). The solvent was removed under reduced pressure and the residue was subjected to a short wash column (silica gel, EtOAc/hexane, 1:4) to provide the pyrazine 29 (1.6 g, 99%). mp: 91–92 °C; IR νmax (NaCl): 2945, 1688, 1613 cm−1; 1H NMR (300 MHz, CDCl3) δ 0.63 (d, 3H, J = 6.8 Hz), 0.95 (m, 15H), 0.98 (d, 3H, J = 6.9 Hz), 1.14 (t, 3H, J = 7.1 Hz), 1.23 (t, 3H, J = 7.1 Hz), 2.23 (m, 1H), 2.80 (dd, 1H, J = 14.04 Hz and 4.53 Hz), 3.45 (dd, 1H, J = 14.02 and 3.51 Hz), 3.73 (s, 3H), 3.84 (s, 3H), 3.85 (m, 1H), 3.90–4.15 (m, 5H), 6.65 (m, 2H), 7.50 (d, 1H, J= 9.2 Hz); 13C NMR (75.5 MHz, CDCl3) δ 4.8, 7.6, 14.3, 14.4, 16.7, 19.1, 31.6, 31.9, 33.1, 55.7, 59.2, 60.4, 60.5, 60.7, 91.9, 108.2, 121.2, 124.2, 124.6, 132.3, 140.6, 156.7, 162.7, 163.9. MS (CI, CH4) m/e (relative intensity) 500 (M+ + 1, 100), 470 (16), 386 (14), 288(21). Anal. Calcd for (C28H45N3O3Si) C, H, N. This material was used directly in a later step.
Na-Isoprenyl-(2S,3R)-3,6-diethoxy-5-[6-methoxy)-3-indolyl]methyl-2,5-dihydropyrazine(30)
Indole 30 (2.1 g) was prepared in 75% yield from 28 and isoprenyl bromide under conditions described above for the preparation of 29. 30: 1H NMR (300 MHz, CDCl3) δ 0.64 (d, 3H, J = 6.78 Hz), 0.92 (d, 3H, J = 6.87 Hz), 1.32 (m, 7H), 1.81 (d, 6H, J = 12.21 Hz), 2.06 (s, 2H), 3.24 (dd, 3H, J = 3.24 Hz and 2,34 Hz), 3.87 (s, 3H), 4.14 (m, 3H), 4.30 (s, 1H), 4.55 (d, 2H, J = 6.93 Hz), 5.34 (s, 1H), 6.74 (m, 2H), 7.50 (d, 1H, J = 2.64 Hz). This material was used directly in a later step.
Na-Benzyl-(2S,5R)-3,6-diethoxy-5-[6-methoxy-2-(triethyl-silyl)-3-indolyl]methyl-2,5-dihydropyrazine (31)
Indole 31 (1.65 g) was prepared from 28 and benzyl bromide in 72% yield under conditions described above for the preparation of 29. 31: 1H NMR (300 MHz, CDCl3) δ 0.88–0.96 (m, 13H), 1.09 (d, 3H, J = 6.87 Hz), 1.21–1.36 (m, 9H), 2.09 (s, 1H), 2.30–2.40 (m, 1H), 3.02 (dd, 1H, J = 14.22 Hz and 9.21 Hz)), 3.59 (dd, 1H, J = 16.61 Hz and 5.46 Hz), 3.88 (s, 3H), 3.93 (t, 1H, J = 3.33 Hz), 4.05–4.34 (m, 5H), 5.48(br, 2H), 6.51 (d, 1H, J = 2.1 Hz), 6.74 (dd, 1H, J = 8.67 Hz and 2.19 Hz), 6.95 (d, 2H, J = 6.96 Hz), 7.20–7.30 (m, 2H), 7.63 (d, 1H, J = 8.67 Hz); 13C NMR (75.5 MHz, CDCl3) δ 4.57, 7.52, 14.17, 14.30, 16.62, 19.13, 20.94, 31.34, 31.40, 31.53, 34.62, 49.57, 55.46, 58.71, 60.38, 60.67, 92.98, 108.38, 121.01, 124.67, 125.01, 125.66, 126.80, 127.55, 127.70, 128.40, 128.94, 132.48, 138.49, 140.41, 156.67, 162.76, 163.95. This material was used directly in a later step.
Na-Allyl-(2S,5R)-3,6-diethoxy-5-[6-methoxy)-3-indolyl]methyl-2,5-dihydropyrazine(32)
Indole 32(1.9 g) was prepared from 28 and allyl bromide in 80% yield under conditions described above for the preparation of 29. 32: 1H NMR (300 MHz, CDCl3) δ 0.64 (d, 4H, J = 6.78 Hz), 0.92 (d, 3H, J = 6.87 Hz), 1.33 (m, 6H), 2.16 (t, 1H, J = 3.60 Hz), 3.32 (m, 3H), 3.86 (s, 3H), 4.16 (m, 3H), 4.60 (m, 2H), 5.02 (s, 1H), 5.18 (d, 1H, J = 1.35), 5.31 (s, 1H), 5.96 (m, 1H), 6.72 (m, 3H), 7.50 (d, 1H, J = 8.58). EIMS m/e (relative intensity) 411(M+, 46). This material was used directly in a later step.
Na-Methyl-6-methoxy-L-tryptophan ethyl ester (33)
Ester 33 (700 mg) was prepared in 84% yield from 29 (1.5 g, 3 mmol) as described above for the preparation of 20. 33: IR νmax (NaCl) 3374, 3311, 2980, 1736, 1623cm−1; 1H NMR (300 MHz, CDCl3) δ 1.30 (t, 3H, J = 7.1 Hz), 1.62 (s, br, 2H), 2.99 (dd, 1H, J = 14.4 Hz and 7.7 Hz), 3.24 (dd, 1H, J = 14.3 Hz and 4.7 Hz), 3.65 (s, 3H), 3.79 (dt, 1H, J = 12.6 Hz and 7.4 Hz), 3.89 (s, 3H), 4.18 (q, 2H, J = 7.1 Hz), 6.75–6.83 (m, 3H), 7.48 (d, 1H, J = 8.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 14.1, 30.6, 32.5, 55.2, 55.8, 60.6, 93.1, 108.9, 109.8, 119.6, 122.6, 126.5, 137.8, 156.6, 175.0. EIMS m/e (relative intensity) 276 (M+, 4), 174 (100), 159 (11). Anal. Calcd for (C15H20N2O3) C, H, N. This material was used directly in a later step.
Na-Isoprenyl-6-methoxytryptophan ethyl ester (34)
Ester 34 (850 mg) was prepared from 30 in 85% yield under conditions described above for the preparation of 20. 34: 1H NMR (300 MHz, CDCl3) δ 1.24–1.30 (m, 4H), 1.83 (t, 3H, J = 18.21 Hz), 2.06 (s, 3H), 3.04 (dd, 1H, J = 14.37 Hz and 7.68 Hz), 3.22 (d, 1H, J = 4.8 Hz), 3.87 (s, 3H), 4.13–4.21 (m, 4H), 5.78 (br, 1H), 4.59 (d, 2H, J= 6.54 Hz), 5.37 (s, 1H), 6.82 (m, 3H), 7.48 (d, 1H, J = 8.04 Hz). This material was used directly in a later step.
Na-Benzyl-6-methoxytryptophan ethyl ester (35)
Ester 35 (1.0 g) was prepared from 31 in 85% yield under conditions described above for the preparation of 20. 35: 1H NMR (300 MHz, CDCl3) δ 1.16–1.26 (m, 3H), 1.87 (s, 1H), 2.01 (s, 1H), 3.02 (dd, 1H, J = 14.28 Hz and 7.32 Hz), 3.18–3.25 (m, 1H), 3.87 (s, 3H), 4.06–4.18 (m, 3H), 5.17 (s, 1H), 6.71 (s, 1H), 6.77–6.80 (m, 1H), 6.87 (s, 1H), 7.08 (d, 1H, J = 7.38 Hz), 7.22–7.28 (m, 2H), 7.50 (d, 1H, J = 8.61 Hz); 13C NMR (75.5 MHz, CDCl3) δ 14.1, 30.6, 32.5, 55.2, 55.8, 60.6, 93.1, 108.9, 109.8, 119.6, 122.6, 126.5, 137.8, 156.6; 13C NMR (75.5 MHz, CDCl3) δ 13.27, 20.06, 29.87, 48.89, 54.26, 54.66, 59.41, 59.92, 92.54, 108.13, 109.53, 118.81, 121.82, 124.99, 125.88, 126.64, 127.82, 136.55, 136.63, 155.62, 174.25. Anal. Calcd for (C21H24N2O3•H2O) C, H, N. This material was used directly in a later step.
Na-Allyl-6-methoxytryptophan ethyl ester (36)
Ester 36 (640 mg) was prepared from 32 in 85% yield under conditions described above for the preparation of 20. 36: 1H NMR (300 MHz, CDCl3) δ 1.23–1.29 (m, 4H), 1.72 (s, 2H), 2.06 (s, 1H), 3.04 (d, 1H, J = 7.56 Hz), 3.87 (s, 3H), 4.12–4.18 (m, 2H), 4.63 (d, 1H, J = 1.5 Hz), 5.18 (dd, 2H, J = 17.2 Hz and 8.7 Hz), 5.98–6.00 (m, 1H), 6.75–6.87 (m, 3H), 7.50 (d, 1H, J = 8.43 Hz). 13C NMR (75.5 MHz, CDCl3) δ 14.09, 30.80, 48.55, 55.02, 55.60, 60.45, 93.24, 108.85, 110.06, 117.09, 119.56, 122.53, 125.39, 133.27, 137.10, 156.32, 175.19. This material was used directly in a later step.
3-(6-Methoxy-1-methyl-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (37)
Indole 37 (280 mg) was prepared as described above for the preparation of 1 in 82% yield. The starting material used here was L-tryptophan derivative 33 (360 mg, 1.3 mmol) and Fmoc-L-pro-Cl 24 (690 mg, 2.05 mmol). 37: IR νmax (NaCl) 3430, 1610, 1550, 1390 cm−1; 1H NMR (300 MHz, CDCl3) Z δ 2.02–2.05 (m, 4H), 2.89 (dd, 1H, J = 15.03 Hz and 10.92 Hz), 3.59–3.74 (m, 5H), 3.88 (s, 3H), 4.08 (dd, 1H, J = 14.8 Hz and 7.14 Hz), 4.32 (d, 2H, J= 10.92 Hz), 5.78 (br, 1H), 6.77–6.91 (m, 3H), 7.48 (d, 1H, J=14.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 22.54, 26.66, 28.20, 32.66, 45.29, 54.44, 55.65, 59.13, 93.01, 108.25, 109.36, 119.20, 121.37, 126.71, 138.18, 156.77, 165.49, 169.21. EIMS m/e (relative intensity) 327(M+, 14), 174(100). Anal. Calcd for (C18H21N3O3) C, H, N.
3-(6-Methoxy-1-(3-methyl-but-2-enyl)-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (38)
Indole 38 (190 mg) was prepared from 34 in 80% yield under conditions described above for the preparation of 1. 38: 1H NMR (300 MHz, CDCl3) δ 1.76–1.85 (m, 3H), 2.00–2.03 (s, 3H), 2.05–2.14 (s, 3H), 2.32 (d, 1H, J = 6.84 Hz), 2.87 (dd, 1H, J = 15.09 Hz and 11.04 Hz), 3.58–3.70 (m, 3H), 3.88 (s, 3H), 4.08 (dd, 1H, J = 14.80 Hz and 7.14 Hz), 4.32 (d, 2H, J = 10.92 Hz), 4.61 (d, 1H, J = 6.87 Hz), 5.38 (t, 1H, J = 1.38 Hz), 5.78 (br, 1H), 6.80–6.82 (m, 2H), 6.90 (s, 1H), 7.45 (d, 1H, J = 6.33 Hz); 13C NMR (75.5 MHz, CDCl3) δ 17.99, 22.53, 25.59, 26.82, 28.22, 44.00, 45.29, 53.34, 54.52, 55.63, 59.13, 93.47, 109.22, 119.20, 119.46, 121.66, 125.27, 136.50, 137.47, 156.55, 165.50, 169.19. EIMS m/e (relative intensity) 381 (M+, 16), 160 (100), 228 55). Anal. Calcd for (C22H27N3O3) C, H, N.
Na-Benzyl-3-(6-methoxy-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (39)
Indole 39 (130 mg) was prepared from 35 in 74% yield under conditions described above for the preparation of 1. 39: mp 124–126 °C; 1H NMR (300 MHz, CDCl3) δ 1.70–1.81 (m, 4H), 2.03–2.06 (m, 2H), 2.82 (dd, 1H, J = 10.74 Hz and 6.69 Hz), 3.11–3.18 (m, 2H), 3.40 (dd, 1H, J = 14.55 Hz and 5.67 Hz), 3.51–3.61 (m, 1H), 3.82 (s, 3H), 4.22–4.27 (m, 1H), 5.21 (s, 1H), 6.01 (d, 1H, J = 3.09 Hz), 6.72 (s, 1H), 6.80 (d, 1H, J = 8.70 Hz), 6.91 (s, 1H), 7.13–7.16 (m, 2H), 7.28–7.33 (m, 2H), 7.49 (d, 1H, J = 8.70Hz); 13C NMR (75.5 MHz, CDCl3) δ 12.41, 28.74, 30.68, 44.97, 49.98, 55.54, 57.77, 58.44, 93.29, 108.48, 109.34, 119.82, 122.03, 126.77, 126.89, 127.69, 128.77, 137.24, 156.53, 165.31, 169.14. Anal. Calcd for (C24H25N3O3•0.5H2O) C, H, N.
Na-Allyl-3-(6-methoxy-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (40)
Indole 40 (220 mg) was prepared from 36 in 80% yield under conditions described above for the preparation of 1. 40: mp 84–86 °C; 1H NMR (300 MHz, CDCl3) δ 1.20–1.25 (m, 3H), 2.07 (d, 1H, J = 7.56 Hz), 2.25 (d, 1H, J = 22Hz), 3.24 (d, 2H, J = 5.70 Hz), 3.70 (dd, 1H, J= 13.92 Hz and 7.01 Hz), 3.83 (s, 3H), 4.06–4.13 (m, 3H), 4.60 (m, 3H), 5.18 (d, 1H, J= 3.10 Hz), 5.89–5.94 (m, 1H), 6.70–6.83 (m, 3H), 7.42 (d, 1H, J = 5.61 Hz). Anal. Calcd for (C20H23N3O3) C, H, N.
(2S,5R)-3,6-Diethoxy-2-isopropyl-5-[6-methoxy-3-indolyl]methyl-2,5-dihydropyrazine(41)
A solution of 16 (500 mg, 0.93 mmol) in xylene was stirred at reflux for 3 d at which time examination by TLC (silica gel) indicated the disappearance of starting material. After removal of xylenes under reduced pressure, the residue was subjected to flash chromatography (silica gel, hexane/EtOAc, 10:1) to provide 41 (330 mg, 80%): 1H NMR (300 MHz, CDCl3) δ 0.61 (d, 3H, J = 6.75 Hz), 0.91 (d, 3H, J = 6.87 Hz), 1.22 (t, 3H, J = 7.08 Hz), 1.35 (t, 4H, J = 7.14 Hz), 1.77 (d, 5H, J = 7.92), 2.15 (s, 1H), 3.24 (m, 3H), 3.42 (d, 2H, J = 7.20 Hz), 3.83 (s, 3H), 3.98–4.30 (m, 4H), 5.29 (s, 1H), 6.73 (m, 2H), 7.42 (t, 1H, J = 8.61 Hz), 7.65 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 14.29, 16.32, 17.71, 18.99, 25.00, 25.67, 28.88, 30.82, 55.58, 57.23, 59.92, 60.24, 60.45, 93.97, 106.77, 108.10, 119.53, 120.94, 123.99, 125.28, 127.67, 134.16, 135.57, 155.45, 162.74, 163.41. EIMS m/e (relative intensity) 439 (M+, 13), 212 (54), 169 (100). Anal. Calcd for (C26H37N3O3) C, H, N. This material was used directly in a later step.
Na-Benzyl-(2S,5R)-3,6-diethoxy-2-isopropyl-5-[6-methoxy-3-indolyl]methyl-2,5-dihydropyrazine (42)
Indole 42 (550 mg) was prepared in 91% yield from 41 and benzyl bromide as described above for the preparation of 29. 42: 1H NMR (300 MHz, CDCl3) δ 0.62 (d, 3H, J = 6.69 Hz), 0.93 (d, 3H, J = 6.84 Hz), 1.17–1.22 (m, 7H), 1.62 (t, 7H, J = 9.4 Hz), 2.10 (m, 1H), 3.21–3.39 (m, 4H), 3.77 (s, 3H), 4.10 (m, 4H), 5.04 (s, 1H), 5.22 (s, 1H), 6.60 (s, 1H), 6.70 (d, 1H, J = 8.55 Hz), 6.88 (d, 2H, J = 6.87 Hz), 7.27 (m, 3H), 7.48 (d, 1H, J = 8.55 Hz); 13C NMR (75.5MHz, CDCl3) δ 14.28, 16.33, 17.80, 19.08, 24.02, 25.57, 28.07, 29.14, 30.77, 46.43, 55.59, 57.35, 59.96, 60.17, 60.44, 92.95, 107.46, 107.91, 119.77, 122.23, 123.16, 125.67, 126.82, 128.47, 131.87, 136.55, 137.08, 138.30, 155.61, 162.67, 163.3. This material was used directly in a later step.
Na-Allyl-(2S,5R)-3,6-diethoxy-2-isopropyl-5-[6-methoxy-3-indolyl]methyl-2,5-dihydropyrazine (43)
Indole 43 (135 mg) was prepared in 82% yield from 41 (150 mg, 0.34 mmol) and allyl bromide (50 mg, 0.40 mmol) as described above for the preparation of 29. 43: 1H NMR (300 MHz, CDCl3) δ 0.60 (t, 3H, J = 6.75 Hz), 0.91 (t, 3H, J = 6.84 Hz), 1.31 (m, 7H), 1.69 (t, 7H, J = 19.7 Hz), 2.10 (m, 1H), 3.14–3.27 (m, 3H), 3.42 (d, 1H, J = 6.45 Hz), 3.85 (s, 3H), 4.12 (m, 4H), 4.58 (s, 2H), 4.78 (d, 1H, J = 8.67 Hz), 5.09 (d, 2H, J = 9.12 Hz), 5.85 (m, 1H), 6.70 (m, 2H), 7.45 (d, 1H, J = 8.58 Hz); 13C NMR (75.5 MHz, CDCl3) δ 14.29, 16.32, 17.83, 18.98, 19.56, 23.87, 25.45, 29.17, 30.79, 45.31, 55.65, 57.41, 59.94, 60.18, 60.43, 92.96, 107.16, 107.67, 115.57, 119.70, 122.45, 123.18, 131.67, 133.62, 136.12, 136.71, 155.46, 162.78, 163.26. EIMS m/e (relative intensity) 479 (M+, 13), 268 (100). This material was used directly in a later step.
Na-Benzyl-2-isoprenyl-6-methoxytryptophan ethyl ester (44)
Ester 44 (330 mg) was prepared in 87% yield from 42, as described above for the preparation of 20. 44: IR νmax (KBr) 3054, 2305, 1733, 1422, 1265 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.18–1.32 (m, 3H), 1.60 (d, 8H, J =15.30 Hz), 3.01 (t, 1H, J = 9.30), 3.40 (d, 2H, J = 6.30 Hz), 3.80 (s, 3H), 4.17 (m, 2H), 5.04 (s, 1H), 5.26 (s, 1H), 6.66 (s, 1H), 6.80 (d, 1H, J = 3.96 Hz), 6.92 (d, 2H, J = 7.17 Hz), 7.25 (d, 3H, J = 7.50 Hz), 7.48 (d, 1H, J = 8.61 Hz); 13C NMR (75.5 MHz, CDCl3) δ 14.06, 17.86, 24.01, 25.40, 30.64, 46.54, 55.52, 55.67, 60.78, 93.51, 106.85, 108.49, 118.89, 121.57, 122.43, 125.72, 126.98, 128.53, 132.68, 136.34, 137.40, 137.92, 155.99, 175.33. EIMS m/e (relative intensity) 420 (M+, 13), 318 (100). This material was used directly in a later step.
Na-Allyl-2-isoprenyl-6-methoxytryptophan ethyl ester (45)
Ester 45 (88 mg) was prepared in 86% yield from 43 (130 mg, 0.27 mmol) as described above for the preparation of 20. 45: IR νmax (NaCl) 3365, 1730, 1625cm−1; 1H NMR (300 MHz, CDCl3) δ 1.26 (m, 3H), 1.78 (t, 10H, J = 10.44), 2.94 (dd, 1H, J = 8.46 and J = 5.82), 3.20 (d, 1H, J = 5.04), 3.44 (d, 1H, J = 6.03), 3.86 (s, 3H), 4.16 (m, 2H), 4.61 (t, 2H, J = 2.43), 4.84 (d, 1H, J = 15.93), 5.12 (m, 2H), 5.90 (s, 1H), 6.75 (t, 2H, J = 6.33), 7.44 (d, 1H, J = 8.55); 13C NMR (75.5 MHz, CDCl3) δ 14.05, 17.91, 23.86, 25.49, 30.54, 45.44, 55.47, 55.71, 60.78, 93.50, 106.40, 108.28, 115.91, 118.81, 121.72, 122.40, 132.54, 133.40, 136.18, 136.95, 155.84, 175.30. EIMS m/e (relative intensity ) 370 (M++1, 12), 268 (100). Anal. Calcd for (C22H30N2O3) C, H, N. This material was used directly in a later step.
Na-Benzyl-2-isoprenyl-3-(6-methoxy-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (46)
Indole 46 (125 mg) was prepared in 87% yield from 44 as described above for the preparation of 1. 46: IR νmax (KBr) 3010, 2408, 1735 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.75 (dd, 3H, J = 7.14 Hz and J = 7.41 Hz), 1.55 (s, 2H), 1.64 (d, 4H, J = 11.82 Hz), 2.06 (d, 2H, J = 2.52), 2.28 (d, 2H, J = 2.70 Hz), 2.75 (m, 1H), 3.00 (m, 1H), 3.38 (m, 2H), 3.82 (s, 3H), 4.14 (m, 3H), 5.15 (d, 1H, J = 7.47 Hz), 5.27 (t, 1H, J = 8.88 Hz), 6.60 (s, 1H), 6.86 (m, 2H), 6.91 (d, 2H, J = 6.18 Hz), 7.05 (d, 2H, J = 2.70 Hz), 7.30 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 13.93, 17.83, 21.31, 23.87, 24.32, 25.38, 28.28, 46.44, 55.69, 59.19, 61.22, 64.48, 84.64, 93.94, 108.52, 118.68, 121.60, 126.09, 126.93, 127.21, 127.88, 128.46, 128.68, 129.40, 137.99, 155.72, 169.02. EIMS m/e (relative intensity) 472 (M+, 5), 318 (48) 91 (100). Anal. Calcd for (C29H33N3O3•H2O) C, H, N.
Na-Allyl-2-isoprenyl-3-(6-methoxy-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (47)
Indole 47 (80 mg) was prepared as described above for the preparation of 1. The starting material used here was L-tryptophan derivative 45 (85 mg, 0.23 mmol) and Fmoc-L-pro-Cl 24 (126 mg, 0.37 mmol) in 82% yield. 47: 1H NMR (300 MHz, CDCl3) δ 1.27 (s, 1H), 1.51–1.76 (m, 9H), 2.37 (t, 1H, J = 3.60 Hz), 3.00 (s, 1H), 3.29 (t, 1H, J = 4.20 Hz), 3.40 (m, 1H), 3.60 (d, 1H, J = 5.70 Hz), 3.92(s, 3H), 4.23 (d, 1H, J = 9.60 Hz), 4.70 (d, 1H, J = 10.2 Hz), 5.14 (s, 1H), 5.54 (s, 1H), 6.95 (d, 1H, J = 7.71 Hz), 7.18 (s, 1H), 7.24 (d, 1H, J = 5.70 Hz). EIMS m/e (relative intensity) 421 (M+, 14), 268 (100). +TOF MS HRMS Calcd for (C25H31N3O3 + Li)+ m/z = 428.2525, found m/z = 428.2519.
(5S,2R)-3,6-Diethoxy-2-bromo-5-[1-t-butyloxycarbonyl-6-methoxy-3-indolyl]methyl-2,5-dihydropyrazine (48)
A solution of NBS (183 mg, 1.03 mmol) which had been dissolved in acetonitrile (10 mL) was syringed into a solution of 28 (500 mg, 1.03 mmol) in acetonitrile (40 mL) at 0 °C. The reaction mixture was allowed to stir at 0 °C for 30 min at which time analysis by TLC (silica gel) indicated the absence of starting material. To this solution was then added 4-dimethylaminopyridine (DMAP, 7 mg, 0.057 mmol) and di-t-butyl-dicarbonate (450 mg, 2.06 mmol) at rt. After the reaction solution was stirred for another 1 h, analysis by TLC (silica gel) indicated the disappearance of the intermediate. The solvent was removed under reduced pressure, and the residue was partitioned between CH2Cl2 (100 mL) and H2O (100 mL). The organic layer was separated, and the aq layer was extracted with CH2Cl2 (2 × 80 mL). The combined organic layers were dried (Na2SO4) and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (silica gel, EtOAc/hexanes, 4: 96) to afford 48 (492 mg) as an oil in 87% yield. 48: IR νmax (NaCl) 2970, 1735, 1690 cm−1; 1H NMR (250 MHz, CDCl3) δ 0.62 (d, 3H, J = 6.8 Hz), 0.97 (d, 3H, J = 6.8 Hz), 1.17 (t, 3H, J = 7.1 Hz), 1.28 (t, 3H, J = 7.1 Hz), 1.67 (s, 9H), 2.20 (m, 1H), 2.92 (dd, 1H, J = 8.1 and 13.9 Hz), 3.28 (dd, 1H, J = 4.6 and 13.9 Hz), 3.68 (t, 1H, J = 3.3 Hz), 3.84 (s, 3H), 3.95–4.25 (m, 5H), 6.80 (dd, 1H, J = 2.2 and 8.6 Hz), 7.38 (d, 1H, J = 8.6 Hz), 7.63 (d, 1H, J = 2.1 Hz); 13C NMR (62.90 MHz, CDCl3) δ 14.31, 14.35, 16.59, 19.09, 28.24, 31.04, 31.46, 55.62, 55.78, 60.49, 60.74, 84.46, 99.59, 107.81,111.61, 119.69, 120.59, 123.66, 137.34, 149.35, 157.65, 162.77, 163.37. EIMS m/e (relative intensity) 551 (M+, 17), 549 (M+, 17), 470 (64), 414 (61), 370 (58), 341 (32), 240 (49), 238 (49), 212 (72), 169 (100), 141 (39). Anal. Calcd for (C26H36N3O5Br) C, H, N. This material was used directly in a later step.
(2S,5R)-3,6-Diethoxy-2-5-[1-t-butyloxycarbonyl-2-benzyl-6-methoxy-3-indolyl]methyl-2,5-dihydropyrazine (49)
Indole 49 (800 mg) was prepared in 85% yield from 29 and benzyl bromide, as described below for the preparation of 50. 49: 1H NMR (300 MHz, CDCl3) δ 0.65 (d, 3H, J = 3.75 Hz), 0.98 (d, 3H, J = 6.87 Hz), 1.20 (t, 3H, J = 7.11 Hz), 1.29 (m, 6H), 1.41 (s, 10H), 2.87 (s, 1H), 3.64 (d, 1H, J = 3.36), 3.88 (s, 3H), 4.00 (m, 1H), 4.56 (s, 2H), 6.89 (d, 1H, J = 8.58 Hz), 7.01 (d, 2H, J = 7.08 Hz), 7.20 (m, 3H), 7.52 (d, 1H, J = 8.58 Hz), 7.75 (s, 1H). EIMS m/e (relative intensity) 561 (M+, 14), 461 (11), 212 (100). Anal. Calcd for (C33H43N3O5) C, H, N. This material was used directly in a later step.
(2S,5R)-3,6-Diethoxy-2-allyl-5-[1-t-butyloxycarbonyl-6-methoxy--3-indolyl]methyl-2,5-dihydropyrazine (50)
A solution of n-BuLi (2.5 M in hexane, 0.22 mL, 0.54 mmol) was added dropwise to a solution of 2-bromopyrazine 29 (250 mg, 0.46 mmol) in dry THF (8 mL) at −78°C under nitrogen. The mixture which resulted was stirred at −78 °C for 30 min and then warmed to 0 °C for 10 min. Then allyl bromide (82.8 mg, 0.69 mmol) was added quickly at 0 °C. The mixture was stirred at 0 °C for 1 h and allowed to warm to rt overnight. The solvent was removed under reduced pressure. The residue was taken up in CH2Cl2 and washed with a 5% aq solution of NaHCO3. The aq layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried (Na2SO4). After removal of the solvent under reduced pressure, the residue was separated by flash chromatography (silica gel, hexane/EtOAc, 15/1) to provide 50 (198 mg, 85%) as an oil. 50: IR νmax (KBr) 3054, 2306, 1733, 1422 cm−1; 1H NMR (300 MHz, CDCl3) δ 0.65 (d, 3H, J = 6.78 Hz), 0.97 (d, 3H, J = 6.90 Hz), 1.23 (t, 10H, J = 6.54 Hz), 1.66 (d, 8H, J = 7.17 Hz), 2.20 (m, 1H), 3.31 (d, 1H, J = 3.30 Hz), 3.64 (s, 1H), 3.88 (s, 3H), 4.07 (m, 3H), 4.21 (m, 2H), 4.94 (d, 2H, J = 6.36 Hz), 5.98 (m, 1H), 6.84 (d, 1H, J = 2.34 Hz), 7.70 (d, 1H, J = 2.25 Hz), 7.98 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 14.23, 16.46, 18.97, 28.08, 29.19, 30.59, 31.33, 55.54, 60.32, 83.25, 98.73, 99.77, 110.94, 114.75, 119.42, 136.23, 136.70, 157.07. EIMS m/e (relative intensity) 511 (M+, 10), 244 (50), 200 (100). +TOF MS HRMS Calcd for (C29H41N3O5 + H)+ m/z = 512.3124, found m/z = 512.3126. This material was used directly in a later step.
(5R,2S)-3,6-Diethoxy-5-[6-methoxy-2-phenyl-3-indolyl]methyl-2,5-dihydropyrazine (51)
A solution of n-BuLi (2.5 M in hexane, 0.27 mL, 0.68 mmol) was added dropwise to a solution of 2-bromopyrazine 29 (250 mg, −78 °C under nitrogen. The mixture 0.46 mmol) in dry THF (10 mL) at which resulted was stirred at −78 °C for 30 min and then warmed to 0 °C for 10 min. Then dry anhydrous pure zinc chloride (0.68 mL, 0.69 mmol) was added quickly at 0 °C. The mixture which resulted was stirred at 0 °C for 1 h and iodobenzene was added and this was followed by addition of tri-2-furyl phosphine (21 mg, 0.1 mmol) and Pd(OAc)2 (12 mg, 0.05 mmol). The mixture which resulted was then stirred overnight. The solvent was removed under reduced pressure. The residue was taken up in CH2Cl2 and washed with a 5% aq solution of NaHCO3. The aq layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried (Na2SO4). After removal of the solvent under reduced pressure, the residue was separated by flash chromatography (silica gel, hexane/EtOAc, 15/1) to provide 51 (170 mg, 65%) as an oil. 1H NMR (300 MHz, CDCl3) δ 0.95 (d, 3H, J = 3.75 Hz), 1.22 (m, 3H), 1.33 (m, 6H), 1.41 (s, 10H), 1.65 (m, 3H), 2.81 (s, 1H), 3.34 (m, 3H), 3.90 (s, 3H), 6.88 (m, 2H), 7.40 (m, 3H), 7.56 (d, 2H, J = 8.58 Hz), 7.71 (s, 1H), 7.52 (d, 1H, J = 2.10 Hz), 7.75 (s, 1H). EIMS m/e (relative intensity) 547 (M+, 56), 236 (100). Anal. Calcd for (C32H41N3O5•H2O) C, H, N. This material was used directly in a later step.
(S)-1-t-Butyloxycarbonyl-2-benzyl-6-methoxytryptophan ethyl ester (52)
Ester 52 (185 mg) was prepared in 87% yield from 49 as described above for the preparation of 20. 52: 1H NMR (300 MHz, CDCl3) δ 1.22 (m, 3H), 1.39 (s, 9H), 1.67 (s, 3H), 2.96 (d, 1H, J = 8.28 Hz), 3.18 (d, 1H, J = 5.46 Hz), 3.89 (s, 3H), 4.10 (m, 2H), 4.49 (s, 2H), 6.90 (d, 1H, J = 2.34 Hz), 7.02 (d, 2H, J = 6.93 Hz), 7.23 (m, 3H), 7.44 (d, 1H, J = 8.58 Hz), 7.80 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 14.10, 27.66, 28.09, 30.13, 32.20, 54.86, 55.58, 64.16, 83.61, 100.01, 111.64, 115.22, 119.38, 122.52, 125.81, 127.44, 128.03, 130.13, 136.79, 139.84, 150.13, 157.60, 174.92. This material was used directly in a later step.
2-Benzyl-3-(6-methoxy-1H-indol-3-ylmethyl)-hexahydropyrrolo[1,2-a] pyrazine-1,4-dione (55)
Indole 55 (95 mg) was prepared in 81% yield from 52, as described above for the preparation of 1. 55: 1H NMR (300 MHz, CDCl3) δ 1.26 (t, 2H, J = 5.16 Hz), 1.64 (s, 1H), 2.01 (m, 2H), 2.33 (m, 1H), 3.03 (t, 1H, J = 11.25 Hz), 3.58–3.73 (m, 2H), 3.83 (s, 3H), 4.09 (m, 2H), 4.32 (d, 1H, J = 10.80 Hz), 5.65 (s, 1H), 6.78 (s, 2H), 7.18 (d, 2H, J = 6.90 Hz), 7.28 (t, 2H, J = 5.70 Hz), 7.40 (d, 2H, J = 9.60 Hz), 7.80 (s, 1H); 13C NMR (75.7 MHz, CDCl3) δ 22.53, 25.65, 28.24, 32.20, 45.32, 54.54, 55.64, 59.15, 94.79, 105.76, 109.56, 118.46, 122.03, 126.48, 128.43, 128.70, 128.89, 136.49, 138.03, 156.53, 165.60, 169.24. EIMS m/e (relative intensity) 403 (M+, 25), 335 (8), 250 (100), 218 (6). Anal. Calcd for (C24H25N3O3) C, H, N.
2-Allyl-3-(6-methoxy-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (56)
Indole 56 (88 mg) was prepared in 81% yield from 53, as described above for the preparation of 1. 56: 1H NMR (300 MHz, CDCl3) δ 1.22 (m, 2H), 1.76 (s, 1H), 2.02 (m, 2H), 2.34 (s, 1H), 3.01 (t, 1H, J = 5.73 Hz), 3.48 (t, 1H, J = 6.30 Hz), 3.67 (s, 2H), 4.05–4.11 (m, 3H), 4.32 (d, 1H, J = 9.60 Hz), 5.15 (m, 1H), 5.88 (m, 1H), 6.78 (t, 2H, J = 3.30 Hz), 7.38 (m, 1H), 8.08 (s, 1H). EIMS m/e (relative intensity) 353 (M+, 8), 200 (100). +TOF MS HRMS Calcd for (C20H23N3O3 + Li)+ m/z = 360.1899, found m/z = 360.1896.
(2S,5R)-3,6-Diethoxy-2-isopropyl-5-[1-t-butyloxycarbonyl-2-methyl-acrylate-6-methoxy-3-indolyl]methyl-2,5-dihydropyrazine (58)
Indole 29 (200 mg, 0.36 mmol) and Pd(PPh3)4 (21 mg, 0.018 mmol) were placed in a round-bottom flask (50 mL) and purged with argon. Toluene (10 mL) was added and this was followed by methyl acrylate (0.16 mL) and dicyclohexylmethylamine (0.09 mL, 0.43 mmol). The reaction was heated to 95 °C for 48 h, cooled and then filtered through celite. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography to afford 58 (190 mg, 94%). 58: 1H NMR (300 MHz, CDCl3) δ 0.77 (d, 3H, J = 6.78 Hz), 0.89 (t, 3H, J = 3.48 Hz), 1.07 (d, 3H, J = 6.87 Hz), 1.25 (m, 6H), 1.67 (s, 9H), 2.18 (m, 1H), 2.89 (t, 1H, J = 9.27 Hz), 3.46 (d, 1H, J = 10.74 Hz), 3.81 (s, 3H), 3.90 (s, 3H), 4.14 (m, 3H), 6.50 (d, 1H, J = 11.46 Hz), 6.86 (d, 1H, J = 2.64 Hz), 7.54 (d, 1H, J = 8.67 Hz), 7.73 (s, 1H), 8.02 (d, 1H, J = 13.53 Hz); 13C NMR (75.5 MHz, CDCl3) δ 14.00, 14.15, 14.28, 16.58, 18.97, 22.54, 28.06, 30.30, 31.48, 31.67, 51.34, 55.48, 56.57, 60.53, 60.70, 60.85, 84.17, 99.08, 112.27, 118.04, 119.50, 122.68, 124.06, 131.18, 137.94, 150.24, 158.86, 162.31, 163.29, 167.39. EIMS m/e (relative intensity) 555 (M+, 64), 455 (37), 412 (22), 288 (18), 244 (100), 212 (92). Anal. Calcd for (C30H41N3O7) C, H, N. This material was used directly in a later step.
2-Methyl acrylyl-6-methoxytryptophan ethyl ester (59)
Indole 59 (180 mg) was prepared in 87% yield from 58 as described above for the preparation of 20. 59: 1H NMR (300 MHz, CDCl3) δ 1.23 (m, 3H), 1.58 (s, 2H), 1.67 (m, 10H), 3.04 (d, 1H, J = 8.67 Hz), 3.23 (d, 1H, J = 4.83 Hz), 3.82 (s, 3H), 3.90 (s, 3H), 4.17 ( d, 2H), 6.38 (d, 1H, J = 15.0 Hz), 6.91 (d, 1H, J = 2.16 Hz), 7.48 (d, 1H, J = 8.67 Hz), 7.75 (s, 1H), 8.01 (d, 1H, J = 15.09 Hz); 13C NMR (75.5 MHz, CDCl3) δ 13.96, 22.09, 28.06, 30.68, 51.55, 55.04, 55.52, 61,13, 84.54, 99.34, 112.66, 118.63, 120.19, 120.88, 123.40, 131.37, 136.29, 137.77, 150.06, 158.99, 164.46, 167.17, 174.88. EIMS m/e (relative intensity) 446 (M+, 35), 244 (100). This material was employed directly in the next step.
2-Methyl acrylyl-3-(6-methoxy-1H-indol-3-yl-methyl)-hexahydro-pyrrolo[1,2 a]pyrazine-1,4-dione (60)
Compound 60 (110 mg) was prepared in 87% yield from 59 as described above for the preparation of 1. 60: mp: 135–137 °C; 1H NMR (300 MHz, CDCl3) δ 2.24 (m, 4H), 3.54 (m, 2H), 3.80 (s, 3H), 3.91 (s, 3H), 4.14 (m, 2H), 6.02 (s, 1H), 6.70 (d, 1H, J = 2.82 Hz), 7.08 (d, 1H, J = 2.79 Hz), 7.38 (s, 1H), 7.68 (d, 1H, J = 12.76 Hz), 8.25 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 14.12, 21.24, 24.35, 28.36, 44.27, 51.64, 55.36, 61.41, 64.01, 93.64, 102.72, 111.40, 119.39, 121.00, 127.26, 128.53, 138.36, 138.61, 159.00, 167.32, 175.08. EIMS m/e (relative intensity) 397 (M+, 34), 324 (77), 293 (100). HRMS Calcd for C21H23N3O5 m/z = 397.1638, found m/z = 397.1657. 6-Methoxy-L-tryptophan ethyl ester (61). Ester 61 (700 mg) was prepared in 86% yield from 28 (1.5 g, 31 mmol) as described above for the preparation of 20. 61: IR νmax (NaCl) 3365, 1730, 1625 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.23 (t, 3H, J = 7.1 Hz), 1.60 (s, br, 2H), 3.0 (dd, 1H, J = 14.3 Hz and 7.7 Hz), 3.22 (dd, 1H, J = 14.4 Hz and 4.8 Hz), 3.78 (m, 1H), 3.81 (s, 3H), 4.15 (q, 2H, J = 7.2 Hz), 6.80 (m, 2H), 6.90 (d, 1H, J= 2.0 Hz), 7.45 (d, 1H, J = 8.5 Hz), 8.15 (s, br, 1H); 13C NMR (75.5 MHz, CDCl3) δ 14.0, 30.7, 54.5, 54.9, 60.8, 94.7, 109.2, 111.0, 119.1, 121.8, 121.9, 136.5, 156.1, 175.2. MS (CI) m/e 263 (M++1, 100). Anal. Calcd for (C14H18N2O3) C, H, N. This material was used directly in a later step.
3-(6-Methoxy-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (62)
Indole 62 (280 mg) was prepared as described above for the preparation of 1 in 82% yield. The starting material used here was L-tryptophan derivative 61 (360 mg, 1.37 mmol) and Fmoc-L-pro-Cl 24 (690 mg, 2.05 mmol). mp 133–135 °C; IR νmax (KBr) 3100, 1700, 1400, 1050 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.98–2.06 (m, 3H), 2.29–2.36 (m, 1H), 2.94 (dd, 1H, J = 15.01 and 10.93 Hz), 3.55–3.77 (m, 3H), 3.87 (s, 3H), 4.10–4.15 (m, 1H), 4.36 (d, 1H, J = 11.8 Hz), 5.76 (br, 1H), 6.83 (dd, 1H, J = 8.6 and 2.2 Hz), 6.90 (d, 1H, J = 2.0 Hz), 6.98 (s, 1H), 7.47 (d, 1H, J = 8.6 Hz), 8.03 (br, 1H); 13C NMR (75.5 MHz, CDCl3) δ 21.41, 28.80, 30.54, 44.94, 55.52, 57.76, 58.19, 94.58, 109.01, 109.58, 119.48, 121.40, 123.12, 136.93, 156.42, 165.66, 169.80. EIMS m/e (relative intensity) 313 (M+, 14), 160 (100). Anal. Calcd for (C17H19N3O3) C, H, N. This material was used directly in a later step.
3-(2-Bromo-6-methoxy-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (63)
A solution of NBS (28.5 mg) which had been dissolved in THF (1 mL) was syringed into a solution of 62 (50 mg, 0.16 mmol) in THF (10 mL) at −78 °C. The reaction mixture which resulted was allowed to stir at rt overnight at which time analysis by TLC (silica gel) indicated the absence of starting material. The solvent was removed under reduced pressure. The residue was purified by preparative TLC (silica gel, 5% EtOH/CH2Cl2) to afford 63 as a powder in 80% yield. 1H NMR (300 MHz, CDCl3) δ 1.76–1.89 (m, 2H), 2.11 (dd, 2H, J = 16.2 Hz and 8.8 Hz), 2.97 (dd, 1H, J = 10.44 and 6.6 Hz), 3.12–3.37 (m, 3H), 3.52–3.60 (m, 1H), 3.83 (s, 3H), 4.27 (d, 1H, J = 4.2 Hz), 6.37 (br, 1H), 6.75–6.84 (m, 2H), 7.39 (d, 1H, J = 8.6 Hz), 8.92 (br, 1H); 13C NMR (75.5 MHz, CDCl3) δ 21.52, 28.95, 29.77, 45.18, 55.54, 57.78, 58.06, 94.32, 108.61, 108.70, 110.00, 118.88, 121.30, 136.71, 156.59, 165.33, 168.96. EIMS m/e (relative intensity) 391 (M+, 18), 393 (M+, 18), 154 (100), 240 (78).
3-(2-Chloro-6-methoxy-1H-indol-3-ylmethyl)-hexahydropyrrolo[1,2-a]pyrazine-1,4-dione (64)
Indole 64 (55 mg) was prepared in 85% yield from 62 under conditions described above for the preparation of 63. 64: 1H NMR (300 MHz, CDCl3) δ 1.81–1.90 (m, 2H), 2.15 (dd, 2H, J = 16.2 Hz and 8.8 Hz), 2.97 (dd, 1H, J = 10.44 Hz and 6.6 Hz), 3.17–3.32 (m, 3H), 3.52–3.60 (m, 1H), 3.82 (s, 3H), 4.26 (t, 1H, J = 4.2 Hz), 6.43 (br, 1H), 6.74–6.84 (m, 2H), 7.35 (d, 1H, J= 9.4 Hz), 9.08 (s, br, 1H); 13C NMR (75.5 MHz, CDCl3) δ 18.30, 21.49, 28.96, 45.13, 55.54, 58.00, 58.32, 94.44, 105.26, 109.94, 118.96, 121.12, 121.42, 135.13, 156.57, 165.40, 169.11. EIMS m/e (relative intensity) 347 (M+, 14), 194 (100), 154 (91). +TOF MS HRMS Calcd for (C17H18N3O3 + Na)+ m/z = 370.0934, found m/z = 370.0935.
3-Benzyl-6-(6-methoxy-1H-indol-3-ylmethyl)-hexahydropyrrolo[1,2-a] pyrazine-1,4-dione (66)
Indole 66 (78 mg) was prepared from 61 in 73% yield under conditions described above for the preparation of 1. 66: 1H NMR (300 MHz, CDCl3) δ 1.99–2.05 (m, 2H), 2.60–2.63 (m, 2H), 3.23–3.29 (m, 3H), 3.50 (d, 1H, J = 6.80 Hz), 3.82 (s, 3H), 4.14–4.16 (m, 2H), 4.92 (s, 1H), 6.74–6.83 (m, 3H), 7.22–7.29 (m, 2H), 7.76 (s, 1H), 8.42 (br, 1H). MS (EI) m/e (relative intensity) 363(M+, 100), 287(62.5). Anal. Calcd for (C21H21N3O3•0.5H2O) C, H, N.
(3S,6S)-3-Isopropyl-6-[6-methoxy-2-(3-methylbut-2-enyl)-1H-indol-3-yl)methyl)piperazine-2,5-dione (67)
Indole 67 (63 mg) was prepared from 20 in 85% yield under conditions described above for the preparation of 1. 67: 1H NMR (300 MHz, CDCl3) δ 0.95–0.97 (d, 3H, J = 6.8 Hz), 1.06–1.08 (d, 3H, J = 7.1 Hz), 1.77–1.80 (d, 6H, J = 9.8 Hz), 2.42 (m, 1H), 2.91–2.99 (dd, 1H, J = 11.0 and 14.4 Hz), 3.43–3.46 (d, 2H, J = 7.2 Hz), 3.58–3.64 (dd, 1H, J = 3.3 and 14.5 Hz), 3.85 (s, 3H), 3.92 (s, 1H), 4.25–4.29 (d, 1H, J = 9.4 Hz), 5.29–5.32 (t, 1H, J = 8.2 Hz), 5.79 (s, 1H), 6.00 (s, 1H), 6.77–6.81 (dd, 1H, J = 8.6 and 2.2 Hz), 6.84 (d, 1H, J = 2.1 Hz), 7.38–7.41 (d, 1H, J = 8.6 Hz), 7.83 (br, 1H). MS (EI) m/e (relative intensity) 383(M+). +TOF MS HRMS Calcd for (C22H29N3O3 + H)+ m/z = 384.2287, found m/z = 384.2282.
(3S,6S)-3-Benzyl-6-[6-methoxy-2-(3-methylbut-2-enyl)-1H-indol-3-yl)methyl)piperazine-2,5-dione (68)
Indole 68 (45 mg) was prepared from 20 in 75% yield under conditions described above for the preparation of 1. 68: 1H NMR (300 MHz, CDCl3) δ 1.81 (d, 6H, J = 9.8 Hz), 1.90–1.98 (m, 1H), 3.12–3.26 (m, 2H), 3.41–3.43 (d, 2H, J = 7.5 Hz), 3.85 (s, 3H), 4.05–4.08 (d, 1H, J = 10.1 Hz), 4.25 (m, 2H), 5.32–5.35 (t, 1H, J = 8.4 Hz), 5.53 (s, 1H), 5.75 (s, 1H), 6.78–6.85 (m, 4H), 7.25–7.28 (m, 3H), 7.43–7.46 (d, 1H, J = 9.3 Hz), 7.83 (br, 1H). MS (EI) m/e (relative intensity) 431(M+, 100). +TOF MS HRMS Calcd for (C26H29N3O3 + H)+ m/z = 432.2287, found m/z = 432.2292.
2-Isoprenyl-3-(6-nitro-1H-indol-3-ylmethyl)-hexahydropyrrolo[1,2-a] pyrazine-1,4-dione (69)
Tryprostatin B 2 (20 mg, 0.056 mmol) was dissolved in anhydrous THF (5 mL) and trifluoroacetic acid (2 mL) was added, after which the mixture was cooled to −78 °C. A solution of NaNO2 (20 mg, 0.28 mmol) in TFA (2 mL) was slowly added to the cooled solution via a syringe over a 10 min period. The reaction mixture was stirred for an additional 30 min and then allowed to warm to −20 °C for 30 min, after which CH2Cl2 (10 mL) and cold aq NH4OH (14%) were added until pH 8. The layers were separated and the aq layer was washed with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL) and dried (Na2SO4). After removal of the solvent under reduced pressure, the residue was purified by flash chromatography (silica gel, CH2Cl2/ethanol 20/1) to provide 69 (17.5 mg, 76%) as a yellow powder. 69: IR νmax (KBr) 3249, 2923, 1650, 1540, 1451,1304.5 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.60 (s, 3H), 1.76 (s, 3H), 1.84–2.06 (m, 3H), 2.34 (s, 2H), 3.04 (dd, 1H, J = 10.86 Hz and J = 4.32 Hz), 4.12 (m, 2H), 4.35 (d, 1H, 8.74 Hz), 5.34 (s, 1H), 5.53 (s, 1H), 7.53 (d, 1H, J = 8.82 Hz), 8.03 (d, 1H, J = 6.00 Hz), 8.29 (s, 1H), 8.53 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 14.08, 17.96, 22.47, 24.30, 25.66, 28.28, 45.38, 54.57, 59.15, 95.47, 106.08, 107.60, 115.52, 117.38, 118.34, 128.05, 129.42, 132.82, 143.15, 165.78, 169.19. EIMS m/e (relative intensity) 396 (M+, 22), 355 (10), 381 (23), 243 (100). Anal. Calcd for (C21H24N4O4) C, H, N.
2-Isoprenyl-3-(6-amino-1H-indol-3-ylmethyl)-hexahydropyrrolo[1,2-a] pyrazine-1,4-dione (70)
Iron (III) chloride hexahydrate (25 mg, 0.063 mmol) and active carbon (2 mg, 0.16 mmol) were added to a solution of 69 in MeOH (4 mL). After the reaction mixture was heated to reflux, hydrazine monohydrate (0.1 mL) was added dropwise. The reaction mixture was allowed to stir at reflux until analysis by TLC (silica gel) indicated the disappearance of starting material (3 h). The reaction was then cooled to rt and the catalysts were removed by filtration through a pad of celite. The CH3OH was removed under vacuum and the residue was dissolved in CH2Cl2. The organic layer was then washed with water, brine, and dried (Na2SO4). After the CH2Cl2 was removed under reduced pressure, the residue was purified by a short wash column (silica gel, CH2Cl2/ethanol, 20:1) to provide 70 as an oil (20.6 mg, 88%). 70: 1H NMR (300 MHz, CDCl3) δ 1.77 (m, 4H), 1.99–2.06 (m, 5H), 2.33 (s, 1H), 2.89 (t, 1H, J = 14.04 Hz), 3.41 (m, 3H), 3.59–3.67 (m, 4H), 4.12 (s, 1H), 4.34 (d, 1H, J = 9.03 Hz), 5.29 (dd, 1H, J = 4.80 Hz and J = 7.20 Hz), 5.67 (s, 1H), 6.55 (d, 1H, J = 6.60 Hz), 6.64 (s, 1H), 7.28 (d, 1H, J = 3.60 Hz), 7.69 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 17.85, 22.55, 24.94, 25.63, 27.47, 28.26, 45.29, 54.46, 59.17, 96.49, 104.42, 110.30, 118.34, 120.04, 121.27, 134.02, 134.90, 136.74, 142.04, 165.78, 169.20. EIMS m/e (relative intensity) 366 (M+, 10), 198 (62), 165 (100). Anal. Calcd for (C21H26N4O2•H2O) C, H, N. This material was converted into the hydrochloride salt for storage purposes.
2-Isoprenyl-3-(6-isothiocyanato-1H-indol-3-ylmethyl)-hexahydro-pyrrolo[1,2-a]pyrazine-1,4-dione (71)
6-Aminotryprostatin B 70 (20 mg, 0.055 mmol) was dissolved in CHCl3 (4 mL) and thiophosgene (0.2 mL, 0.003 mmol) was added dropwise at rt. The reaction mixture was stirred for 4 h and then the solution was treated with triethylamine (2 mL). After the solvent was removed under reduced pressure, the residue was purified by a short wash column (silica gel, CH2Cl2/ethanol, 20:1) to provide 71 as an oil (20.0 mg, 91%). 71: IR νmax (KBr) 2961, 2120, 1615 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.59 (s, 3H), 1.75 (s, 3H), 1.82 (s, 3H), 2.34 (m, 1H), 2.97 (dd, 1H, J = 10.86 Hz and J = 11.16 Hz), 3.50 (s, 2H), 3.63 (m, 3H), 4.14 (s, 1H), 4.32 (d, 1H, J = 6.96 Hz), 5.31 (s, 1H), 5.61 (s, 1H), 7.03 (d, 1H, J = 6.72 Hz), 7.28 (s, 1H), 7.42 ( d, 1H, J = 8.52 Hz), 8.07 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 17.95, 22.53, 25.13, 25.49, 25.68, 28.31, 29.62, 45.36, 54.49, 59.19, 105.33, 108.07, 118.41, 118.50, 118.94, 124.68, 127.27, 134.81, 136.20, 138.83, 165.37, 169.26. EIMS m/e (relative intensity) 408 (M+, 18), 255 (100). Anal. Calcd for (C22H24N4O2S•0.3H2O) C, H, N.
2-Isoprenyl-3-(6-azido-1H-indol-3-ylmethyl)-hexahydropyrrolo[1,2-a] pyrazine-1,4-dione (72)
6-Aminotryprostatin B 70 (15 mg, 0.041 mmol) was dissolved in CH2Cl2 (2 mL). Triethylamine (0.1 mL, 0.72 mmol) and a solution of CuSO4 (2.0 mg, 0.014 mmol) in H2O (0.05 mL) were added to the reaction mixture. A freshly prepared solution of TfN3 (21 mg, 0.12 mmol) in CH2Cl2 (1 mL) was then added, and the solution which resulted was brought to homogeneity by adding MeOH (1 mL). The solution which resulted was stirred at rt for 2 h. The reaction solution was then poured into a saturated solution of aq NaHCO3 (5 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL) and dried (Na2SO4). After the solvent was removed under reduced pressure, the residue was purified by a short wash column (silica gel, CH2Cl2/EtOH, 20:1) to provide 72 as an oil (14.5 mg, 90%). 72: IR νmax (KBr) 2960, 2112.2, 1615 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.77 (m, 6H), 2.06 (s, 3H), 2.34 (s, 1H), 3.00 (t, 1H, J = 8.67), 3.47 (s, 2H), 3.60 (m, 3H), 4.12 (t, 1H, J = 7.20), 4.33 (d, 1H, J = 9.63), 5.32 (s, 1H), 5.61 (s, 1H), 6.83 (d, 1H, J = 6.60), 6.99 (s, 1H), 7.38 (d, 1H, J = 12.00), 7.96 (s, 1H). EIMS m/e (relative intensity) 392 (M+, 28). HRMS Calcd for C21H24N6O2 m/z = 392.1961, found m/z = 392.1957.
Topoisomerase II-Mediated DNA Relaxation Assay. 52,53
The DNA relaxation assay tests the ability of a drug to inhibit the Topo II-mediated relaxation of supercoiled DNA. The assay was performed in a total volume of 10 μL and contained 62.5 μg of plasmid pUC18 DNA (from Escherichia coli) and 100 μM drug in an incubation assay buffer (0.05 M Tris-HCl pH 8.0, 0.12 M KCl, 0.01 M MgCl2, 0.5 mM ATP, 0.5 mM DTT, 30 mg/mL BSA). Stock solutions of drug were made up in either DMSO or EtOH and the total percentage of these in the assay mixture was kept to less than 1%. The mixture was allowed to warm to 37 °C and the reaction was initiated by the addition of 2.0 units of Topo II (TopoGEN, Inc.). The reaction was allowed to proceed for 30–45 minutes before being stopped by addition of 2.5 μL decatenation buffer (5% sarkosyl, 25% glycerol, and 0.0025% bromophenol blue). The drugs were then extracted from the incubation with 10 μL of 24: 1 CHCl3: isoamyl alcohol and the samples loaded onto a 1% agarose gel to run for 90 min at 90 volts. The gel was stained with ethidium bromide and destained with H2O. The DNA bands were detected on a UV light box and photographed with Polaroid 525 film. Controls were no enzyme, enzyme, 100 μM m-AMSA, and 1% DMSO.
The gels were analyzed qualitatively by looking for the presence of DNA bands that migrate farther down on the gel than the negative controls. Topo II-mediated relaxation of the DNA prevents the band from migrating down the gel as far as one that is still in a supercoiled form. Therefore, DNA incubated with Topo II inhibitors will migrate farther on the gel than the no-enzyme or DMSO controls.
Microtubule Assembly Assay. 13,54
Tubulin, containing MAPs (microtubule associated proteins), was prepared as described in the literature.59 The tubulin polymerization assay was run at 37 °C by adding to 240 μL of PME buffer (100 mM PIPES, pH 6.9, 2 mM EGTA, 1 mM MgSO4), 8 μL of GTP (50 mM), 32μL of drug in DMSO, and 120 μL of tubulin (added last). The change in absorbance was measured at 351 nm over 10 min. The sample cuvette was zeroed against a reference cuvette containing 360 μL TBE buffer, 8 μL GTP (50 mM), and 32 μL DMSO. The concentration of the drug solution was varied for different runs to obtain a delta absorbance versus concentration curve. Standard curves were prepared on each batch of separately prepared tubulin using colchicine as the standard. Polymerization assays were conducted on the tryprostatins (1–8) and similar derivative (colchicine).
Cytotoxicity Assay
Three human cell lines were purchased from American Type Culture Collection (ATCC) and used in all cytotoxicity assays. The MCF7 breast adenocarcinoma cells, NCI-H520 lung squamous cell carcinoma cells and PC-3 prostate adenocarcinoma cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Cells were subcultured twice a week in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 0.1 mM non-essential amino acids.
Normally-growing cells were plated at 1 × 104 cells/well into 96-well plates and incubated for 24 h at 37 °C. After 24 hours, the cells were drugged for initial screening with 100 μM, 50 μM, and 10 μM drug dissolved in a DMSO or EtOH vehicle (less than 1% in culture medium). Any drug showing < 50% cell survival at 100 μM was further tested using appropriate drug concentrations to determine its growth inhibition-50% (GI50). Drugs were run in quadruplicate or greater and control wells contained an appropriate percentage of DMSO or EtOH, usually 0.2%. Positive controls were either etoposide (ETOP) or amsacrine (m-AMSA).
After incubation with drug for 72 h, the CellTiter 96™ AQueous Non-Radioactive Cell Proliferation Assay (Promega) was used to evaluate cell survival. Cells were treated with a solution of MTS [3-(4,5-dimthylthiozol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium] and an electron coupling reagent, PMS (phenazine methosulfate) diluted in RPMI-1640 medium. MTS (Owen's reagent)60 was bioreduced by viable cells into formazan, and the amount of formazan present can be measured by reading the absorbance at 490 nm.61 The amount of formazan present was proportional to the number of living cells in culture. Vehicle control lanes were assumed to have 100% cell survival and the percentage of cells remaining in the drug-treated wells was calculated as a percentage of these control wells. The absorbance of wells containing only the MTS reagent (the plate blank) was subtracted from all wells.
Cell Culture
A temperature-sensitive cdc2 mutant cell line, tsFT210, which was isolated from the mouse mammary carcinoma cell line FM3A, was a kind gift from Dr. F. Hanaoka (RIKEN).4 The tsFT210 cells were maintained in RPMI 1640 with 10% fetal calf serum at the permissive temperature of 32 °C.
Cell Cycle Analysis
In a synchronous-culture assay, cells were seeded at a density of 1x105 cells/mL in 0.5 mL into a 24-well plate and were preincubated at 32 °C for 1 h. Then, 5 μL of each sample solution was added, and the cells were incubated at 32 °C for 18 h. After incubation, morphological characteristics of the cells were examined by microscopic observation. The cells were subjected to flow cytometric analysis as described below to confirm the DNA contents in cells.
Flow cytometric analysis was performed essentially as described by previous reports.4,58 The harvested cells were stained with solution containing 50 μg/mL propidium iodide, 0.1% sodium citrate, and 0.2% NP-40 and analyzed for DNA contents using a flow cytometer (Coulter Co., Hialeah, FL).
Cell Staining
Carnoy fixation and staining were performed with slight modification. Cells were treated with 0.55% of KCl for 20 min, fixed in Carnoy’s solution and dropped onto a wet glass slide. The chromosomes and intact nuclei were stained with 1 mg/mL of Hoechst 33258, and examined by using fluorescent microscopy (Olympus, Tokyo, Japan).
Proliferation Assay
Exponentially growing tsFT210 cells were treated with test compounds at 32 °C for 48 h. The cell number was evaluated by the subsequent color reaction. The 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H tetrazolium, mono-sodium salt, WST-8™ (Nakalai Tesque, Kyoto, Japan) was added, and the cells were further incubated for 4 h at 37 °C. The absorbance (A450) of each well was measured by a Wallac 1420 multilabel counter (Amersham Biosciences, Piscataway, NJ).
Acknowledgments
We thank the National Institute of Mental Health MH46851 in part, and the Research Growth Initiative of the University of Wisconsin-Milwaukee for support of this work.
Appendix
| Elemental Analyses
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | Formula | Calculated (%) | Found (%) | ||||
| C | H | N | C | H | N | ||
| 1 | C22H27N3O3•1/3H2O | 68.19 | 7.20 | 10.84 | 68.21 | 7.16 | 10.85 |
| 2 | C21H25N3O2•1/4H2O | 70.86 | 7.22 | 11.81 | 70.88 | 7.25 | 11.85 |
| 3 | C22H27N3O3•3/5H2O | 67.36 | 7.25 | 10.71 | 67.33 | 7.26 | 10.75 |
| 4 | C21H25N3O2•3/4H2O | 69.11 | 7.32 | 11.51 | 69.11 | 7.28 | 11.55 |
| 5 | C22H27N3O3•3/4H2O | 66.90 | 7.27 | 10.64 | 66.92 | 7.26 | 10.67 |
| 6 | C21H25N3O2•1/5H2O | 71.04 | 7.21 | 11.84 | 71.06 | 7.24 | 11.79 |
| 7 | C22H27N3O3•3/8H2O | 70.41 | 7.25 | 11.73 | 70.44 | 7.26 | 11.75 |
| 8 | C21H25N3O2•1/8H2O | 71.31 | 7.20 | 11.88 | 71.34 | 7.21 | 11.85 |
| 12 | C26H37N3O5 | 66.22 | 7.91 | 8.91 | 66.23 | 7.90 | 8.55 |
| 16 | C31H45N3O5 | 68.99 | 8.40 | 7.79 | 68.69 | 8.66 | 7.40 |
| 17 | C30H43N3O4•1/4H2O | 70.08 | 8.53 | 8.17 | 70.10 | 8.56 | 8.15 |
| 29 | C28H45N3O3Si | 67.29 | 9.08 | 8.41 | 67.49 | 9.16 | 8.34 |
| 33 | C15H20N2O3 | 65.18 | 7.30 | 10.14 | 64.96 | 7.36 | 10.24 |
| 35 | C21H24N2O3•H2O | 68.11 | 7.03 | 7.58 | 67.71 | 6.68 | 7.81 |
| 37 | C18H21N3O3 | 66.04 | 6.47 | 12.84 | 65.80 | 6.75 | 13.06 |
| 38 | C22H27N3O3 | 69.27 | 7.13 | 11.02 | 69.03 | 7.28 | 11.29 |
| 39 | C24H25N3O3•0.5H2O | 69.90 | 6.31 | 10.19 | 69.54 | 5.89 | 9.87 |
| 40 | C20H23N3O3 | 67.97 | 6.56 | 11.89 | 68.20 | 6.28 | 11.63 |
| 41 | C26H37N3O3 | 71.04 | 8.48 | 9.56 | 71.39 | 8.01 | 9.82 |
| 45 | C22H30N2O3 | 71.32 | 8.16 | 7.56 | 70.90 | 7.81 | 7.89 |
| 46 | C29H33N3O3•H2O | 71.02 | 7.14 | 8.57 | 70.61 | 6.75 | 8.28 |
| 48 | C26H36N3O5Br | 56.73 | 6.59 | 7.63 | 56.97 | 6.47 | 7.48 |
| 49 | C33H43N3O5 | 70.56 | 7.72 | 7.48 | 70.23 | 7.98 | 7.14 |
| 51 | C32H41N3O5•H2O | 67.96 | 7.61 | 7.43 | 67.51 | 7.24 | 7.94 |
| 55 | C24H25N3O3 | 71.44 | 6.25 | 10.41 | 71.09 | 6.56 | 10.05 |
| 58 | C30H41N3O7 | 64.85 | 7.44 | 7.56 | 65.22 | 7.10 | 7.98 |
| 61 | C14H18N2O3 | 64.12 | 6.87 | 10.69 | 63.96 | 6.98 | 10.54 |
| 62 | C17H19N3O3 | 65.16 | 6.11 | 13.41 | 64.93 | 6.36 | 13.68 |
| 66 | C21H21N3O3•0.5H2O | 67.74 | 5.91 | 11.29 | 67.36 | 5.49 | 11.73 |
| 69 | C21H24N4O4 | 63.62 | 6.10 | 14.13 | 63.96 | 5.73 | 14.47 |
| 70 | C21H26N4O2•H2O | 65.62 | 7.29 | 14.58 | 65.20 | 14.26 | 15.02 |
| 71 | C22H24N4O2S•0.3H2O | 63.86 | 5.95 | 13.55 | 63.52 | 5.54 | 13.13 |
|
| |||||||
| High-Resolution Mass Spectra (HRMS) (EI) | |||||||
|
| |||||||
| Compd | Formula | Calcd mass | Found Mass | ||||
|
| |||||||
| 1 | C22H27N3O3 | 381.2052 | 381.2044 | ||||
| 13 | C25H35N3O4 | 441.2628 | 441.2536 | ||||
| 14 | C26H37N3O5 | 471.2733 | 471.2739 | ||||
| 15 | C25H35N3O4 | 441.2628 | 441.2634 | ||||
| 20 | C24H34N2O5 | 430.2468 | 430.2481 | ||||
| 47 | C25H31N3O3 + Li+ | 428.2525 | 428.2519 | ||||
| 50 | C29H41N3O5 + H+ | 512.3124 | 512.3126 | ||||
| 56 | C20H23N3O3 + Li+ | 360.1899 | 360.1896 | ||||
| 60 | C21H23N3O5 | 397.1638 | 397.1657 | ||||
| 64 | C21H23N3O5 + Na+ | 370.0934 | 370.0935 | ||||
| 67 | C22H29N3O3 + H+ | 384.2287 | 384.2282 | ||||
| 68 | C26H29N3O3 + H+ | 432.2287 | 432.2292 | ||||
| 72 | C21H24N6O2 | 392.1961 | 392.1957 | ||||
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Nasmyth K. Science. 1996;274:1643. doi: 10.1126/science.274.5293.1643. [DOI] [PubMed] [Google Scholar]
- 2.Paulovich AG, Toczyski DP, Hartwell LH. Cell. 1997;88:315. doi: 10.1016/s0092-8674(00)81870-x. [DOI] [PubMed] [Google Scholar]
- 3.Pardee AB. PNAS. 1974;71:1286. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osada H, Cui C-B, Onose R, Hanaoka F. Bioorg Med Chem. 1997;5:193. doi: 10.1016/s0968-0896(96)00207-6. [DOI] [PubMed] [Google Scholar]
- 5.Osada H. J Antibiotics. 1998;51:973. doi: 10.7164/antibiotics.51.973. [DOI] [PubMed] [Google Scholar]
- 6.Cui CB, Kakeya H, Okada G, Onose R, Ubukata M, Takahashi I, Isono K, Osada H. J Antibiotics. 1995;48:1382. doi: 10.7164/antibiotics.48.1382. [DOI] [PubMed] [Google Scholar]
- 7.Cui CB, Kakeya H, Osada H. J Antibiotics. 1996;49:534. doi: 10.7164/antibiotics.49.534. [DOI] [PubMed] [Google Scholar]
- 8.Cui CB, Kakeya H, Okada G, Onose R, Osada H. J Antibiotics. 1996;49:527. doi: 10.7164/antibiotics.49.527. [DOI] [PubMed] [Google Scholar]
- 9.Gan T, Cook JM. Tetrahedron Lett. 1997;38:1301. [Google Scholar]
- 10.Ma C, Liu X, Li X, Flippen-Anderson J, Yu S, Cook JMJ. Org Chem. 2001;66:4525. doi: 10.1021/jo001679s. [DOI] [PubMed] [Google Scholar]
- 11.Zhao S, Gan T, Yu P, Cook JM. Tetrahedron Lett. 1998;39:7009. [Google Scholar]
- 12.Zhao S, Smith KS, Deveau AM, Dieckhaus CM, Johnson MA, Macdonald TL, Cook JM. J Med Chem. 2002;45:1559. doi: 10.1021/jm0155953. [DOI] [PubMed] [Google Scholar]
- 13.Usui T, Kondoh M, Cui CB, Mayumi T, Osada H. Biochemical J. 1998;333:543. doi: 10.1042/bj3330543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amos LA. Curr Opin Struct Biol. 2000;10:236. doi: 10.1016/s0959-440x(00)00070-1. [DOI] [PubMed] [Google Scholar]
- 15.Maiato H, Sampaio P, Sunkel CE. Int Rev Cytol. 2005;241:53. doi: 10.1016/S0074-7696(04)41002-X. [DOI] [PubMed] [Google Scholar]
- 16.Hamel E, Campo AAd, Lowe MC, Waxman PG, Lin CM. Biochemistry. 1982;21:503. doi: 10.1021/bi00532a014. [DOI] [PubMed] [Google Scholar]
- 17.Zhou J, Giannakakou P. Anti-Cancer Agents Med Chem. 2005;5:65. doi: 10.2174/1568011053352569. [DOI] [PubMed] [Google Scholar]
- 18.Shi Q, Chen K, Morris-Natschke SL, Lee KH. Curr Pharm Des. 1998;4:219. [PubMed] [Google Scholar]
- 19.Mani S, Macapinlac M, Goel S, Verdier-Pinard D, Fojo T, Rothenberg M, Colevas D. Anti-Cancer Drugs. 2004;15:553. doi: 10.1097/01.cad.0000131681.21637.b2. [DOI] [PubMed] [Google Scholar]
- 20.Beckers T, Mahboobi S. Drugs of the Future. 2003;28:767. [Google Scholar]
- 21.Rowinsky E, Donehower RC. In: Cancer Principles and Practice of Oncology. DeVita VT, Hellman S, Rosenburg SA, editors. Lippincott-Raven; Philadelphia: 2001. pp. 431–452. [Google Scholar]
- 22.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Experientia. 1989;45:209. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 23.Blokhin AV, Yoo HD, Geralds RS, Nagle DG, Gerwick WH, Hamel E. Mol Pharmacol. 1995;48:523. [PubMed] [Google Scholar]
- 24.Damayanthi Y, Lown JW. Curr Med Chem. 1998;5:205. [PubMed] [Google Scholar]
- 25.Flörsheimer A, Altmann KH. Expert Opin Ther Patents. 2001;11:951. [Google Scholar]
- 26.Poncet J. Curr Pharm Design. 1999;5:139. [PubMed] [Google Scholar]
- 27.Simpson D, Wagstaff AJ. Am J Cancer. 2003;2:373. [Google Scholar]
- 28.Mazumdar M, Parrack PK, Mukhopadhyay K, Bhattacharyya B. Biochemistry. 1992;31:6470. doi: 10.1021/bi00143a016. [DOI] [PubMed] [Google Scholar]
- 29.Lage H, Dietel M. Lancet Oncol. 2000;1:169. doi: 10.1016/s1470-2045(00)00032-2. [DOI] [PubMed] [Google Scholar]
- 30.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. PNAS. 1998;95:15665. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abbott BL. Hematol Oncol. 2003;21:115. doi: 10.1002/hon.714. [DOI] [PubMed] [Google Scholar]
- 32.Allen JD, Schinkel AH. Mol Cancer Ther. 2002;1:427. [PubMed] [Google Scholar]
- 33.Doyle LA, Ross DD. Oncogene. 2003;22:7340. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 34.Benderra Z, Faussat AM, Sayada L, Perrot JY, Chaoui D, Marie JP, Legrand O. Clin Cancer Res. 2004;10:7896. doi: 10.1158/1078-0432.CCR-04-0795. [DOI] [PubMed] [Google Scholar]
- 35.Uggla B, Stahl E, Wagsaeter D, Paul C, Karlsson MG, Sirsjoe A, Tidefelt U. Leukemia Res. 2005;29:141. doi: 10.1016/j.leukres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Candeil L, Gourdier I, Peyron D, Vezzio N, Copois V, Bibeau F, Orsetti B, Scheffer GL, Ychou M, Khan QA, Pommier Y, Pau B, Martineau P, Del Rio M. Int J Cancer. 2004;109:848. doi: 10.1002/ijc.20032. [DOI] [PubMed] [Google Scholar]
- 37.Ross DD. Leukemia. 2000;14:467. doi: 10.1038/sj.leu.2401694. [DOI] [PubMed] [Google Scholar]
- 38.Sauerbrey A, Sell W, Steinbach D, Voigt A, Zintl F. Br J Haematol. 2002;118:147. doi: 10.1046/j.1365-2141.2002.03550.x. [DOI] [PubMed] [Google Scholar]
- 39.Ross DD, Yang W, Abruzzo LV, Dalton WS, Schneider E, Lage H, Dietel M, Greenberger L, Cole SP, Doyle LA. J Natl Cancer Inst. 1999;91:429. doi: 10.1093/jnci/91.5.429. [DOI] [PubMed] [Google Scholar]
- 40.Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, Senderowicz AM, Ross DD, Bates SE. Clin Cancer Res. 2001;7:145. [PubMed] [Google Scholar]
- 41.Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, Traxler P. Cancer Res. 2004;64:2333. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- 42.Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Cancer Res. 2000;60:47. [PubMed] [Google Scholar]
- 43.Woehlecke H, Osada H, Herrmann A, Lage H. Int J Cancer. 2003;107:721. doi: 10.1002/ijc.11444. [DOI] [PubMed] [Google Scholar]
- 44.Maliepaard M, van Gastelen MA, Tohgo A, Hausheer FH, van Waardenburg RCAM, de Jong LA, Pluim D, Beijnen JH, Schellens JHM. Clin Cancer Res. 2001;7:935. [PubMed] [Google Scholar]
- 45.Larock RC, Yum EK, Refvik MD. J Org Chem. 1998;63:7652. [Google Scholar]
- 46.Larock RC, Yum EKJ. Am Chem Soc. 1991;113:6689. [Google Scholar]
- 47.Feldman KS, Eastman KJ, Lessene G. Org Lett. 2002;4:3525. doi: 10.1021/ol026694t. [DOI] [PubMed] [Google Scholar]
- 48.Dieck HA, Heck FRJ. Organomet Chem. 1975;93:259. [Google Scholar]
- 49.Lifer S. Master thesis. University of Wisconsin-Milwaukee; Milwaukee, WI: 1987. [Google Scholar]
- 50.Nyffeler PT, Liang CH, Koeller KM, Wong CHJ. Am Chem Soc. 2002;124:10773. doi: 10.1021/ja0264605. [DOI] [PubMed] [Google Scholar]
- 51.Liu Q, Tor Y. Org Lett. 2003;5:2571. doi: 10.1021/ol034919+. [DOI] [PubMed] [Google Scholar]
- 52.Spitzner JR, Muller MT. Nucleic Acids Res. 1988;16:5533. doi: 10.1093/nar/16.12.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muller MT, Spitzner JR, DiDonato JA, Mehta VB, Tsutsui K, Tsutsui K. Biochemistry. 1988;27:8369. doi: 10.1021/bi00422a012. [DOI] [PubMed] [Google Scholar]
- 54.Kondoh M, Usui T, Mayumi T, Osada H. J Antibiotics. 1998;51:801. doi: 10.7164/antibiotics.51.801. [DOI] [PubMed] [Google Scholar]
- 55.Depew KM, Danishefsky SJ, Rosen N, Sepp-Lorenzino L. J Am Chem Soc. 1996;118:12463. [Google Scholar]
- 56.We thank the Developmental Therapeutics Program of the National Cancer Institute for performing the in vitro anticancer evaluation.
- 57.Turner JG, Gump JL, Zhang C, Cook JM, Marchion D, Hazlehurst L, Munster P, Schell MJ, Dalton WS, Sullivan DM. Blood. 2006;108:3881. doi: 10.1182/blood-2005-10-009084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kakeya H, Onose R, Liu PC, Onozawa C, Matsumura F, Osada H. Cancer Res. 1998;58:704. [PubMed] [Google Scholar]
- 59.Bulman AL. Ph D Thesis. University of Virginia; Charlottesville, VA: 1998. Kinases and Drugs Targeting the microtubule System. [Google Scholar]
- 60.Barltrop JA, Owen TC, Cory AH, Cory JG. Bioorg Med Chem Lett. 1991;1:611. [Google Scholar]
- 61.Cory AH, Owen TC, Barltrop JA, Cory JG. Cancer Commun. 1991;3:207. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]