

Abstract
Ataxia telangiectasia mutated (ATM) is a PI3-kinase-like kinase (PIKK) associated with DNA double-strand break (DSB) repair and cell cycle control. We have previously reported comparable efficiencies of DSB repair in nuclear extracts from both ATM deficient (A-T) and control (ATM+) cells; however, the repair products from the A-T nuclear extracts contained deletions encompassing longer stretches of DNA compared to controls. These deletions appeared to result from end-joining at sites of microhomology. These data suggest that ATM hinders error-prone repair pathways that depend on degradation of DNA ends at a break. Such degradation may account for the longer deletions we formerly observed in A-T cell extracts. To address this possibility we assessed the degradation of DNA duplex substrates in A-T and control nuclear extracts under DSB repair conditions. We observed a marked shift in signal intensity from full-length products to shorter products in A-T nuclear extracts, and addition of purified ATM to A-T nuclear extracts restored full-length product detection. This repression of degradation by ATM was both ATP-dependent and inhibited by the PIKK inhibitors wortmannin and caffeine. Addition of pre-phosphorylated ATM to an A-T nuclear extract in the presence of PIKK inhibitors was insufficient in repressing degradation, indicating that kinase activities are required. These results demonstrate a role for ATM in preventing the degradation of DNA ends possibly through repressing nucleases implicated in microhomology-mediated end-joining.
Keywords: ATM, DNA degradation, Double-strand break repair, Microhomology-mediated, end-joining, PI-3-kinase-like kinases
1. Introduction
Preserving genomic integrity is critical to the vitality of an organism and the continuity of any species. The gravity of this task is perhaps best reflected in the number of pathways and mediators involved in maintaining the genetic code and the fidelity of its perpetuation. The repair of a DNA double-strand break (DSB) is one facet of the genomic maintenance tale with one key player being the ataxia telangiectasia mutated (ATM) protein. An ATM deficiency results in ataxia telangiectasia, a neurodegenerative disorder accompanied by immunological malfunctions and a propensity for cancer development. ATM, a PI3-kinase-like kinase (PIKK), is present in the nucleus in the form of inactive dimers and oligomers that undergo trans-autophosphorylation and dissociate upon DSB occurrence. Activated ATM then modulates the activity of a plethora of proteins involved in repair and cell cycle control [1–3]. Although a role for ATM in DSB repair and cell cycle regulation is well documented, the particular defect in DNA repair emanating from an ATM dysfunction is not well characterized.
We have previously reported comparable DSB repair efficiencies in A-T and control nuclear extracts [4]. The fidelity of repair, however, was defective in the A-T nuclear extracts. To demonstrate this we assessed the repair of a circular plasmid linearized with a restriction enzyme-induced DSB. Both A-T and control nuclear extracts had equivalent potentials of repairing a DSB and rejoining the plasmid. On the other hand, the mutation frequency was significantly higher in A-T nuclear extracts than in controls. A number of mutant plasmids generated from these experiments were sequenced and all revealed deletions spanning the repaired DSB site. Small sequences of microhomology (one to six nucleotide repeats) were involved in 95% of the deletion events. That is, rejoining occurred at sequences of microhomology that flanked both ends of the break more often than random expectation. Deletion stretches were longer in A-T than in control extracts. The repair fidelity of blunt-end DSBs and those with short (<4 nt) overhangs was significantly less in A-T than in control nuclear extracts. Differences in the fidelity of repairing DSBs with 4 nt overhangs were not statistically significant. This data indicated a potential role for ATM in repressing degradation at DSB ends thereby preventing error-prone repair.
We report here a greater extent of degradation of DNA ends in A-T than in control nuclear extracts. Degradation levels declined when purified ATM was added into repair reactions with an A-T nuclear extract background. Prevention of DNA end-degradation was ATP-dependent and was inhibited by the PIKK inhibitors wortmannin and caffeine. Addition of pre-phosphorylated ATM in the presence of PIKK inhibitors did not repress DNA end-degradation in an A-T nuclear extract. This excessive DNA end-degradation in nuclear extracts from A-T cells probably accounts for the longer deletion mutations and repair defects we observed in our previous study.
2. Materials and methods
2.1. Cell culture
Cell lines AT5BIVA, GM16666 and GM16667 were obtained from the Coriell Cell Repository (Coriell Institute of Medical Research, Camden, NJ). The WI-38VA13 cell line was obtained from ATCC (American Type Culture Collection, Manassas, VA). AT5BIVA is a SV40-transformed fibroblast cell line derived from a patient afflicted with ataxia telangiectasia. WI-38VA13 is a SV-40 transformed lung fibroblast line used as an ATM positive control for AT5BIVA. GM16666 and GM16667 are matched lines derived from the AT22IJE-T A-T cell line which was transfected with either an ATM expression construct (GM16667) or an empty vector (GM16666) and maintained under hygromycin selection to generate A-T-corrected and A-T stable cell lines [5].
All cells lines were grown at 37 °C in 5% CO2 in Dulbecco’s modified Eagle medium (GIBCO, Invitrogen Corporation, Carlsbad, CA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 100 U/ml penicillin, and 100 μg/ml streptomycin (penicillin–streptomycin–glutamine, GIBCO, Invitrogen Corporation, Carlsbad, CA). Medium for both GM16666 and GM16667 additionally contained 100 μg/ml hygromycin (Invitrogen Corporation, Carlsbad, CA) to maintain stable cell line selection.
2.2. Nuclear extract preparation
Cells grown to 80% confluency in 250 mm2 tissue culture flasks were washed three times with 20 ml of ice cold hypotonic buffer (0.25 mM EDTA pH 7.4, 0.2 mM PMSF, 0.5 mM DTT), collected using a cell lifter (Fisher Scientific Co., NJ) and centrifuged at 1850 × g for 10 min. Cells were resuspended in five times the pellet volume of hypotonic buffer and incubated for 30 min at 4 °C. Cells were then collected by centrifugation at 1850 × g for 30 min and intact nuclei were released using a Dounce homogenizer using a loose fitting (Type B) pestle. Following concentration by centrifugation at 3300 × g for 30 min, nuclei were resuspended in one-half the packed nuclear volume of resuspension buffer (20 mM HEPES pH 7.9, 25% glycerol, 1.5 mM MgCl2, 0.02 M KCl, 0.2 mM EDTA, 0.2 mM PMSF, 0.5 mM DTT). Nuclear lysis buffer (resuspension buffer with a final concentration of 1.2 M KCl) equivalent to one-half the packed nuclear volume was then added. Nuclei were incubated for 30 min at 4 °C and subjected to three cycles of snap-freezing in liquid nitrogen and rapid thawing at 37 °C. After lysis by Dounce homogenization, nuclear lysates were centrifuged at 25,000 × g for 30 min and the supernatant was dialyzed for 18 h at 4 °C against dialysis buffer (50 mM Tris–Cl pH 7.5, 0.1 mM EDTA, 10 mM 2-mercaptoethanol, 0.1 mM PMSF, 10% glycerol). Aliquots of the samples were snap-frozen in liquid nitrogen and stored at −80 °C. The protein concentration of the nuclear extracts was determined by the Bradford protein assay using the Bradford reagent (Sigma, St. Louis, MO) and BSA as a standard.
2.3. Purification of ATM
The purification of ATM was based on the procedure of Goodarzi and Lees-Miller [6]. HeLa cells (6 l) were grown to log phase (4 × 105 cells/ml) and collected by sedimentation at 10,000 × g for 15 min at 4 °C. The resulting cell pellet was washed twice with 10 ml low salt buffer (10 mM HEPES pH 7.4, 25 mM KCl, 10 mM NaCl, 1 mM MgCl2, and 0.1 mM EDTA). The cells were collected and resuspended in 7 ml of high salt buffer (50 mM Tris–Cl pH 8.0, 5% glycerol, 1 mM EDTA, 10 mM MgCl2, 400 mM KCl). This buffer and all subsequent buffers were supplemented with the protease inhibitors PMSF (0.1 mM), leupeptin (1 μg/ml) and pepstatin (1 μg/ml). After disruption using a Dounce homogenizer; the lysate was centrifuged at 10,000 × g for 30 min and the supernatant (S1) was saved. The pellet was extracted with 3 ml of high salt buffer and centrifuged generating a second supernatant (S2). S1 and S2 were combined (termed P10) and immediately diluted with TB buffer (50 mM Tris, 5% glycerol, 0.2 mM EDTA, 1 mM DTT) to a final conductivity equal to 75 mM KCl.
P10 (170 mg) was applied onto a 10 ml DEAE-Sepharose fast flow (GE Healthcare, Princeton, NJ) column equilibrated in TB–75 mM KCl at a rate of 2 ml/min. After the column was washed with 10-column volumes of TB–75 mM KCl, bound protein including ATM was eluted with 5-column volumes of TB–200 mM KCl. The eluted protein (32 mg) was pooled, immediately diluted to a conductivity equal to 75 mM KCl, and applied to a 5 ml SP-Sepharose fast flow column (Amersham Pharmacia Biotech, Piscataway, NJ). Again the column was washed with 10-column volumes of TB–75 mM KCl, and eluted with 5-column volumes of TB–200 mM KCl. The eluted protein (3.5 mg) containing ATM was diluted in TB buffer to a conductivity equal to 125 mM KCl and applied onto a 0.5 ml single-strand DNA-cellulose column (Sigma, St. Louis, MO) at 0.2 ml/min.
The flow-through fraction (2 mg), containing the majority of the ATM protein, was collected, diluted with TB buffer to a conductivity equal to 100 mM KCl and loaded onto a 2 ml Macroprep-Q column (Bio-Rad Laboratories, Hercules, CA) equilibrated in TB–100 mM KCl. Protein was eluted with a 15 ml linear salt gradient from 0.1 to 1 M KCl at 0.5 ml/min. Fractions containing ATM were pooled (0.3 mg) and stored at −80 °C. Fractions containing ATM were identified by SDS-PAGE. Protein concentration was determined by the Bradford assay using BSA as a standard.
2.4. Western immunoblotting
Samples (20 μg of nuclear extract or 1 μg of the purified ATM preparation) were incubated at 100 °C for 5 min in Laemmli sample buffer and then electrophoresed on 6% (for DNA-PKcs, ATR and ATM) or 12% (for Ku80, Mre11, Ku70 and RPA2) denaturing-polyacrylamide gels. Proteins were transferred to Trans-Blot Medium nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA), probed and then visualized with the SuperSignal West Dura Extended Duration Substrate (Pierce Biotechnology, Inc., Milwaukee, WI). The FluorChem system (Alpha Innotech Corporation, San Leandro, CA) was used for gel documentation. The DNA-PKcs (1:10,000), ATM (1:1000), Ku80 (1:10,000), Ku70 (1:10,000) and Mre11 (1:40,000) primary antibodies were obtained from Abcam, Inc. (Cambridge, MA). The ATR (1:10,000) primary antibody was from Novus Biologicals, Inc. (Littleton, CO) while the RPA2 (1:10,000) primary antibody was from Bethyl, Inc. (Montgomery, TX).
2.5. Autophosphorylation of ATM
To pre-phosphorylate ATM, 0.34 pmol of purified ATM were incubated with 0.83 pmol of ATP or [γ-32P]ATP in 15 μl phosphorylation buffer (20 mM Tris–Cl pH 7.5, 20 mM MgCl2, 10 mM MnCl2, 1 μM fostriecin).
2.6. Duplex oligonucleotide substrates
A series of duplex DNA oligonucleotide substrates (Integrated DNA Technologies, Inc., IA) were generated and used to measure degradation of DNA ends in different cellular extracts (Table 1). A 71 nt oligonucleotide (Template/5′Cy3 Template) was hybridized to a Top Strand of variable lengths resulting in substrates with different 5′-end overhangs or a blunt end. Alternatively, where indicated, a 45 nt Template was hybridized to a 50 nt 3′Cy3Sp Top Strand. Template (0.9 nmol) and Top Strand (0.9 nmol) oligonucleotides were incubated in 100 μl of hybridization buffer (10 mM Tris–Cl pH 7.9, 50 mM NaCl and 10 mM MgCl2) for 10 min at 100 °C and then slowly cooled to 25 °C. The resulting substrates had either a blunt end or 5′-end overhang corresponding to 5′AATTC, 5′TAGC, 5′CGCG, 5′TAT, or 5′CG. Assays were designed to examine degradation at the overhang end of the duplexes; therefore, the final six bases at the 3′-end of each Top Strand were linked with phosphorothioate linkages to prevent nuclease digestion. Similarly, the first six nucleotides at the 5′-end of the Template were linked by phosphorothioate linkages for the same purpose. In addition, a 5′Cy3-labeled 71 nt Template protected from nuclease digestion by phosphorothioate linkages at its 5′-end was used to measure the 3′-end-degradation of the non-overhang-presenting strand in the duplex.
Table 1.
Oligonucleotides used to assess DNA degradation
| Name | Overhang | Length (nt) | Sequence |
|---|---|---|---|
| Top Strand | 5′AATTC | 76 | 5′AATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCACTGGCCGTCGTTTTAC |
| 5′TAGC | 75 | 5′TAGCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCACTGGCCGTCGTTTTAC | |
| 5′CGCG | 75 | 5′CGCGGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCACTGGCCGTCGTTTTAC | |
| 5′TAT | 74 | 5′TATGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCACTGGCCGTCGTTTTAC | |
| 5′CG | 73 | 5′CGGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCACTGGCCGTCGTTTTAC | |
| Blunt | 71 | 5′GAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCACTGGCCGTCGTTTTAC | |
| Template | 71 | 3′CTCGAGCCATGGGCCCCTAGGAGATCTCAGCTGGACGTCCGTACGTTCGAACCGTGACCGGCAGCAAAATG | |
| Cy3 Template | 71 | 3′CTCGAGCCATGGGCCCCTAGGAGATCTCAGCTGGACGTCCGTACGTTCGAACCGTGACCGGCAGCAAAATG/Cy3 | |
| Cy3Sp Top Strand | 5′AATTC | 50 | 5′AATTCAGTCGACCTGCAGGCATGCAAGCTTGGCACTGGCCGTCGTTTTAC/Cy3Sp |
| 45 nt Template | 45 | 3′TCAGCTGGACGTCCGTACGTTCGAACCGTGACCGGCAGCAAAATG | |
| Extension primer | 20 | 3′CCGTGACCGGCAGCAAAATG/Cy3 |
Underlined nucleotides are unpaired in the final substrate. Bold nucleotides are linked by phosphorothioate bonds.
2.7. DNA end processing assay
Measurement of DNA end-protection was accomplished by incubating the oligonucleotide substrates defined above in control or A-T extracts, followed by DNA extraction and primer extension to detect the length of DNA products. The in vitro assay conditions simulated those used for DNA DSB repair. Reactions (50 μl) containing 50 μg of nuclear extract and 90 pmol of a DNA duplex in reaction buffer (65.5 mM Tris–Cl pH 7.5, 10 mM MgSO4, 10 mM MnSO4, 91 nM EDTA, 9.1% glycerol) were assembled on ice and then incubated for 10 min at 30 °C. Reaction buffer was supplemented with Complete, Mini, EDTA-free Protease Inhibitor Cocktail (Roche Diagnostics GmbH, Mannheim, Germany) used according to the manufacturer’s instructions. Reactions were stopped by adding 50 μl of phenol. Where indicated in the text, ATP (1 mM), the phosphatase inhibitor fostriecin (1 μM) and the PIKK inhibitors wortmannin (13 nM) and caffeine (71.85 μM) were included in the assay. When used, purified ATM or pre-phosphorylated purified ATM was incorporated into reactions containing AT nuclear extracts as indicated in the text. The DNA duplex was recovered from the assay reactions by phenol phase separation and subsequent ethanol precipitation with 120 μg of glycogen (Fermentas, Hanover, MD) and 10 μl of 3 M sodium acetate pH 5.2.
2.8. Primer extension assay
The lengths of the Top Strands of DNA duplexes retrieved from the end processing reactions were determined by a primer extension assay using a 5′Cy3-labeled extension primer (Table 1). This primer anneals to the 3′-end of Top Strands used to generate the DNA duplexes. Reactions (20 μl) contained total DNA extracted from the end processing reactions, 12.3 pmol of the extension primer and 0.5 units of Taq DNA polymerase (Roche Diagnostics GmbH, Mannheim, Germany) in extension assay buffer (200 μM of each dNTP, 50 mM Tris–Cl pH 8.3, 10 mM KCl, 5 mM (NH4)2SO4, and 2 mM MgCl2). The population of Top Strands was amplified by PCR in an Eppendorf Mastercycler Gradient (Eppendorf AG, Hamburg, Germany) thermocycler. Following an initial denaturation step at 94 °C for 20 min, reactions were incubated for five cycles of 1 min at 94 °C, 1 min at 58 °C and 1 min at 72 °C with a final extension at 72 °C for 10 min. The 20 μl extension reactions were stopped by the addition of 5 μl formamide buffer (95% formamide, 10 mM EDTA pH 7.6, 0.1% xylene cyanol, 0.1% bromophenol blue), heated to 95 °C for 10 min and then brought down to room temperature prior to product analysis.
2.9. Product analysis
Products from primer extension reactions and from end-processing assays employing a 5′Cy3-labeled Template were separated on 12% acrylamide/7 M urea sequencing gels (Sequagel Sequencing System reagents, National Diagnostics, Atlanta, GA). Reaction products were visualized using a Typhoon 9410 Variable Mode Imager (GE Healthcare, Princeton, NJ) and analyzed using ImageQuant v5.2 software (Molecular Dynamics, Amersham Biosciences, Princeton, NJ). Product intensities were determined, corrected for background and then converted into percent intensities where percent intensity = (product intensity/total lane intensity) × 100.
3. Results
We have previously reported a decrease in the fidelity of DSB repair in A-T nuclear extracts when compared to controls [4]. The most prominent type of mutations observed were deletion events associated with sites of microhomology flanking a break. The deletions encompassed one of the two sites of microhomology in addition to the region between the two sites. To assess whether these events were the result of DNA end-degradation, we employed an in vitro system that simulates DSB repair conditions (Fig. 1). This system was used to assess the role of ATM in repressing degradation at DSB ends.
Fig. 1.
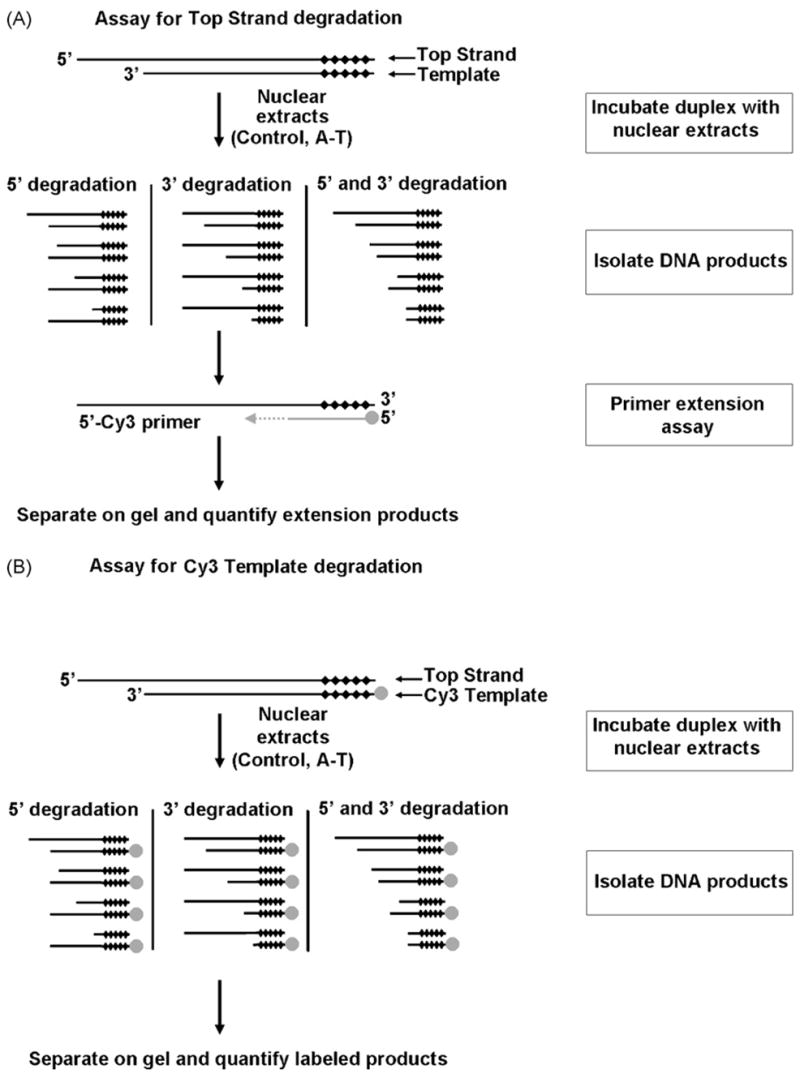
Assays used to measure degradation of DNA strands in a duplex incubated with control or A-T nuclear extracts under DNA double-strand break repair conditions. (A) PCR-based primer extension assay for measuring degradation of the overhang (top) strand. (B) Assay for measuring degradation of the 3′-recessed (Template) strand. Black diamonds (◆) represent nuclease-resistant phosphorothioate linkages while gray circles (●) a 5′Cy3 label. See text for details.
3.1. Increased levels of DNA end-degradation in A-T nuclear extracts
We used DNA duplex substrates with a single nuclease-susceptible end in an in vitro DSB repair reaction. Substrates were designed to limit degradation to the 5′-end of the overhang-presenting strand and the 3′-end of the 3′-recessed strand, here forth referred to as the Top Strand and the Template, respectively. DNA was extracted from the repair reactions after incubation with the extracts and subjected to a primer extension assay that allowed examination of degradation levels of the Top Strand (Fig. 1A). The extension assay employed a 5′Cy3-labeled primer that annealed to the 3′-end of the Top Strand. The inclusion of phosphorothioate linkages at the blunt end of the duplex prevented nuclease-mediated degradation of the primer annealing site on the Top Strand.
The potential role of ATM in repressing DNA end-degradation was tested using a substrate harboring a 5′AATTC overhang. The 5′AATTC substrate was incubated with A-T or control nuclear extracts under in vitro DSB repair conditions. The AT5BIVA and GM16666 cell lines were used as sources of A-T nuclear extracts whereas the WI-38VA13 and GM16667 cell lines were used as their respective controls (Fig. 2A). The expected length of the product obtained from a fully extended non-degraded strand was 76 nt. Extension products were clustered into four groups for quantification purposes: full-length, long, medium-sized, short and un-extended primer. Product intensities were determined, corrected for background and then converted into percent intensities where percent intensity = (product intensity/total lane intensity) × 100. Intensities of the full-length product from the WI-38VA13 and GM16667 control nuclear extracts were 22 and 13%, respectively. In comparison, the intensities of the full-length product retrieved from the AT5BIVA and GM16666 A-T nuclear extracts were both 1% (Table 2A). Hence, an elevated level of degradation of DNA ends is detected in both types of A-T nuclear extracts; this is strongly indicated by an approximate 10-fold decrease in full-length product intensities. The shift in intensity from the full-length product in the A-T extracts was mostly towards the un-extended primer.
Fig. 2.
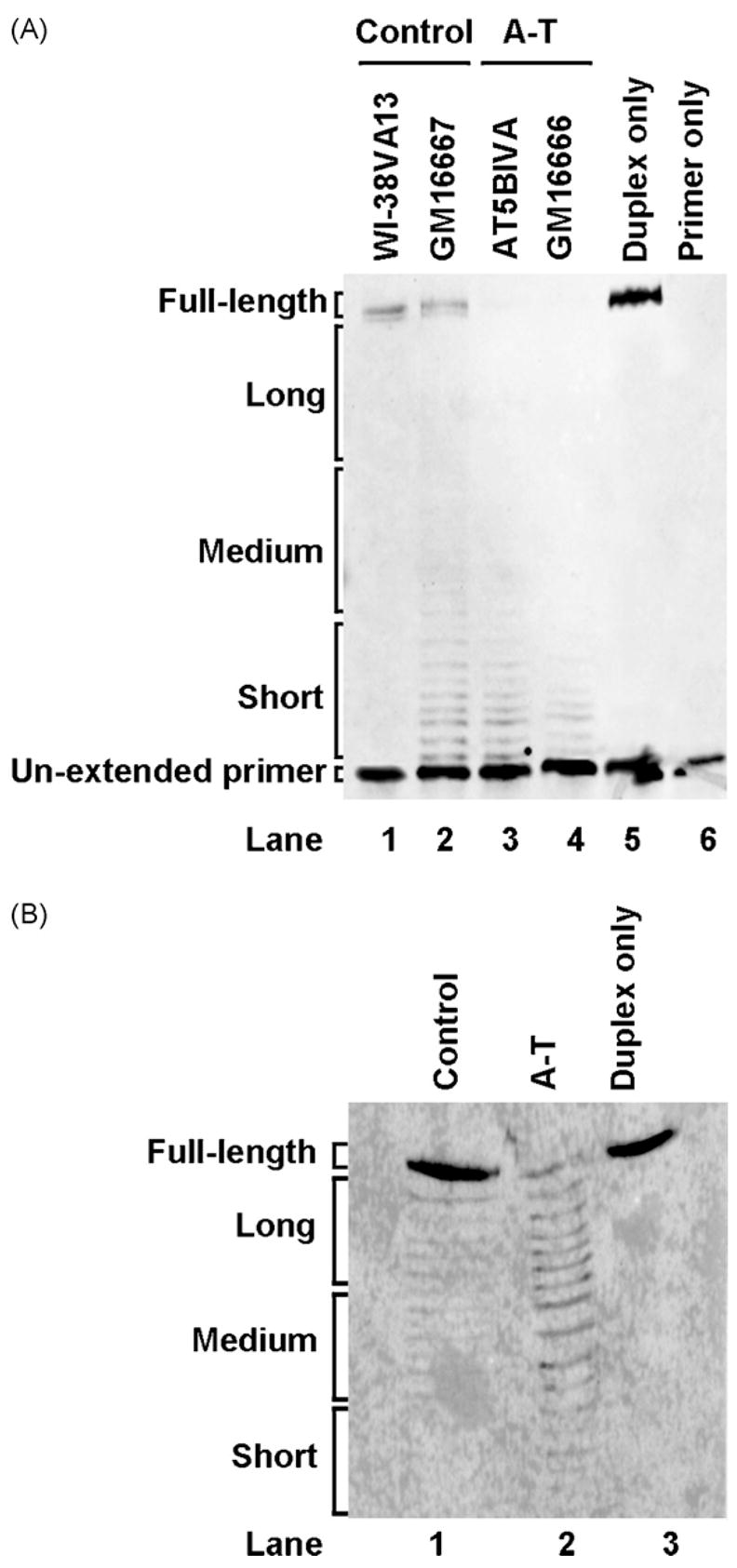
A-T nuclear extracts show increased DNA end-degradation. (A) Top Strand primer extension products following gel separation. The 5′AATTC substrate was incubated with control (WI-38VA13, GM16667) or A-T (AT5BIVA, GM16666) nuclear extracts, isolated and then subjected to a primer extension assay as described in Section 2. The duplex (lane 5) and the 5′Cy3-labeled primer (lane 6) were incubated in absence of nuclear extract as controls. (B) 5′Cy3-labeled Template degradation products following gel separation. The 5′AATTC substrate with a 5′Cy3-labeled Template strand was first incubated with WI-38VA13 (control) and AT5BIVA (A-T) nuclear extracts and then extracted from the reaction. Reactions with duplex only (lane 3) in the absence of nuclear extract were included as controls.
Table 2.
Percent intensity of end-processing assay products using a 5′AATTC substrate
| Nuclear extract
|
Duplex only | Primer only | ||||
|---|---|---|---|---|---|---|
| Control
|
A-T
|
|||||
| WI-38VA13 | GM16667 | AT5BIVA | GM16666 | |||
| (A) Top Strand primer extension products | ||||||
| Product | ||||||
| Full-length | 22 | 13 | 1 | 1 | 44 | 2 |
| Long | 6 | 8 | 0 | 2 | 5 | 5 |
| Medium | 6 | 13 | 13 | 0 | 0 | 0 |
| Short | 9 | 19 | 10 | 2 | 0 | 10 |
| Un-extended primer | 57 | 47 | 76 | 95 | 51 | 83 |
| Lane (Fig. 2A) | 1 | 2 | 3 | 4 | 5 | 6 |
| Nuclear extract
|
Duplex only | |||||
| Control WI-38VA13 | A-T AT5BIVA | |||||
|
| ||||||
| (B) 5′Cy3 Template degradation products | ||||||
| Product | ||||||
| Full-length | 73 | 9 | 93 | |||
| Long | 12 | 42 | 7 | |||
| Medium | 15 | 38 | 0 | |||
| Short | 0 | 11 | 0 | |||
| Lane (Fig. 2B) | 1 | 2 | 3 | |||
In parallel with the reactions described above, the duplex and the labeled primer were incubated under repair reaction conditions in absence of nuclear extract, subjected to DNA extraction and then the primer extension assay (Fig. 2A, lanes 5 and 6). This was performed to ensure that the repair buffer, the DNA extraction and the primer extension procedures did not bias the results by affecting degradation or by adding background signal.
Since the chemistry of the primer extension assay only allows for examination of the Top Strand, a different strategy was employed to assess the degradation of the 3′-end of the Template (Fig. 1B). Duplex substrates contained a Template labeled itself with a 5′Cy3 moiety. Following incubation with nuclear extracts, products were isolated, separated on a gel and then quantified.
A 5′AATTC substrate with a 5′Cy3-labeled Template was incubated with A-T and control extracts as described above for Fig. 2A. Subsequent to incubation with WI-38VA13 and AT5BIVA nuclear extracts, the duplex was extracted, products were separated (Fig. 2B) and then quantified (Table 2B). In addition, the duplex substrate was incubated under repair reaction conditions in the absence of nuclear extract as a control (Fig. 2B, lane 3). Intensity of the full-length labeled Template retrieved from the control nuclear extract was 73% of the total intensity whereas it was 9% in the A-T nuclear extract. Hence, degradation of both strands in the duplex was elevated in A-T extracts.
To validate the primer extension assay described above and utilized in subsequent experiments, we assessed the degradation of a Top Strand labeled itself at the 3′-end with a Cy3 moiety and incorporated into a 5′AATTC duplex (Fig. 3A). This substrate was incubated under repair conditions in control and A-T nuclear extracts. Products were retrieved, gel-separated and then analyzed. As observed with the primer extension assay, an increase in Top Strand degradation in A-T nuclear extracts was observed over controls (Fig. 3B). Therefore, both assay systems revealed comparable results.
Fig. 3.
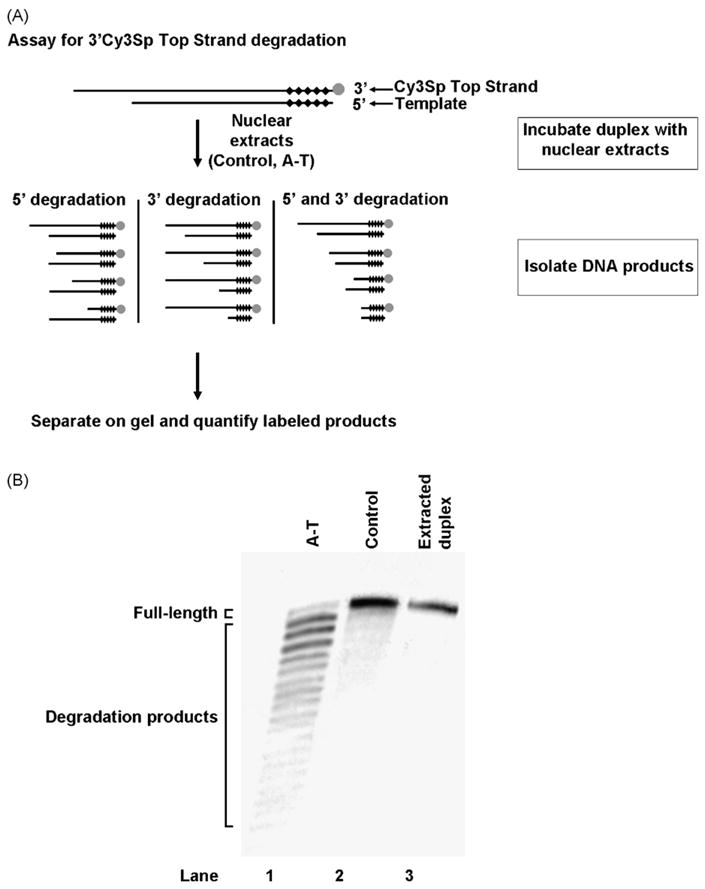
Validation of the primer extension assay used for assessment of Top Strand degradation. (A) Assay for measuring degradation of a 3′Cy3Sp-labeled Top Strand. Black diamonds (◆) represent nuclease-resistant phosphorothioate linkages while gray circles (●) a 3′Cy3Sp label. (B) 3′Cy3Sp-labeled Top Strand degradation products following gel separation. A 5′AATTC substrate with a 3′Cy3Sp-labeled Top Strand was first incubated with WI-38VA13 (control) and AT5BIVA (A-T) nuclear extracts and then extracted from the reaction. Reactions with duplex only (lane 3) in the absence of nuclear extract were included as controls.
3.2. Repression of degradation of various types of DNA ends in control nuclear extracts
To examine whether the length and the sequence of the overhang affects degradation and protection activities, we used various duplex substrates in our in vitro repair system (Table 1). DNA duplexes tested had one blunt end protected from degradation by phosphorothioate linkages and a 5′ overhang-presenting end. Overhang sequences assessed were 5′TAGC, 5′CGCG, 5′TAT, and 5′CG. We also tested a duplex with one blunt end vulnerable to degradation and another protected by phosphorothioate linkages.
These DNA substrates were incubated with control or AT nuclear extracts under appropriate DSB repair conditions. DNA duplexes were then extracted and subjected to primer extension for the Top Strand population retrieved as described in Section 2. Marked degradation in A-T nuclear extracts was observed for the different substrates tested (Fig. 4A). A decrease of around 10-fold in full-length product intensity was observed in A-T nuclear extracts when compared to controls (Fig. 4B). Average intensities of the full-length extension products for the substrates tested ranged from 12 to 19% in the control nuclear extracts. In comparison, their intensities in the A-T nuclear extracts were all less than 1%. The shift in intensity was again mostly towards the un-extended primer. Despite minor variability in the degradation trends observed for the various substrates, the data presented consistently demonstrate enhanced DNA end-protection in control extracts over A-T extracts (compare lanes 1, 3, 5, 7, and 9 to lanes 2, 4, 6, 8, and 10 in Fig. 4A). This protection is also independent of the nature of the DNA end.
Fig. 4.
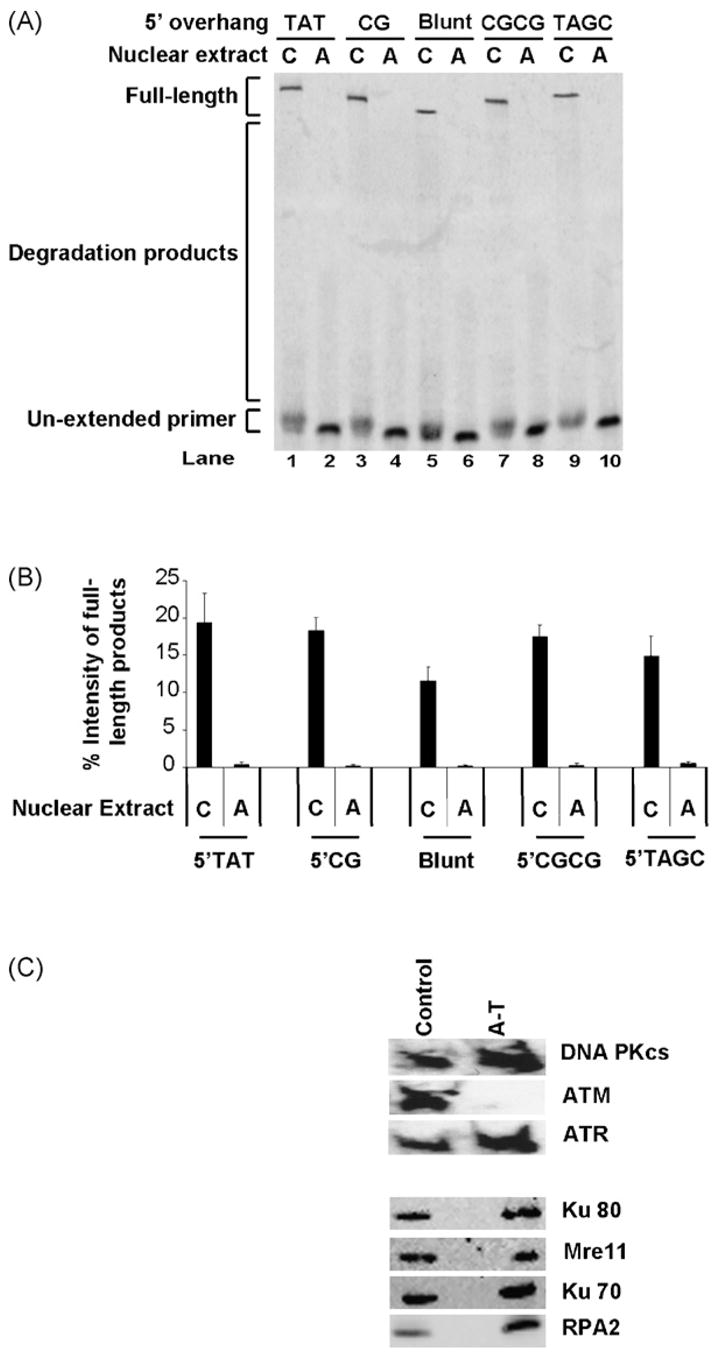
Increased DNA end-degradation in A-T nuclear extracts is independent of overhang sequence and length. (A) Extension products of Top Strands in various duplexes following gel separation. Substrates with different types of overhangs were first incubated with WI-38VA13 (C) or AT5BIVA (A) nuclear extracts, isolated and then subjected to a primer extension assay. (B) Full-length products were quantified for indicated overhangs and expressed as percent intensity (% intensity = (product intensity/total intensity) × 100). Data were generated from three independent experiments. (C) Western immunoblots for key double-strand break repair proteins in WI-38VA13 (C) and AT5BIVA (A) nuclear extracts. Nuclear extracts (20 μg) were separated by SDS-PAGE, transferred to PVDF membranes and incubated with various antibodies to detect indicated proteins as described in Section 2.
Since we made extensive use of the WI-38VA13 (control) and AT5BIVA (A-T) nuclear extracts in this and all subsequent experiments, we ensured that levels of key DSB repair proteins, besides ATM, were relatively equivalent in both types of extracts (Fig. 4C). Western immunoblotting for DNA-PKcs, ATR, Ku80, Mre11, Ku70 and RPA2 revealed comparable levels of these proteins in our nuclear extract preparations from both cell lines. We were unable to detect ATM in the AT5BIVA nuclear extracts.
3.3. ATP is required for prevention of end-degradation
To assess the ATP-requirement for the enhanced DNA end-stability phenomenon observed in the control extracts, we examined the degradation of the Top Strand in a duplex with a 5′AATTC overhang in the presence or absence of ATP (Fig. 5). In the presence of ATP, average intensities of the full-length product were 18 and 1% in WI-38VA13 (control) and in AT5BIVA (A-T) nuclear extracts, respectively (Table 3). Removing ATP from the repair reaction resulted in ablation of this difference; in ATP-deficient conditions both A-T and control extracts displayed a low intensity of the full-length product (<3%). Although we observed variations in the intensities of the long, medium-sized and short products generated by different control and A-T nuclear extract batches, the trend of elevated degradation in the A-T nuclear extracts was consistent. Moreover, ATP was required for hindering degradation in multiple independently prepared control nuclear extracts.
Fig. 5.
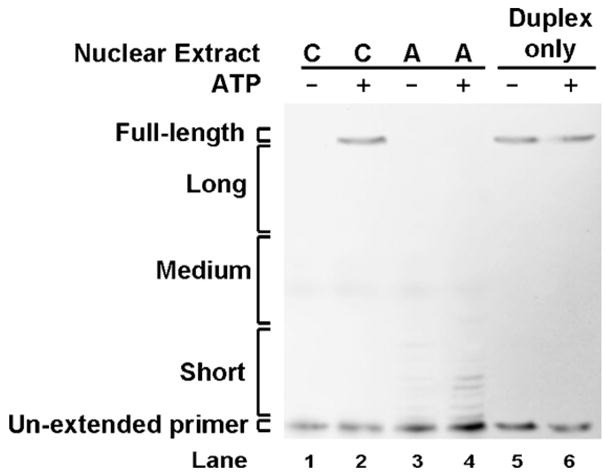
ATP is required for DNA end-protection. Top Strand primer extension products following gel separation. The 5′AATTC substrate was first incubated with WI-38VA13 (C) or AT5BIVA (A) nuclear extracts in the presence or absence of ATP, isolated and then subjected to a primer extension assay. The duplex (lanes 5 and 6) was also incubated with the reaction buffers (±ATP) in absence of nuclear extract as a control.
Table 3.
Percent intensity of primer extension products of the Top Strand in a 5′AATTC substrate in reactions with or without ATP
| Nuclear extract
|
Duplex only
|
|||||
|---|---|---|---|---|---|---|
| Control WI-38VA13
|
A-T AT5BIVA
|
− | + | |||
| ATP | − | + | − | + | ||
| Product | ||||||
| Full-length | 1 ± 1 | 18 ± 5 | 3 ± 1 | 1 ± 1 | 17 ± 3 | 18 ± 3 |
| Long | 3 ± 2 | 5 ± 4 | 3 ± 3 | 4 ± 4 | 6 ± 1 | 6 ± 2 |
| Medium | 4 ± 4 | 2 ± 3 | 0 ± 3 | 3 ± 3 | 4 ± 1 | 4 ± 3 |
| Short | 0 ± 0 | 0 ± 1 | 0 ± 0 | 3 ± 6 | 0 ± 0 | 0 ± 0 |
| Un-extended primer | 92 ± 5 | 75 ± 5 | 94 ± 1 | 89 ± 5 | 73 ± 4 | 72 ± 7 |
3.4. Addition of purified ATM to A-T nuclear extracts restores end-protection
We examined if addition of purified ATM would restore DNA end-protection to A-T nuclear extracts. Purified ATM was added to AT5BIVA (A-T) nuclear extracts and DNA end-degradation of the Top Strand in a duplex with a 5′AATTC overhang was assessed (Fig. 6A). The intensity of the full-length product detected in the absence of purified ATM in an A-T nuclear extract was 1.82% (Fig. 6A, lane 14). Addition of increasing amounts of purified ATM (Fig. 6A, lane 11 (0.05 nM), lane 12 (0.1 nM) and lane 13 (0.2 nM)) increased the amount of full-length product intensity (to 2.01, 10.78 and 28.45%, respectively). Full-length product intensity detected with 0.2 nM purified ATM was comparable to the 27.44% intensity detected in the WI-38VA13 (control) nuclear extract in this experiment (Fig. 6A, lane 15). Hence, a dose–response in protection from degradation was observed with increasing concentrations of ATM. The use of a reaction buffer lacking ATP eliminated the prevention of substrate degradation conferred by the purified ATM (Fig. 6A, lanes 5–7). This again demonstrates the dependency on ATP for repressing degradation. To ensure that our purified ATM preparation did not contain other DSB-associated PIKKs that may affect restoration of DNA end-protection we used immunoblotting to assay for DNA-PKcs and ATR (Fig. 6B); neither DNA-PKcs nor ATR was detected in the ATM preparation.
Fig. 6.
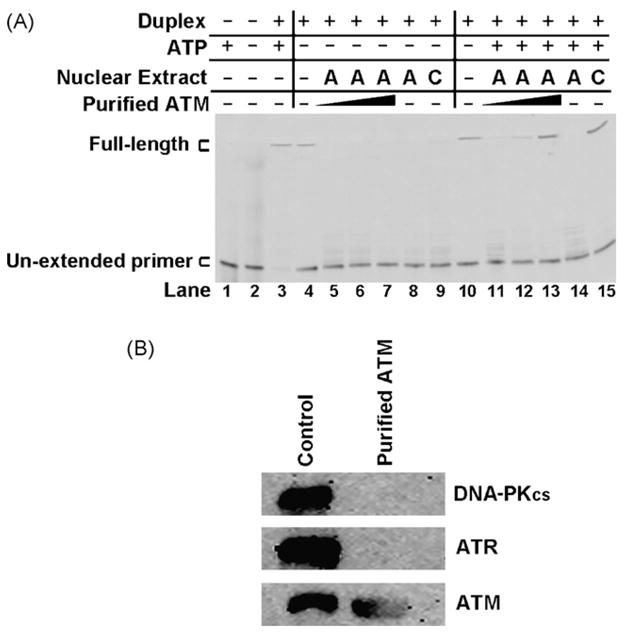
Purified ATM restores DNA end-protection to A-T nuclear extracts. (A) Top Strand primer extension products following gel separation. The 5′AATTC substrate was first incubated with WI-38VA13 (C) or AT5BIVA (A) nuclear extracts. Purified ATM was added to AT5BIVA extracts at 0.05, 0.1 and 0.2 nM (lanes 5–7 and 11–13). The 5′Cy3-labeled primer (lanes 1 and 2) and duplex (lanes 3 and 4) were also incubated with the reaction buffers (±ATP) as controls. Products were isolated and then subjected to a primer extension assay. (B) Western immunoblots for the DNA repair-associated PI-3-kinase-like kinases DNA-PKcs, ATR and ATM performed on the purified ATM preparation. WI-38VA13 (control) nuclear extract was used as a positive control. Purified protein (1 μg) and nuclear extract (20 μg) were separated by SDS-PAGE, transferred to PVDF membranes and incubated with various antibodies to detect indicated proteins as described in Section 2.
3.5. Caffeine and wortmannin inhibit end-protection
Prevention of end-degradation was ATP and ATM-dependent. With ATM being a PIKK kinase, we tested whether inhibition of its kinase activity would affect end-protection (Fig. 7). The PIKK inhibitors caffeine and wortmannin were added to the end processing reactions at concentrations previously shown to inhibit the kinase activity of ATM [6]. Both inhibitors were capable of abolishing the protective effects of 0.2 nM purified ATM (Fig. 7, lanes 2 and 3) and of the control nuclear extract (Fig. 7, lanes 4 and 5) in the presence of ATP. This was evident by the sharp decline in the intensity of full-length products (Fig. 7).
Fig. 7.
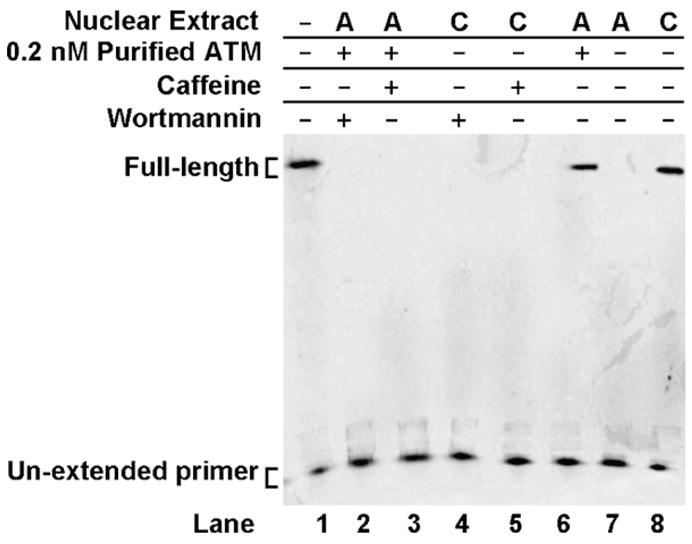
PI-3-kinase-like kinase inhibitors prevent DNA end-protection. Top Strand primer extension products following gel separation. The 5′AATTC substrate was first incubated with WI-38VA13 (C) or AT5BIVA (A) nuclear extracts. Purified ATM (0.2 nM), wortmannin or caffeine were incorporated into the reaction where indicated. Products were isolated and then subjected to a primer extension assay.
3.6. ATM autophosphorylation is not sufficient for end-protection
The dependency on ATP to repress degradation and the inhibition of this repression by wortmannin or caffeine reflects the requirement for kinase activity for DNA end-protection. This requirement could reflect a dependence on ATM autophosphorylation alone; or it could indicate the need for phosphorylation of a downstream substrate by ATM or by another component of the system.
Hence, to examine whether an ATM autophosphorylation event was sufficient to confer protection to DNA ends without the need for subsequent kinase activities, we incubated pre-phosphorylated purified ATM with a duplex presenting a 5′AATTC overhang in an A-T nuclear extract along with wortmannin or caffeine (Fig. 8A). This was done in the presence of the phosphatase inhibitor fostriecin to ensure that ATM remained phosphorylated during the reaction. We used fostriecin at a concentration previously shown to inhibit ATM dephosphorylation by PP2A [7]. The addition of fostriecin had no effect on end-protection by purified ATM (Fig. 8A, lane 5) or by a control nuclear extract (Fig. 8A, lane 4). Pre-phosphorylated ATM was capable of repressing DNA end-degradation. However, it was unable to do so in the presence of either wortmannin or caffeine as reflected by a sharp decline in detectable full-length product and an increase in intensities of shorter products (compare Fig. 8A, lane 9 to lanes 10 and 11). These data indicate that autophosphorylation of ATM is necessary but not sufficient and that downstream kinase activities are probably needed to prevent degradation of DNA ends. We ensured that ATM remained phosphorylated in the extract via parallel monitoring of 32P-labeled ATM incubated with A-T nuclear extract, wortmannin, fostriecin and DNA duplex under typical repair reaction conditions (Fig. 8B).
Fig. 8.
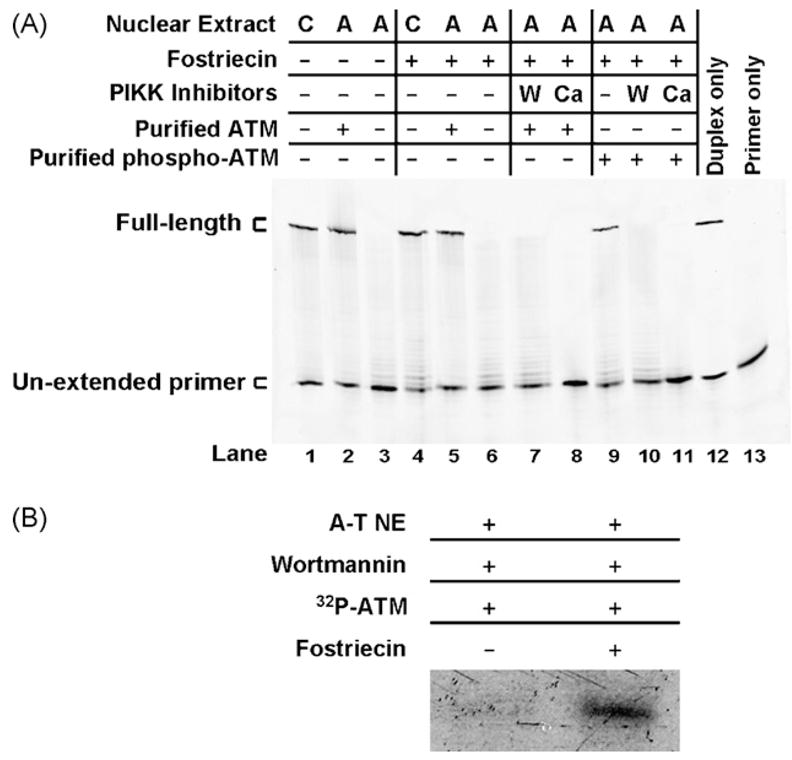
Autophosphorylation of ATM is insufficient for prevention of DNA end-degradation. (A) Top Strand primer extension products following gel separation. The 5′AATTC substrate was first incubated with WI-38VA13 (C) or AT5BIVA (A) nuclear extracts. Pre-phosphorylated ATM, wortmannin (W), caffeine (C) and the phosphatase inhibitor fostriecin were added where indicated. Products were isolated and then subjected to a primer extension assay. (B) Autoradiogram of 32P-ATM incubated with AT5BIVA (A-T NE) nuclear extract, wortmannin and a duplex with a 5′AATTC overhang under repair reaction conditions with or without fostriecin. 32P-ATM was monitored in parallel with reactions in Fig. 7A to ensure that pre-phosphorylated ATM remained phosphorylated in the reactions.
4. Discussion
Non-homologous end-joining (NHEJ) is believed to be the major DNA DSB repair mechanism in mammalian cells during G0, G1 and early S-phase of the cell cycle. Proteins involved in the NHEJ pathway include the Ku70/Ku80 heterodimer, DNA-PKcs, XRCC4, DNA Ligase IV and Artemis. Microhomology-mediated NHEJ, on the other hand, may involve the MRN complex (discussed below). NHEJ-deficient cells fail to repair up to 60% of induced DSBs [8]. On the other hand, cells with ATM deficiencies, or A-T cells, display levels of residual un-repaired DSBs that are similar to those detected in controls [9–11] or at most slightly elevated [12,13]. We have previously reported comparable efficiencies of DSB repair in A-T and control nuclear extracts; however, repair in the A-T extracts resulted in a higher level of mutations, mostly deletion events [4]. These events involved rejoining at sequences of microhomology flanking a DSB. We report here increased levels of DNA end-degradation in A-T nuclear extracts. These data, along with our previous findings, support that the repair defect in A-T cells is based on the failure to protect DNA ends at a break from erroneous degradation. Such degradation probably leads to improper end-ligation and deletions which culminate in the genetic instability phenotype associated with defects in ATM. Our data is consistent with other studies indicating that the fidelity of repair rather than efficiency is primarily affected in A-T cells [4,14,15,10,16]. These studies report an elevated level of deletions and rearrangements in the repair of plasmids harboring DSBs by A-T cells or their respective extracts.
In our former study [4], we used SupF22 plasmids harboring endonuclease-induced DSBs to evaluate the repair of different types of ends at a break. Plasmids were subjected to DSB repair reactions in A-T and in control nuclear extracts; then they were isolated and used to transform competent bacterial cells. We observed an increased level of mutations in the repair of DSBs with short (<3 nt) overhangs and blunt ends in A-T nuclear extracts. However, fidelity did not significantly vary from controls in the repair of DSBs with 4 nt overhangs. In the present study, we report an increased level of DNA end-degradation in A-T nuclear extracts for various types of DNA ends including those with 4 nt overhangs. Disparity in data regarding the repair of breaks with 4 nt overhangs is probably due to differences in the experimental systems utilized. It is conceivable that the use of a 5553 bp plasmid with cohesive 4 nt overhangs in our former study may have promoted intramolecular interactions resulting in plasmid circularization. This would have limited the duration of exposure of plasmid ends to nucleases in either type of extract hence resulting in greater end-stability and higher repair fidelity.
In their 1993 paper, Powell et al. [10] concluded that nuclease-mediated degradation of DNA ends is probably not the sole repair defect in A-T cells. This was based on observing deletions and sequence-insertions affecting linearized plasmids at and around the break site in A-T cells. Moreover, they reported rearrangements involving multiple sites along an intact circular plasmid transfected into A-T cells. However, their analysis of the data did not include assessing whether a subset of those mutations was non-random or rather directed by the presence of microhomologies.
A possible link between loss of ATM function and illegitimate recombination may be deduced from the interaction between ATM and Mre11, a nuclease that has been implicated in microhomology-mediated end-joining and whose role in recombination is well documented. Mre11 is a member of the Mre11–Rad50–Nbs1 (MRN) complex that participates in end-resection at DNA DSBs. This process precedes the strand invasion step observed during meiotic recombination and homologous recombination repair. The role of Nbs1 has not been fully elucidated whereas resection seems to mostly depend on the Mre11–Rad50 complex. Rad50 is an ATPase related to the structural maintenance of chromosome (SMC) proteins [2] and distantly related to the ATP binding cassette (ABC) family of transporters [17]. Mre11, on the other hand, is a nuclease [18] whose role in NHEJ is under debate. Studies in budding yeast indicate that all three components of the complex are required for end-joining in vivo [19] and in vitro [20]. On the other hand, while some in vitro studies in mammalian extracts support that the MRN complex is required for NHEJ [21,22] others conclude that it is dispensable regardless of the type of DNA substrate [23]. Insight into a possible role for this complex in a microhomolgy-dependent form of NHEJ comes from studies by Paull and Gellert [24,25] demonstrating that recombinant human Mre11 can degrade duplex DNA substrates up to sequences of microhomology in vitro. End-degradation by Mre11 was stimulated by the addition of DNA with non-homologous ends but inhibited by ends capable of base pairing. Moreover, during degradation, the Mre11 nuclease activity stalled upon encountering cohesive sequences.
Mre11 is phosphorylated in an ATM-dependent manner in response to DNA damage [26]. Whether this phosphorylation is direct by ATM [27] or indirect through a downstream kinase [28] remains debatable. Nbs1 is another member of the MRN complex that is phosphorylated by ATM [29,30]. These interactions provide the means through which ATM could regulate degradation at DNA ends. Hence, we envisage a model in which activated ATM is recruited to DNA ends by MRN which is then phosphorylated by ATM at sites that regulate its resection-related activities. We found ATP to be a requirement for prevention of substrate degradation in non-A-T control nuclear extracts. Moreover, this protection was inhibited by the PI-3 kinase-like kinase inhibitors caffeine and wortmannin. These pieces of evidence, although not conclusive, lend support to this model. Alternatively, ATM could be activating a downstream effector that in turn represses degradation. A myriad of proteins interacts with ATM and could play a role in enhancing DNA end-stability. The list of candidates includes multiple kinases (Chk1, Chk2, DNA-PKcs, etc.) and repair-associated factors (BRCA1, MDC1, 53BP, etc.). The scope of protection mediated by ATM is probably not limited to Mre11 but also extends to other nucleases; however, our knowledge of the Mre11 nuclease and its activities places it as the primary candidate for microhomology-mediated end-joining. Worth noting is that the levels of non-full-length products detectable in A-T nuclear extracts were slightly higher in reactions containing ATP than those lacking ATP. Although these differences are very subtle, they may signify an alternate, albeit less efficient, non-ATM-dependent DNA end-protection mechanism.
When examining the repair of a plasmid with a bleomycin-induced DSB, Dar et al. [15] did not observe illegitimate recombinational repair in A-T extract, in contrast to predictions of the model delineated above. One possible explanation is that in the repair of ends generated by bleomycin in A-T cells, other pathways predominate over microhomology-mediated end-joining. Bleomycin induces oxidative damage and is believed to produce DSBs that resemble those induced by ionizing radiation [31]. By virtue of their chemistry (3′-phosphoglycolate and 5′-phosphate termini), such ends may be resistant to the degradation process we observed in our assays.
To recapitulate, we have assessed the degradation of DNA substrates bearing various overhangs in A-T and control nuclear extracts. These substrates resemble DNA ends at a double-strand break and similar substrates were previously shown to activate ATM [32–34]. We observed greater extents of degradation in A-T extracts, a phenomenon that was repressed by the addition of purified ATM. This repression of degradation was ATP-dependent and was inhibited by the PI3-kinase-like kinase inhibitors wortmannin and caffeine. Pre-phosphorylated ATM was incapable of hindering degradation in the presence of PI3-kinase-like kinase inhibitors. These pieces of data conform to a model in which ATM prevents the degradation of DNA ends via its kinase activity. Future exploration of this model will include assessing the actual involvement of the ATM kinase activity in the process and mediators, such as the MRN complex, it may be acting upon to repress degradation.
Acknowledgments
We thank members of the Genomics Maintenance Group at the University of Arizona for helpful discussions and Eric G. Thompson, Muriel Brengues, Hope Jones and Helen F. Smith for critical review of the manuscript. This work was supported by NIH grant R01-NS34782 to Kathleen Dixon.
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author's institution, sharing with colleagues and providing to institution administration. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- 1.Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 2.Abraham RT, Tibbetts RS. Guiding ATM to broken DNA. Science. 2005;308:510–511. doi: 10.1126/science.1112069. [DOI] [PubMed] [Google Scholar]
- 3.Taylor AMR, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58:1009–1015. doi: 10.1136/jcp.2005.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Carty MP, Oakley GG, Seidman MM, Medvedovic M, Dixon K. Expression of ATM in ataxia telangiectasia fibroblasts rescues defects in DNA double-strand break repair in nuclear extracts. Environ Mol Mutagen. 2001;37:128–140. doi: 10.1002/em.1020. [DOI] [PubMed] [Google Scholar]
- 5.Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ, Tsarfati I, Shiloh Y. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15:159–167. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- 6.Goodarzi AA, Lees-Miller SP. Biochemical characterization of the ataxia-telangiectasia mutated (ATM) protein from human cells. DNA Repair. 2004;3:753–767. doi: 10.1016/j.dnarep.2004.03.041. [DOI] [PubMed] [Google Scholar]
- 7.Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23:4451–4461. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeggo PA. DNA breakage and repair. In: Hall JC, Dunlap JC, Friedmann T, Gianelli F, editors. Advances in Genetics. Vol. 38. pp. 185–218. [DOI] [PubMed] [Google Scholar]
- 9.Lehmann AR, Stevens S. The production and repair of double-strand breaks in cells from normal humans and from patients with ataxia-telangiectasia. Biochim Biophys Acta. 1977;474:49–60. doi: 10.1016/0005-2787(77)90213-1. [DOI] [PubMed] [Google Scholar]
- 10.Powell S, Whitaker S, Peacock J, McMillan T. Ataxia telangiectasia: an investigation of the repair defect in the cell line AT5BIVA by plasmid reconstitution. Mutat Res. 1993;294:9–20. doi: 10.1016/0921-8777(93)90053-j. [DOI] [PubMed] [Google Scholar]
- 11.Thierry D, Rigaud O, Duranton I, Moustacchi E, Magdelenat H. Quantitative measurement of DNA strand breaks and repair in gamma-irradiated human leukocytes from normal and ataxia telangiectasia donors. Radiat Res. 1985;102:347–358. [PubMed] [Google Scholar]
- 12.Foray N, Arlett CF, Malaise EP. Dose-rate effect on induction and repair rate of radiation-induced DNA double-strand breaks in a normal and an ataxia telangiectasia human fibroblast cell line. Biochimie. 1995;77:900–905. doi: 10.1016/0300-9084(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 13.Foray N, Priestley A, Alsbeih G, Badie C, Capulas EP, Arlett CF, Malaise EP. Hypersensitivity of ataxia telangiectasia fibroblasts to ionizing radiation is associated with a repair deficiency of DNA double-strand breaks. Int J Radiat Biol. 1997;72:271–283. doi: 10.1080/095530097143266. [DOI] [PubMed] [Google Scholar]
- 14.Cox R, Debenham PG, Masson WK, Webb MB. Ataxia-telangiectasia: a human mutation giving high-frequency misrepair of DNA double-stranded scissions. Mol Biol Med. 1986;3:229–244. [PubMed] [Google Scholar]
- 15.Dar ME, Winters TA, Jorgensen TJ. Identification of defective illegitimate recombinational repair of oxidatively-induced DNA double-strand breaks in ataxia-telangiectasia cells. Mutat Res. 1997;384:169–179. doi: 10.1016/s0921-8777(97)00021-9. [DOI] [PubMed] [Google Scholar]
- 16.Powell SN, Mills J, McMillan TJ. Radiosensitive human tumour cell lines show misrepair of DNA termini. Br J Radiol. 1998;71:1178–1184. doi: 10.1259/bjr.71.851.10434913. [DOI] [PubMed] [Google Scholar]
- 17.Davidson AL. Not just another ABC transporter. Science. 2002;296:1038–1040. doi: 10.1126/science.1072484. [DOI] [PubMed] [Google Scholar]
- 18.Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulton SJ, Jackson SP. Components of the Ku-dependent nonhomologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998;17:1819–1828. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J, Dynan WS. Reconstitution of the mammalian DNA double strand break end-joining reaction reveals a requirement for an Mre11/Rad50/NBS1-containing fraction. Nucleic Acids Res. 2002;30:667–674. doi: 10.1093/nar/30.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong Q, Boyer TG, Chen PL, Lee WH. Deficient nonhomologous end-joining activity in cell-free extracts from Brca1-null fibroblasts. Cancer Res. 2002;62:3966–3970. [PubMed] [Google Scholar]
- 23.Di Virgilio M, Gautier J. Repair of double-strand breaks by nonhomologous end joining in the absence of Mre11. J Cell Biol. 2005;171:765–771. doi: 10.1083/jcb.200506029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 25.Paull TT, Gellert M. A mechanistic basis for Mre11-directed DNA joining at microhomologies. PNAS. 2000;97:6409–6414. doi: 10.1073/pnas.110144297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan SS, Chang HL, Hou MF, Chan TF, Kuo YH, Wu YC, Su JH. Neocarzinostatin induces Mre11 phosphorylation and focus formation through an ATM- and NBS1-dependent mechanism. Toxicology. 2002;177:123–130. doi: 10.1016/s0300-483x(02)00220-2. [DOI] [PubMed] [Google Scholar]
- 27.Kim ST, Lim DS, Canman CS, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–37543. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 28.Kim ST. Protein kinase CK2 interacts with Chk2 and phosphorylates Mre11 on serine 649. Biochem Biophys Res Commun. 2005;331:247–252. doi: 10.1016/j.bbrc.2005.03.162. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, Ranganathan V, Weisman DS, Heine WF, Ciccone DN, O’Neill TB, Crick KE, Pierce KA, Lane WS, Rathbun G, Livingston DM, Weaver DT. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000;405:477–482. doi: 10.1038/35013089. [DOI] [PubMed] [Google Scholar]
- 30.Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 31.Murugesan N, Xu C, Ehrenfeld GM, Sugiyama H, Kilkuskie RE, Rodriguez LO, Chang LH, Hecht SM. Analysis of products formed during bleomycin-mediated DNA degradation. Biochemistry. 1985;24:5735–5744. doi: 10.1021/bi00342a008. [DOI] [PubMed] [Google Scholar]
- 32.You Z, et al. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 2005;13:5363–5379. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J, et al. ATM activation by DNA double-strand breaks through the Mre11–Rad50–Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 34.You Z, et al. Rapid activation of ATM on DNA flanking double-strand breaks. Nat Cell Biol. 2007;9:1311–1318. doi: 10.1038/ncb1651. [DOI] [PubMed] [Google Scholar]