

Abstract
The XPC–hHR23B complex (XPC–hHR23B) is a heterodimeric protein required for the initial step of DNA damage recognition in the global nucleotide excision repair (NER) pathway. A strong preference of XPC–hHR23B for UV- and cisplatin-damaged DNA has previously been demonstrated using equilibrium binding assays. To better understand the molecular mechanism of damage recognition by XPC–hHR23B, we carried out the pre-steady-state kinetic analysis of the XPC–hHR23B–DNA interactions using a stopped-flow fluorescence assay. XPC–hHR23B displays a faster kon for cisplatin-and UV-damaged duplex DNA than for undamaged DNA, with additional, minor effects on the koff rates. XPC–hHR23B has a high affinity for undamaged single-stranded DNA compared to duplex DNA, which can be largely attributed to a high rate of association. However, cisplatin damage on single-stranded DNA reduced the overall level of binding by a factor of 7, with nearly equal contributions from changes to the kon and koff rates. Together, these results support a model for initial damage recognition by XPC–hHR23B that is dependent on structural changes in the DNA, and not adduct chemistry.
Nucleotide excision repair (NER)1 is a versatile DNA repair pathway that exists to remove helix-distorting DNA lesions that would otherwise interfere with essential DNA-dependent processes, such as DNA replication and transcription. Together, more than 30 eukaryotic proteins are involved in four distinct steps that include initial damage recognition, cleavage of the damaged strand, displacement of the strand, and resynthesis and ligation, and the entire process has been reconstituted using purified proteins (1, 2). The initial step of DNA damage recognition can occur by either of two distinct mechanisms. The first couples NER to transcription, ensuring that the transcribed strand of genes is efficiently repaired (3). The second targets genomic DNA and achieves damage recognition through proteins that have a higher affinity for damaged DNA than for undamaged DNA. To date, several proteins have shown damage-specific binding, including the XPC–hHR23B complex (XPC–hHR23B), replication protein A (RPA), XPA, and DDB (4). Of these, only XPC–hHR23B is dispensable for transcription-coupled repair (TCR) in the presence of a stalled RNA polymerase II (5) but is specifically required for global genomic DNA repair (GGR) (6).
The 144 kDa XPC protein is found in a heterotrimeric complex with hHR23B, the 44 kDa human homologue of the yeast protein Rad23 (7) and centrin 2, an 18 kDa centrosome component (8). However, XPC–hHR23B is sufficient for in vitro reconstitution of NER (9–11). Abundant evidence suggests that XPC–hHR23B is the first complex to recognize and bind to damaged DNA during GGR. For example, XPC–hHR23B is required for the earliest detectable open complex formation around a lesion at the beginning of NER (12, 13) and is the only NER protein with sufficient specificity and affinity to produce a distinct DNase footprint on DNA (14). A recent study used highly purified NER proteins and permanganate footprinting to confirm that XPC–hHR23B is the first complex to recognize damaged DNA and initiate the opening of nine bases of the helix around a site of damage (15).
Although XPC–hHR23B is likely the initiator of GGR, the role of XPC in damage recognition has not been without controversy. Previous reports of the binding activity of XPC note that although XPC binds to a wide variety of lesions such as UV-induced photoproducts, cisplatin adducts, and AAF adducts, XPC also binds with high affinity to undamaged DNA (16). XPC–hHR23B is additionally able to bind with equal affinity to substrates containing bubbles with or without damage, although only the damaged substrates are able to be repaired (17). However, the preference of XPC–hHR23B for damaged versus undamaged DNA has been shown to increase up to 400-fold with the addition of undamaged competitor DNA (10). Notably, the large preference observed by XPC–hHR23B for damaged DNA in equilibrium binding experiments is consistent with either an increased rate of association or a decreased rate of dissociation from damaged DNA; however, these studies are unable to distinguish between these possibilities.
In this report, we have examined the pre-steady state kinetics of binding of XPC–hHR23B to undamaged and damaged DNA using the intrinsic fluorescence of XPC–hHR23B and stopped-flow analysis to better understand the mechanism of damage-specific DNA binding. Equilibrium binding experiments were performed with the same DNA substrates to provide additional insight into the binding of XPC–hHR23B to damaged and undamaged DNA. The results revealed that damage-specific binding by XPC–hHR23B was mostly attributable to a dramatically increased kon for cisplatin and UV-damaged duplex DNA substrates compared to that for the undamaged duplex substrate, with very little difference in the dissociation rate. XPC–hHR23B displayed a 3-fold higher affinity for a 1,3 d(GpXpG) cisplatin adduct than for a 1,2 d(GpG) adduct, which was due to an increased rate of dissociation from the 1,2 d(GpG) adduct. Notably, although XPC–hHR23B binds with high affinity to single-stranded DNA and damaged duplex DNA, the presence of cisplatin damage on single-stranded DNA resulted in a decrease in the kon and an increase in the koff compared to those of undamaged single-stranded DNA, suggesting that XPC–hHR23B likely binds to the undamaged strand or adjacent to the adduct but not to the adduct itself. These data provide additional evidence that recognition and binding by XPC–hHR23B are predominantly a function of altered DNA structure.
MATERIALS AND METHODS
Materials
Oligonucleotides (Table 1) were purchased from Integrated DNA Technologies (Coralville, IA) and purified by electrophoresis through 12% polyacrylamide, 7 M urea, preparative denaturing gels.
Table 1.
DNA Oligonucleotides
| DNA | Sequence (5′-3′)a |
|---|---|
| LH10 | TACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTTTGTTCCCTTTAGTGAGGGTTAATTCCGAGCT |
| C-LH10b | AGCTCGGAATTAACCCTCACTAAAGGGAACAAAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTA |
| 1,2d(GpG) | CCCTTCTTTCTCTTCCCCCTCTCCTTCTTGGCCTCTTCCTTCCCCTTCCCTTTCCTCCCC |
| C-1,2d(GpG)b | GGGGAGGAAAGGGAAGGGGAAGGAAGAGGCCAAGAAGGAGAGGGGGAAGAGAAAGAAGGG |
| 1,3d(GpXpG) | CCCTTCTTTCTCTTCCCCCTCTCCTTCTTGCGCTCTTCCTTCCCCTTCCCTTTCCTCCCC |
| C-1,3d(GpXpG)b | GGGGAGGAAAGGGAAGGGGAAGGAAGAGCGCAAGAAGGAGAGGGGGAAGAGAAAGAAGGG |
The position of the cisplatin modification is indicated by the underlined bases.
The DNA substrates were synthesized with and without a 5′-fluorescein label.
Purification of XPC–hHR23B
XPC–hHR23B protein was purified from Sf9 insect cells infected with recombinant baculovirus expressing XPC and hHR23B proteins (18). During the purification, the protein concentration was monitored using a Bradford dye-based assay (Bio-Rad, Hercules, CA). Sf9 cells (300 mL) were infected with recombinant baculovirus at an input of multiplicity of 10 PFU per cell and incubated at 27 °C for 48 h. Cells were collected by low-speed sedimentation, and cell free extract was prepared as described previously (19), except that supernatant was dialyzed into buffer A [25 mM Tris (pH 7.5), 1 mM EDTA, 1 mM DTT, 0.01% Triton X-100, and 10% glycerol] containing 0.3 M NaCl. Dialyzed extract was centrifuged at 4 °C for 10 min at 12000g to remove particulates. The clarified supernatant was applied to a 10 mL cellulose phosphate column equilibrated in the same buffer. The column was washed with 50 mL of buffer A and 0.3 M NaCl and eluted with buffer A containing 1 M NaCl. Eluted protein was pooled (P-cell fraction) and diluted with buffer A to achieve a final salt concentration of 0.6 M NaCl. Protein was applied to a 4 mL ssDNA cellulose column equilibrated in buffer A and 0.6 M NaCl. The column was washed with 35 mL of buffer A and 0.6 M NaCl and eluted with 20 mL of buffer A and 1.5 M NaCl. Eluted protein was pooled (ss-DNA fraction) and diluted with buffer A to 0.3 M NaCl and applied to a 2 mL heparin–Sepharose column. The column was washed with 25 mL of buffer A and 0.3 M NaCl and eluted with a 20 mL linear salt gradient from 0.3 to 1.0 M NaCl in buffer A. Peak fractions containing XPC–hHR23B were pooled and dialyzed into buffer B [25 mM HEPES (pH 7.8), 0.2 M KCl, 1 mM EDTA, and 1 mM DTT] containing 50% glycerol, at 4 °C for 16 h (H–S pool). The purified proteins were stored at −80 °C in small aliquots and thawed immediately prior to being used. The purity of XPC–hHR23B-containing fractions was analyzed by SDS–PAGE followed by silver staining, and the XPC–hHR23B protein complex was judged to be greater than 95% pure, noted by the lack of contaminating polypeptides (Figure 1). Protein preparations were at least 50% active as determined by EMSA binding analysis (data not shown). The molar concentration of protein represents the active protein concentration and was calculated using a molecular mass of 187,000 Da for the heterodimeric XPC–hHR23B complex. It should be noted that the stoichiometry of the complex cannot be determined on the basis of the band intensity observed in silver-stained gels. The purification procedure that was employed, however, results in the majority of the protein being in a 1:1 complex (18).
Figure 1.
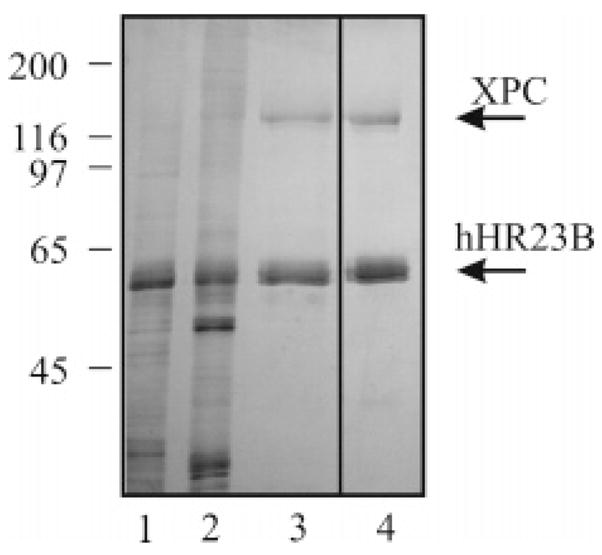
Purity of the XPC–hHR23B protein preparation. XPC–hHR23B was expressed in insect cells infected with recombinant baculovirus and purified by column chromatography as described in Materials and Methods. Samples from each step of the purification (2 μg of total protein) were subjected to SDS–PAGE and were detected by silver stain analysis: lane 1, cell extract; lane 2, P-cell fraction; lane 3, ssDNA fraction; and lane 4, H–S fraction.
Electrophoretic Mobility Shift Assays (EMSAs)
Oligo-nucleotide LH10 (75-mer) was labeled at the 5′ end with [γ-32P]ATP. DNA was either left undamaged or treated with cisplatin at a drug:molecule ratio of 40:1 in buffer containing 1 mM NaHPO4 (pH 7.5) and 3 mM NaCl at 37 °C for 40 h. Undamaged and cisplatin-damaged DNA were annealed to complement C-LH10, and duplex DNA was purified by nondenaturing PAGE. EMSA reactions (20 μL) were performed in buffer containing 20 mM HEPES (pH 7.8), 0.001% Nonidet P-40, 50 mM NaCl, 1 mM DTT, 0.05 mg/mL BSA, and 2 mM MgCl2. Reactions that included 20 fmol of DNA were initiated by the addition of XPC–hHR23B, and the mixtures were incubated for 15 min at 25 °C. The reactions were stopped by the addition of glutaraldehyde (0.25%) and buffer containing 10 mM EDTA, 10% glycerol, 0.01% bromophenol blue, and 0.01% xylene cyanol. The samples were then separated on 4% native polyacrylamide gels for 1 h at 170 V. Gels were dried under vacuum, and the product was quantified using a PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Stopped-Flow Kinetic Experiments
Undamaged and platinum-damaged single-stranded and duplex DNA substrates were prepared as described for EMSA binding experiments, except that the DNA was unlabeled. Duplex DNA was UV-damaged by exposure to 5 kJ/m2 light at 254 nm. Duplex DNA substrates containing site-specific cisplatin lesions [1,2 d(GpG) and 1,3 d(GpXpG)] were produced by treating the 1,2 d(GpG) or 1,3 d(GpXpG) DNA with cisplatin in a 10:1 ratio of drug to GG (or GXG) site. The platinated 1,2 d(GpG) and 1,3 GpXpG DNA substrates were annealed to C-1,2 GpG and C-1,3 GpXpG, respectively, restriction enzyme digested to remove unplatinated substrate, and purified by nondenaturing PAGE. Stopped-flow reactions were performed in buffer B containing 10% glycerol. Equal volumes of XPC–hHR23B and DNA from separate syringes were rapidly mixed at 24 °C using an SX.18MV stopped-flow reaction analyzer (Applied Photophysics, Leatherhead, U.K.). Fluorescence was measured following excitation at 290 nm, using a 340 nm cut-on filter. XPC–hHR23B (10 nM) was mixed with varying concentrations of DNA. To ensure that the reactions were performed under pseudo-first-order kinetic conditions, the DNA titrations were designed to have the majority of the points with DNA concentrations greater than 4–5 times the protein concentrations. The traces shown are averages from at least eight individual shots. The kinetic data were fit using ProK (Applied Photophysics) to single-exponential decay equations to calculate the observed rate, kobs. A minimum of three independent experiments were performed for each DNA concentration, and the average kobs and standard deviation were plotted versus DNA concentration. Assuming a one-step mechanism for binding of XPC–hHR23B to DNA and under pseudo-first-order kinetic conditions, kobs = kon[DNA] + koff, where the slope of the line is the bimolecular association rate, kon, and the y-intercept is the unimolecular dissociation rate, koff. The rate constants were calculated using Sigmaplot, and the KD values were calculated using the equation KD = koff/kon.
Fluorescence Polarization
The duplex LH10, 1,2 d(GpG), and 1,3 d(GpXpG) substrates were prepared as noted for stopped-flow analysis, except that C-LH10, C-1,2 d(GpG), and C-1,3 d(GpXpG) DNA substrates contained a 5′-fluorescein modification. Fluorescence polarization experiments were carried out using a Cary Eclipse fluorescence spectrophotometer (Varian) with the fluorescence emission wavelength monitored at 515 nm following excitation at 495 nm. The excitation and emission bandwidths were set at 10 nm, with three data points taken at 3 s intervals for each titration point. Reaction mixtures (0.5 mL) contained buffer B with 5 nM fluorescein-labeled DNA substrate as indicated. XPC–hHR23B was added such that the final volume did not exceed 10% of the reaction volume. r values were calculated as previously described (20). Mean r values from three independent experiments were plotted versus protein concentration and fit to a hyperbolic function.
RESULTS
Affinity of XPC–hHR23B for Undamaged and Cisplatin-Damaged Single-Strand DNA Substrates
A considerable amount of evidence demonstrating that the XPC–hHR23B complex is the initial factor involved in damage recognition for the GGR pathway has accumulated. However, conflicting data regarding the relative ability of purified XPC–hHR23B protein preparations to bind to single-stranded versus duplex undamaged and damaged DNA have emerged (16). Oligonucleotides with fewer than 60 bp demonstrate weakened binding, as well as an “end effect” that reduces the level of damage-specific binding (18, 21). Therefore, a 75-mer DNA (LH10) was selected to determine the affinity of XPC–hHR23B for single-stranded and duplex damaged and undamaged DNA in steady-state and pre-steady-state reactions (Table 1). First, the DNA binding activity of XPC–hHR23B was measured using electrophoretic mobility shift assays (EMSAs) with cisplatin-damaged or undamaged 32P end-labeled single-stranded DNA. Consistent with previous results (10), EMSAs performed in the absence of competitor DNA resulted in the formation of multiple DNA–protein complexes with the majority of the products not entering the separation gel. To combat this, competitor DNA [100 ng of poly(dI/dC)] was added to each reaction mixture to lower the amount of binding observed such that only one complex of XPC–hHR23B was bound (Figure 2A). The results demonstrate that purified XPC–hHR23B was able to bind to the single-stranded DNA substrate in the presence or absence of cisplatin lesions, and the amount bound increased with increasing amounts of protein added. Interestingly, the presence of cisplatin adducts on the single-stranded DNA clearly weakened the ability of XPC–hHR23B to bind. The amount of XPC–hHR23B bound to DNA was quantified by PhosphorImager analysis and plotted as a function of the total amount of XPC–hHR23B protein present (Figure 2B). At the lowest protein concentration, there was a greater than 10-fold decrease in the level of binding to the cisplatin-modified DNA.
Figure 2.
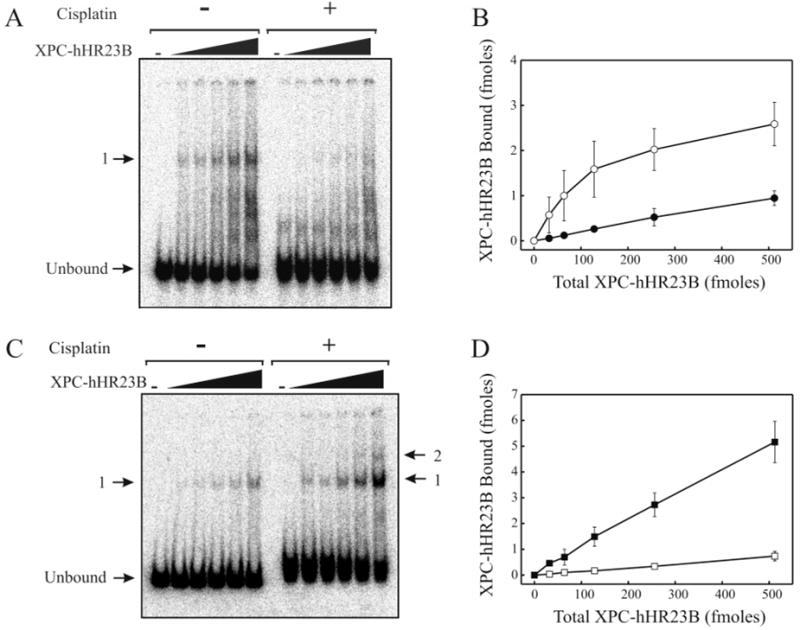
XPC–hHR23B equilibrium binding to undamaged and cisplatin-damaged DNA. Binding assays were performed with varying concentrations of XPC–hHR23B and 20 fmol of undamaged or cisplatin-treated single-stranded (A and B) or duplex DNA (C and D). (A and C) XPC–hHR23B–DNA complexes were separated from unbound DNA by 4% native PAGE. (B and D) XPC–hHR23B bound to undamaged (○) and cisplatin-damaged (●) single-stranded DNA (B) or undamaged (□) and cisplatin-damaged (■) duplex DNA (D) was quantified by PhosphorImager analysis and plotted as a function of total XPC–hHR23B concentration. Error bars represent the mean and standard deviations for three independent experiments.
Affinity of XPC–hHR23B for Undamaged and Cisplatin-Damaged Duplex DNA Substrates
Because XPC–hHR23B is known to prefer damaged duplex DNA over undamaged DNA, it was important to verify the duplex DNA binding activity of our XPC–hHR23B preparations. Therefore, the 32P end-labeled 75-mer single-stranded DNA with and without cisplatin treatment was annealed to undamaged complementary DNA to form undamaged and cisplatin-damaged duplex DNA substrate, and binding reactions were performed as for single-stranded DNA. The results of EMSA binding analysis are shown in Figure 2C and demonstrate that up to two complexes of XPC–hHR23B were able to bind to undamaged and cisplatin-damaged duplex DNA and that there was clearly an increased affinity for damaged compared to undamaged duplex DNA. Quantification of these results revealed a 13-fold increase in the level of binding to damaged DNA at the lowest concentration of XPC–hHR23B, and a 7-fold increase in the level of binding at the highest concentration that was tested (Figure 2D).
Intrinsic Fluorescence of XPC–hHR23B in the Absence and Presence of DNA
Although damage-specific binding by XPC–hHR23B has been previously examined in the steady state, the mechanism of binding is less well understood. Here, stopped-flow analysis was employed to measure the pre-steady-state kinetics of binding of XPC–hHR23B to DNA. This assay has been successfully used to measure the pre-steady-state kinetics of binding of RPA to cisplatin-damaged DNA, and the effect of XPA on this binding (22, 23). These assays require either a purified protein that possesses intrinsic fluorescence which is quenched upon DNA binding or fluorescently labeled DNA that exhibits a change in fluorescence upon protein binding. To this end, purified XPC–hHR23B protein was examined for intrinsic tryptophan fluorescence which might be altered upon DNA binding. First, excitation and emission scans were performed, and XPC–hHR23B demonstrated intrinsic fluorescence with a fairly tight excitation peak at 275 nm and a broad emission from 290 to >350 nm with a peak at 306 nm (data not shown). Fluorescence was reduced 50–70% by the addition of undamaged plasmid DNA, while the excitation and emission spectra remained constant (data not shown). Therefore, quenching of the intrinsic fluorescence of XPC–hHR23B by DNA was employed to measure the pre-steady-state kinetics of binding using stopped-flow analysis.
Pre-Steady-State Kinetics of Binding of XPC–hHR23B to Undamaged and Cisplatin-Damaged Single-Stranded DNA
The kinetics of binding of XPC–hHR23B to undamaged and cisplatin-damaged single-stranded DNA was first assessed. The intrinsic fluorescence of XPC–hHR23B was measured with an excitation wavelength of 290 nm, and the emission was monitored using a 340 nm cut-on filter. The fluorescence of XPC–hHR23B in the absence of DNA did not change over the times measured, and the resulting trace provided the initial fluorescence from which quenching was monitored (Figure 3A, top trace). The addition of undamaged LH10 75-mer single-stranded DNA resulted in fluorescence quenching that fit to a single-exponential decay function, indicative of a single-step binding model (Figure 3A, bottom trace). XPC–hHR23B at a constant concentration was mixed with increasing concentrations of DNA to achieve pseudo-first-order kinetics. The observed rate of quenching, kobs, was plotted versus the DNA concentration for both undamaged and cisplatin-damaged single-stranded DNA (Figure 3B). The results revealed a linear relationship, with the y-intercept being equal to the dissociation rate (koff) and the slope of the line being equal to the rate of association (kon). The kon for the undamaged single-stranded DNA substrate was 0.329 ± 0.007 nM−1 s−1, and the koff was 14.7 ± 1.0 s−1 [Figure 3B (○)]. In contrast, the kon for the cisplatin-damaged single-stranded DNA substrate was 0.103 ± 0.017 nM−1 s−1, and the koff was 34.0 ± 3.6 s−1 [Figure 3B (●)]. These results reveal a 3-fold difference in the kon values and a 2-fold difference in the koff values of binding of XPC–hHR23B to undamaged versus cisplatin-damaged single-stranded DNA. These results were demonstrated to be highly statistically significant (data not shown). The calculated KD values for the two substrates revealed a 7-fold greater affinity for undamaged than for cisplatin-damaged ssDNA (Table 2). It should be noted that in the single-stranded undamaged DNA series, the lowest DNA concentration resulted in a molar DNA:protein ratio that was less than 5:1. Linear regression analysis performed omitting these data, however, did not significantly alter the calculated rate constants (data not shown). These results are consistent with the EMSA binding experiments in the steady state that demonstrated a reduction in the level of binding to the cisplatin-damaged compared to undamaged single-stranded DNA in the presence of competitor DNA (Figure 2A, B).
Figure 3.
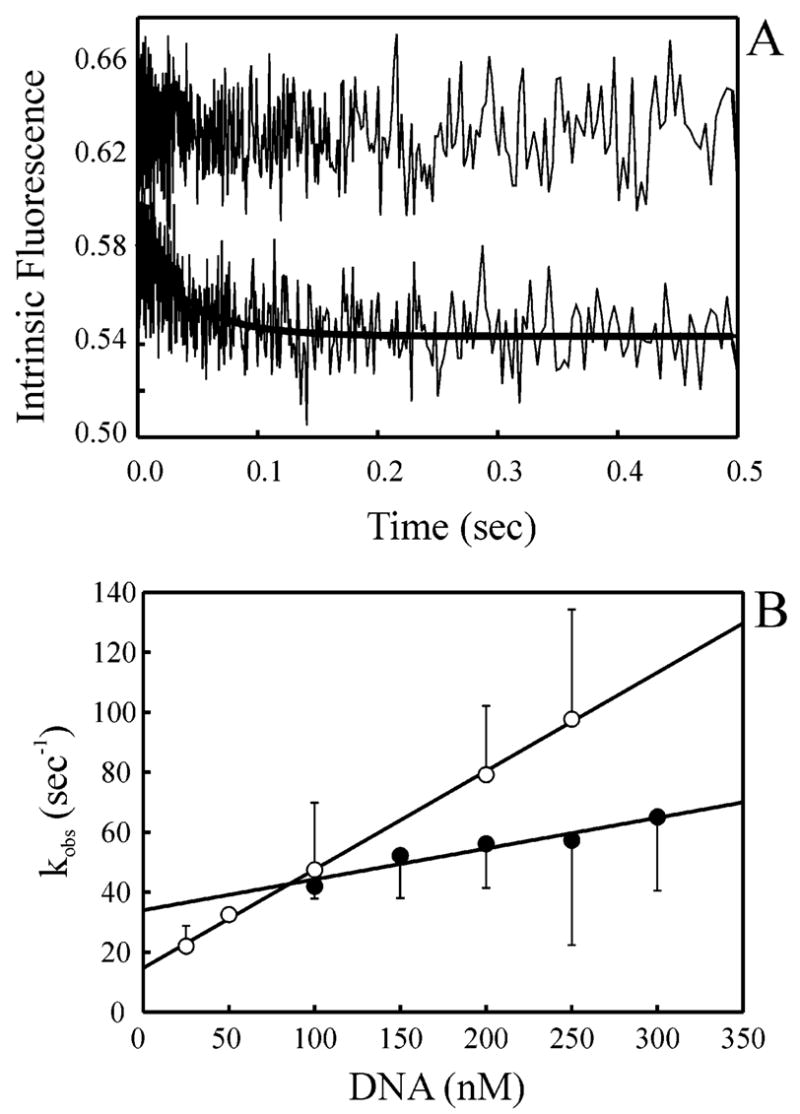
Pre-steady-state kinetics of binding of XPC–hHR23B to undamaged and cisplatin-damaged single-stranded DNA substrates. Reaction mixtures were excited at 290 nm, and fluorescence was monitored via emission at 340 nm. Traces shown are the average of at least eight shots and fit to single-exponential decay functions. (A) XPC–hHR23B (10 nM) was mixed with buffer (top trace) or with 25 nM undamaged 75-mer DNA (bottom trace) and fit to a single-exponential decay function. The top trace was offset by 0.07 for clarity. (B) Undamaged (○) and cisplatin-damaged (●) single-stranded DNA was titrated with 10 nM XPC–hHR23B, and the observed rate of quenching (kobs) was plotted vs the DNA concentration and fit to a straight line. The slope of the line provides the bimolecular rate constant, kon, and the y-intercept provides the rate of dissociation, koff. Each point represents the mean and standard deviation from three independent experiments.
Table 2.
Kinetic Parameters of Binding of XPC–hHR23B to Damaged and Undamaged DNA
| DNA substrate | kon (nM−1 s−1) | koff (s−1) | KD(calcd) (nM) | KD(steady state) (nM) | koff(calcd) (s−1) |
|---|---|---|---|---|---|
| single-stranded | |||||
| LH10 | 0.329 ± 0.007 | 14.7 ± 1.0 | 45 | ||
| LH10 and Pt | 0.103 ± 0.017 | 34.4 ± 3.6 | 334 | ||
| duplex | |||||
| undamaged LH10 | 0.029 ± 0.005 | −2.1 ± 2.0 | 70.7 ± 13.0 | 2.0 | |
| LH10 and Pt | 0.291 ± 0.043 | −11.1 ± 3.7 | |||
| LH10 and UV | 0.103 ± 0.006 | −0.02 ± 0.7 | |||
| 1,2 d(GpG) | 0.318 ± 0.048 | −4.8 ± 4.4 | 27.1 ± 5.1 | 8.6 | |
| 1,3 d(GpXpG) | 0.281 ± 0.020 | −2.0 ± 1.8 | 8.3 ± 1.2 | 2.3 | |
Pre-Steady-State Kinetics of Binding of XPC–hHR23B to Undamaged and Damaged Double-Stranded DNA
Previously, damage-specific binding has been demonstrated for XPC–hHR23B (16). However, while the affinity is higher for damaged duplex DNA, it is unclear whether this is a function of the association or dissociation rates of binding. Therefore, stopped-flow analysis was employed to measure the pre-steady-state kinetics of binding of XPC–hHR23B to undamaged and cisplatin-damaged duplex DNA. To determine if any differences in binding observed between the undamaged and cisplatin-damaged DNA were lesion-specific, we additionally examined the pre-steady-state kinetics of binding to UV-damaged dsDNA. Each DNA substrate was titrated with a constant amount of XPC–hHR23B, and the observed rate of quenching, kobs, was fit to a single-exponential decay function, as observed for the single-stranded substrate–XPC–hHR23B interaction. The DNA concentrations in these experiments are expressed as the molar concentration of potential XPC–hHR23B binding sites. This is based on the results obtained from EMSAs where two distinct complexes were observed in binding reactions with XPC–hHR23B and double-stranded DNA (Figure 2). The kinetic fits for XPC–hHR23B (10 nM) binding to the undamaged or cisplatin- and UV-damaged substrates (100 nM) are shown in Figure 4A. Notably, at this substrate concentration, the quenching observed upon binding to the UV- or cisplatin-modified substrate was 6 or 17 times faster, respectively, than that observed for binding to the undamaged substrate. The kobs values for binding to undamaged or cisplatin- or UV-damaged dsDNA were plotted versus the concentration of XPC–hHR23B binding sites. Under the conditions of these binding reactions, the kinetics follow pseudo-first-order kinetics such that the kon and koff values can be determined from these graphs. The kon for the duplex undamaged DNA substrate was 0.029 ± 0.005 nM−1 s−1, and the y-intercept was −2.1 ± 2.0 s−1. On the other hand, the kon for the duplex cisplatin-damaged DNA substrate was 0.291 ± 0.043 nM−1 s−1, and the y-intercept was −11.1 ± 3.7 s−1. Interestingly, the rate of association (kon) with the cisplatin-damaged duplex DNA (●) was more than 10 times faster than the rate with the undamaged duplex DNA (○) and similar to the rate of binding to undamaged ssDNA (Figure 3B). The rate of association with the UV-damaged substrate was 0.103 ± 0.006 nM−1 s−1, ~4 times the rate of binding to undamaged DNA [Figure 4B (■)]. As with the other duplex DNA substrates, a negative y-intercept of −0.02 ± 0.7 s−1 was calculated. Together, these results indicate that damage recognition by XPC–hHR23B is a function of an increased rate of association with damaged DNA and that this mechanism was observed with both cisplatin- and UV-damaged DNA. In the UV-damaged DNA titration experiment, the lowest DNA concentration resulted in a molar DNA:protein ratio that was less than 5:1. Again, linear regression analysis performed omitting these data did not significantly alter the calculated rate constant (data not shown).
Figure 4.
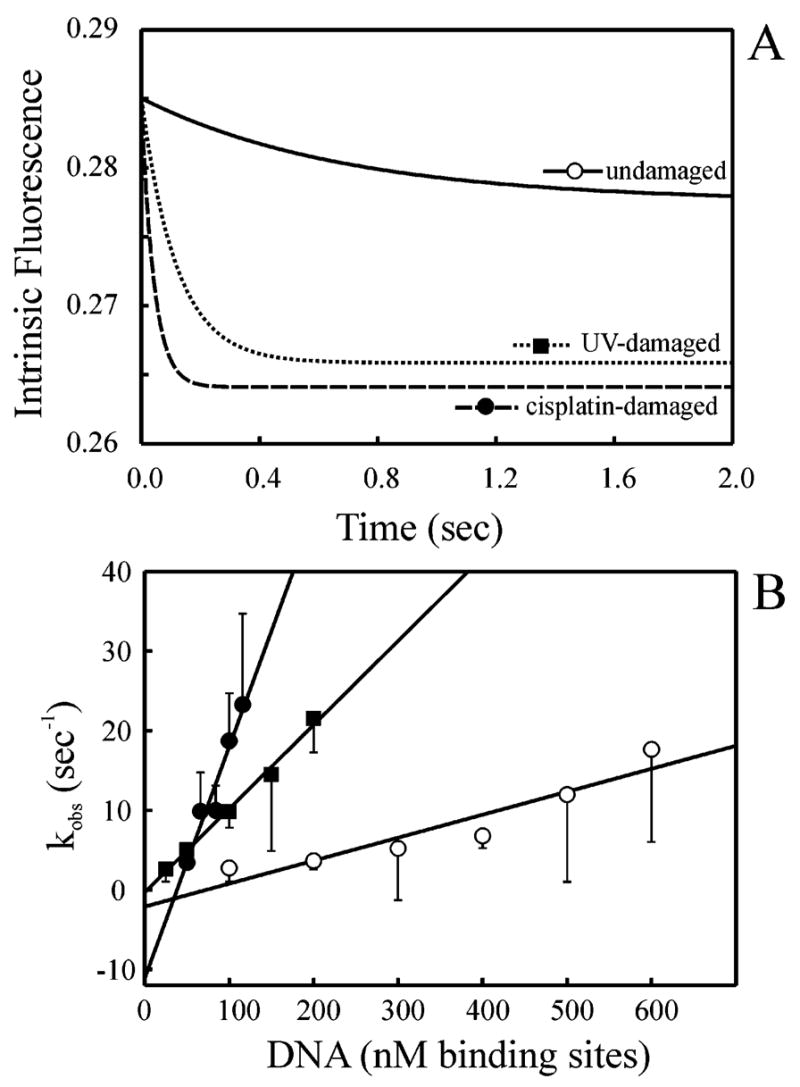
Pre-steady-state kinetics of binding of XPC–hHR23B to undamaged or UV- or cisplatin-damaged duplex DNA substrates. Reaction mixtures were excited at 290 nm, and fluorescence was monitored via emission at 340 nm. (A) XPC–hHR23B (10 nM) was mixed with 100 nM dsDNA that was undamaged (—), cisplatin-damaged (- - -), or UV-damaged (···). Traces are the average of at least eight shots and fit to single-exponential decay functions. For clarity, the intrinsic fluorescence for the UV-damaged and cisplatin-damaged data was offset by 0.0069 and 0.0141, respectively. The observed rates of binding were 1.4 ± 0.5, 23.7 ± 2.6, and 8.4 ± 1.2, respectively. (B) Kinetic traces were measured at a constant XPC–hHR23B concentration (10 nM) and varying concentrations of duplex DNA that was undamaged (○), cisplatin-damaged (●), or UV-damaged (■). The observed rate of quenching (kobs) was plotted vs the DNA concentration and fit to a straight line. Each point represents the mean and standard deviation from at least three independent experiments.
Numerous in vivo and in vitro studies of DNA damage recognition and subsequent repair have correlated the kinetics of repair with the degree of structural distortion (4). Therefore, two structurally distinct cisplatin adducts were employed to examine the relative effect of duplex DNA distortion on the pre-steady-state binding kinetics of XPC–hHR23B. The 1,2 d(GpG) and 1,3 d(GpXpG) cisplatin lesions both cause a localized structural distortion of the DNA; however, the 1,3 adduct additionally results in a two-to four-base localized melting of the duplex DNA surrounding the adduct (24, 25). Thus, duplex 60 bp DNA substrates were constructed to contain either a 1,2 or a 1,3 site-specific cisplatin adduct, and the stopped-flow kinetics of XPC–hHR23B binding were examined (Figure 5). The rates of association with the 1,2 d(GpG) substrate were 0.318 ± 0.048 nM−1 s−1 (●) and 0.281 ± 0.020 nM−1 s−1 for the 1,3 d(GpXpG) substrate (○). Interestingly, these results demonstrate that while the association rates with either the 1,2 or 1,3 adduct were not significantly different, they were approximately 5 times higher than that observed for the duplex undamaged DNA substrate (Table 2). As noted for the other duplex DNA substrates, the y-intercepts for either the 1,2 d(GpG) or 1,3 d(GpXpG) substrate were negative (−4.8 ± 4.4 and −2.0 ± 1.8 s−1, respectively).
Figure 5.
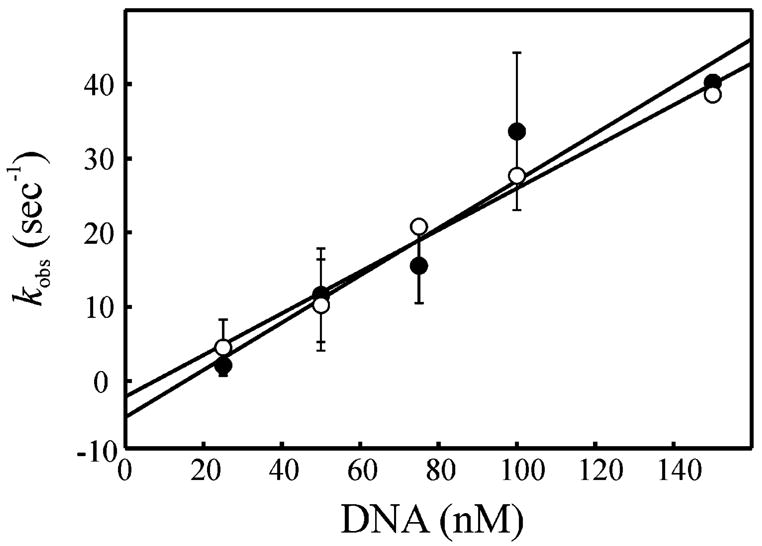
Pre-steady-state kinetics of binding of XPC–hHR23B to duplex damaged DNA containing 1,2 d(GpG) and 1,3 d(GpG) site-specific cisplatin adducts. Kinetic traces were measured at a constant XPC–hHR23B concentration (10 nM) and varying concentrations of duplex DNA with a 1,2 d(GpG) adduct (○) or a 1,3 d(GpXpG) adduct (●). The observed rate of quenching (kobs) was plotted vs the DNA concentration and fit to a straight line.
Steady-State Binding of XPC–hHR23B to Undamaged and Damaged Duplex DNA with Fluorescence Polarization
The direct analysis of dissociation rates was hampered by the fact that the y-intercepts from the kobs versus DNA concentration plots were negative for each duplex DNA substrate (Figures 4B and 5). This is a common observation with molecular interactions where the koff is much less than the kon[DNA] value (25) and required additional analyses for the accurate determination of the rates of dissociation. Therefore, the steady-state binding affinity of XPC–hHR23B for undamaged and damaged duplex DNA substrates was determined using fluorescence polarization. The assay measures the increase in the degree of fluorescence polarization of the DNA upon binding of the relatively large XPC–hHR23B protein complex. Therefore, duplex DNA substrates were prepared with a 5-fluorescein modification and were undamaged or contained a single, site-specific 1,2 d(GpG) or 1,3 d(GpXpG) cisplatin lesion. XPC–hHR23B was titrated into reaction mixtures containing a constant level of DNA substrate, and the anisotropy (r value) was plotted versus protein concentration and fit to hyperbolic functions (Figure 6). The results for the undamaged duplex 75-mer DNA revealed a KD of 70.7 ± 13.0 nM and a calculated koff of 2.0 s−1 (Table 2). In contrast, the dissociation constants for the damaged duplex substrates containing either a 1,2 d(GpG) or a 1,3 d(GpXpG) cisplatin lesion were significantly lower with KD values of 27.1 ± 5.1 and 8.3 ± 1.2 nM, respectively (Table 2). The 3-fold difference in the affinity of XPC–hHR23B for the two cisplatin adducts is in excellent agreement with previous results demonstrating that repair of the 1,3 adduct is 2–3 times faster than that of the 1,2 adduct with either cell-free extracts or reconstituted repair (27, 28). Because the association rates for these substrates were not significantly different (Figure 5), the differences in affinity can be attributed to differences in dissociation rates and are calculated to be 8.6 s−1 for the 1,2 lesion and 2.3 s−1 for the 1,3 lesion (Table 2). Together, these data demonstrate that the lower affinity observed for duplex undamaged DNA than for damaged DNA (Figure 4) is due predominately to a slow rate of association. However, the difference in affinity observed between various types of damage is largely due to a faster rate of dissociation from lesions that distort the DNA to a lesser extent.
Figure 6.
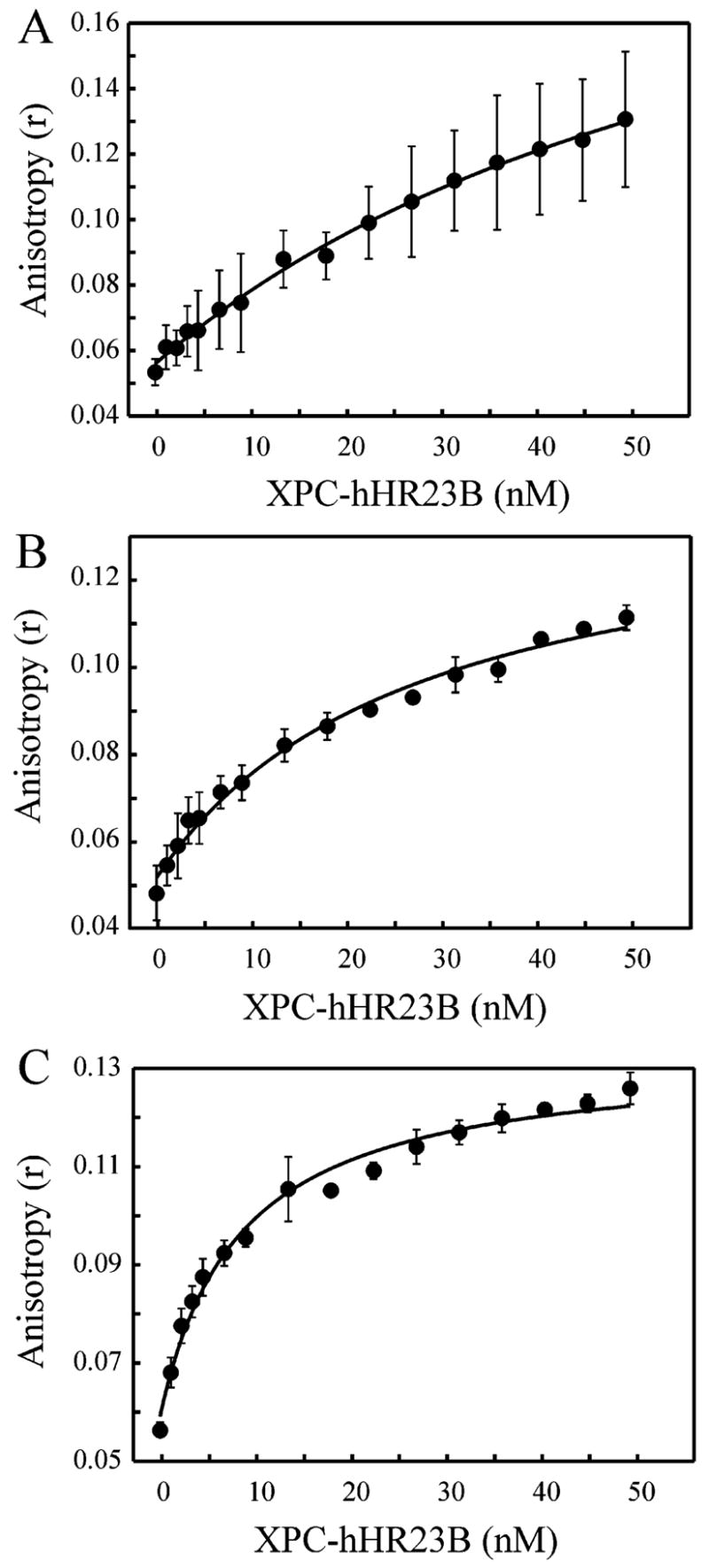
Fluorescence anisotropy measurements of binding of XPC–hHR23B to undamaged and cisplatin-damaged duplex DNA. XPC–hHR23B was titrated with 5 nM fluorescently labeled DNA substrate: (A) undamaged LH10 (75-mer), (B) 1,2 d(GpG) cisplatin adduct (60-mer), and (C) 1,3 d(GpXpG) cisplatin adduct (60-mer). Each point represents the mean and standard deviation from three independent experiments.
DISCUSSION
The process of damage recognition within genomic DNA by NER is vital in ensuring that lesions are processed expediently and efficiently to prevent mutations and ensuing cell death. As such, NER eliminates a wide variety of adducts that differ on the basis of chemical structure, as well as the degree of induced DNA helix distortion. Although XPC–hHR23B is likely responsible for the initiation of this process (14, 15, 29), numerous questions remain regarding its affinity for undamaged and damaged DNA, as well as its ability to recognize the variety of DNA lesions known to be repaired by NER (16). For example, equilibrium binding experiments have demonstrated that XPC binds to undamaged and damaged DNA with little difference in affinity, whereas significant damage specificity can be observed only in the presence of competitor DNA (16). Further, XPC has a restricted ability to bind to lesions that have limited DNA distortion, such as cyclopyrimide dimers, CPDs (17, 21, 30, 31), yet the affinity of XPC for DNA substrates containing single-stranded bubble regions is unchanged by the presence of a 2AAF–DNA adduct within the bubble region (17). While these data demonstrate that XPC–hHR23B clearly has an increased affinity for a variety of DNA adducts, the mechanistic details of damage recognition have yet to be completely elucidated. Here, a stopped-flow assay was developed to directly measure the association and dissociation rates of binding of XPC–hHR23B to a variety of DNA substrates. These analyses, along with equilibrium binding experiments, provide a more comprehensive examination of the interaction of XPC–hHR23B with damaged and undamaged DNA.
The pre-steady-state kinetic data reveal a dramatically increased rate of association with cisplatin or UV-damaged duplex DNA compared to that with undamaged DNA. These data suggest that the increased affinity for damaged duplex DNA observed in equilibrium binding studies is primarily a result of an increased rate of association with damaged DNA. Steady-state binding experiments have noted a high-affinity binding to single-stranded DNA by XPC–hHR23B. However, the rate of dissociation from single-stranded DNA was significantly greater than that observed for damaged or undamaged duplex DNA substrates. Therefore, the observed increased affinity for single-stranded DNA compared to duplex undamaged DNA is primarily a result of an increased association rate for single strands. The difference in binding kinetics observed between single-stranded undamaged DNA and duplex damaged DNA supports previous studies suggesting that XPC–hHR23B may in fact preferentially bind to the junctions formed between single-stranded and double-stranded DNA (32). The structural distortion caused by the UV and cisplatin adducts clearly provides this preferred binding substrate, as indicated by the high kon and low koff rates.
Interestingly, previous studies have not addressed whether XPC preferentially contacts the damaged or undamaged strand of the DNA. Our data demonstrating that XPC–hHR23B has a 3-fold decrease in association rate as well as a 2-fold increase in dissociation rate with cisplatin-damaged single-stranded DNA are consistent with XPC–hHR23B likely binding to the undamaged strand or adjacent to the adduct, but not directly to the adduct itself. The observed reduction in the level of binding to the cisplatin-modified single-stranded DNA likely is not a result of the presence of massive structural distortions, because the cisplatin-damaged substrate was able to bind the same number of XPC–hHR23B molecules as the unmodified DNA in EMSA binding studies in the absence of a DNA competitor. Damage recognition based upon structural changes in the DNA and not specific lesion chemistry would at least partially explain the ability of the NER apparatus to recognize and repair an extremely wide variety of DNA lesions.
Although NER eliminates a wide variety of DNA lesions, numerous in vivo and in vitro studies of DNA damage recognition and subsequent repair have correlated the kinetics of repair with the degree of structural distortion (4). This has been associated with the reduced affinity of XPC–hHR23B for lesions that do not distort the DNA as measured in the steady state (17, 21). In this study, we employed two structurally distinct cisplatin adducts to examine the relative effect of duplex DNA distortion on the pre-steady-state binding kinetics of XPC–hHR23B. Interestingly, the results suggest that XPC–hHR23B associates with the 1,2 d(GpG) and 1,3 d(GpXpG) cisplatin lesions with similar rates. The more rapid rate of dissociation of XPC–hHR23B from the 1,2 d(GpG) compared to the 1,3 d(GpXpG) cisplatin–DNA lesion is likely a result of the lack of localized melting of the duplex DNA containing the 1,2 d(GpG) adduct. Surprisingly, the rate of dissociation from the DNA containing the 1,2 d(GpG) cisplatin adduct was even greater than the rate of dissociation from the undamaged duplex DNA substrate, suggesting that damage-specific binding is primarily a consequence of a dramatically increased association rate. In addition, the finding that dissociation from the undamaged DNA is relatively slow compared to that of DNA containing a 1,2 d(GpG) cisplatin lesion suggests that the damage verification step is crucial in establishing a functional NER complex. This verification step would then be envisioned to accelerate the rate of dissociation of XPC–hHR23B from the undamaged DNA, while potentially stabilizing the protein on damaged DNA. Alternatively, the increased rate of association drives the recognition process as efficient dissociation of XPC–hHR23B is required from both damaged and undamaged DNA once TFIIH, RPA, and XPA assemble at the damaged site. The kinetic data obtained in this study also clarify our understanding of the inability of XPC–hHR23B to recognize nondistorting adducts from the surrounding undamaged DNA. The data suggest that the lack of observed binding in steady-state experiments is a result of a rapid rate of dissociation from DNA containing these adducts coupled with a slower association rate.
While initial damage recognition is likely a function of XPC–hHR23B binding, numerous other NER proteins have demonstrated damage specificity, and their role in modulating the binding of XPC is not well understood. For example, while DDB is not required for the recognition of all DNA adducts processed by NER, it can stimulate repair of certain DNA lesions (33). Our results also suggest that XPC does not recognize specific lesion chemistry and is similar to what we have observed for RPA binding damaged DNA (22, 34). In contrast, both XPA alone and the RPA–XPA complex require a chemical lesion for efficient binding to duplex DNA and subsequent repair (13, 23, 35–37). This suggests that XPA will perform the lesion verification step for DNA lesions recognized by XPC–hHR23B and repaired via GGR NER. The mechanism by which DDB stimulates DNA damage recognition by XPC–hHR23B and how the RPA–XPA complex participates in lesion recognition and verification process clearly warrants further investigation.
Acknowledgments
We thank Aziz Sancar for kindly providing the recombinant baculovirus expressing XPC and hHR23B. We additionally thank members of the Turchi lab for critical reading of the manuscript.
Footnotes
This work was supported by a grant CA82741 from the NIH, National Cancer Institute.
Abbreviations: XPC, xeroderma pigmentosum group C; hHR23B, human homologue of Rad23; NER, nucleotide excision repair; RPA, replication protein A; XPA, xeroderma pigmentosum group A; CPD, cyclobutane pyrimidine dimer; TCR, transcription-coupled repair; GGR, global genomic repair.
References
- 1.Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 2.Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem. 1995;270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]
- 3.Hanawalt PC. DNA damage and repair. Volume 2: DNA repair in higher eukaryotes Overview. DNA Damage Repair. 1998;2:1–8. [Google Scholar]
- 4.Batty DP, Wood RD. Damage recognition in nucleotide excision repair of DNA. Gene. 2000;241:193–204. doi: 10.1016/s0378-1119(99)00489-8. [DOI] [PubMed] [Google Scholar]
- 5.Christians FC, Hanawalt PC. Repair in ribosomal-RNA genes is deficient in xeroderma pigmentosum group-C and in cockaynes-syndrome cells. Mutat Res. 1994;323:179–187. doi: 10.1016/0165-7992(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 6.Shivji MK, Eker AP, Wood RD. DNA repair defect in xeroderma pigmentosum group C and complementing factor from HeLa cells. J Biol Chem. 1994;269:22749–22757. [PubMed] [Google Scholar]
- 7.Masutani C, Sugasawa K, Yanagisawa J, Sonoyama T, Ui M, Enomoto T, Takio K, Tanaka K, van der Spek PJ, Bootsma D. Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. EMBO J. 1994;13:1831–1843. doi: 10.1002/j.1460-2075.1994.tb06452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Araki M, Masutani C, Takemura M, Uchida A, Sugasawa K, Kondoh J, Ohkuma Y, Hanaoka F. Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem. 2001;276:18665–18672. doi: 10.1074/jbc.M100855200. [DOI] [PubMed] [Google Scholar]
- 9.Sugasawa K, Ng JM, Masutani C, Maekawa T, Uchida A, van der Spek PJ, Eker AP, Rademakers S, Visser C, Aboussekhra A, Wood RD, Hanaoka F, Bootsma D, Hoeijmakers JH. Two human homologs of Rad23 are functionally interchangeable in complex formation and stimulation of XPC repair activity. Mol Cell Biol. 1997;17:6924–6931. doi: 10.1128/mcb.17.12.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batty D, Rapic’-Otrin V, Levine AS, Wood RD. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J Mol Biol. 2000;300:275–290. doi: 10.1006/jmbi.2000.3857. [DOI] [PubMed] [Google Scholar]
- 11.Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD. Nucleotide excision repair of DNA with recombinant human proteins: Definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 2000;14:349–359. [PMC free article] [PubMed] [Google Scholar]
- 12.Evans E, Moggs JG, Hwang JR, Egly J, Wood RD. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997;35:2157–2167. doi: 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003;22:5293–5303. doi: 10.1093/emboj/cdg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 15.Tapias A, Auriol J, Forget D, Enzlin JH, Scharer OD, Coin F, Coulombe B, Egly JM. Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J Biol Chem. 2004;279:19074–19083. doi: 10.1074/jbc.M312611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thoma BS, Vasquez KM. Critical DNA damage recognition functions of XPC-hHR23B and XPA-RPA in nucleotide excision repair. Mol Carcinog. 2003;38:1–13. doi: 10.1002/mc.10143. [DOI] [PubMed] [Google Scholar]
- 17.Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15:507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reardon JT, Mu D, Sancar A. Overproduction, purification, and characterization of the XPC subunit of the human DNA repair excision nuclease. J Biol Chem. 1996;271:19451–19456. doi: 10.1074/jbc.271.32.19451. [DOI] [PubMed] [Google Scholar]
- 19.Manley JL, Fire A, Cano A, Sharp PA, Gefter ML. DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc Natl Acad Sci USA. 1980;77:3855–3859. doi: 10.1073/pnas.77.7.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrews BJ, Turchi JJ. Development of a high-throughput screen for inhibitors of replication protein A and its role in nucleotide excision repair. Mol Cancer Ther. 2004;3:385–391. [PubMed] [Google Scholar]
- 21.Hey T, Lipps G, Sugasawa K, Iwai S, Hanaoka F, Krauss G. The XPC–HR23B complex displays high affinity and specificity for damaged DNA in a true-equilibrium fluorescence assay. Biochemistry. 2002;41:6583–6587. doi: 10.1021/bi012202t. [DOI] [PubMed] [Google Scholar]
- 22.Patrick SM, Turchi JJ. Stopped-flow kinetic analysis of replication protein A-binding DNA: Damage recognition and affinity for single-stranded DNA reveal differential contributions of kon and koff rate constants. J Biol Chem. 2001;276:22630–22637. doi: 10.1074/jbc.M010314200. [DOI] [PubMed] [Google Scholar]
- 23.Patrick SM, Turchi JJ. Xeroderma pigmentosum complementation group A protein (XPA) modulates RPA-DNA interactions via enhanced complex stability and inhibition of strand separation activity. J Biol Chem. 2002;277:16096–16101. doi: 10.1074/jbc.M200816200. [DOI] [PubMed] [Google Scholar]
- 24.Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature. 1995;377:649–652. doi: 10.1038/377649a0. [DOI] [PubMed] [Google Scholar]
- 25.Anin MF, Leng M. Distortions induced in double-stranded oligonucleotides by the binding of cis- or trans-diammine-dichloroplatinum(II) to the d(GTG) sequence. Nucleic Acids Res. 1990;18:4395–4400. doi: 10.1093/nar/18.15.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kozlov AG, Lohman TM. Stopped-flow studies of the kinetics of single-stranded DNA binding and wrapping around the Escherichia coli SSB tetramer. Biochemistry. 2002;41:6032–6044. doi: 10.1021/bi020122z. [DOI] [PubMed] [Google Scholar]
- 27.Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc Natl Acad Sci USA. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamble DB, Mu D, Reardon JT, Sancar A, Lippard SJ. Repair of cisplatin–DNA adducts by the mammalian excision nuclease. Biochemistry. 1996;35:10004–10013. doi: 10.1021/bi960453+. [DOI] [PubMed] [Google Scholar]
- 29.Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JHJ, van Driel R, van Zeeland AA, Mullenders LHF. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–224. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 30.Reardon JT, Sancar A. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 2003;17:2539–2551. doi: 10.1101/gad.1131003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang QE, Zhu Q, Wani MA, Wani G, Chen J, Wani AA. Tumor suppressor p53 dependent recruitment of nucleotide excision repair factors XPC and TFIIH to DNA damage. DNA Repair. 2003;2:483–499. doi: 10.1016/s1568-7864(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 32.Sugasawa K, Shimizu Y, Iwai S, Hanaoka F. A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair. 2002;1:95–107. doi: 10.1016/s1568-7864(01)00008-8. [DOI] [PubMed] [Google Scholar]
- 33.Tang J, Chu G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair. 2002;1:601–616. doi: 10.1016/s1568-7864(02)00052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patrick SM, Turchi JJ. Replication Protein A (RPA) Binding to Duplex Cisplatin-damaged DNA Is Mediated through the Generation of Single-stranded DNA. J Biol Chem. 1999;274:14972–14978. doi: 10.1074/jbc.274.21.14972. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Liu YY, Yang ZG, Utzat C, Wang GZ, Basu AK, Zou Y. Cooperative interaction of human XPA stabilizes and enhances specific binding of XPA to DNA damage. Biochemistry. 2005;44:7361–7368. doi: 10.1021/bi047598y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rademakers S, Volker M, Hoogstraten D, Nigg AL, Mone MJ, van Zeeland AA, Hoeijmakers JHJ, Houtsmuller AB, Vermeulen W. Xeroderma pigmentosum group A protein loads as a separate factor onto DNA lesions. Mol Cell Biol. 2003;23:5755–5767. doi: 10.1128/MCB.23.16.5755-5767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hess MT, Schwitter U, Petretta M, Giese B, Naegeli H. Bipartite substrate discrimination by human nucleotide excision repair. Proc Natl Acad Sci USA. 1997;94:6664–6669. doi: 10.1073/pnas.94.13.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]