

Abstract
The Diels-Alder reactions of o-quinone methides generated from OBOC-salicylic aldehydes and alcohols are described, allowing for the synthesis of various substituted benzopyrans. The low temperatures employed for this procedure enable high diastereoselectivity in reactions with β-substituted o-quinone methides.
Introduction
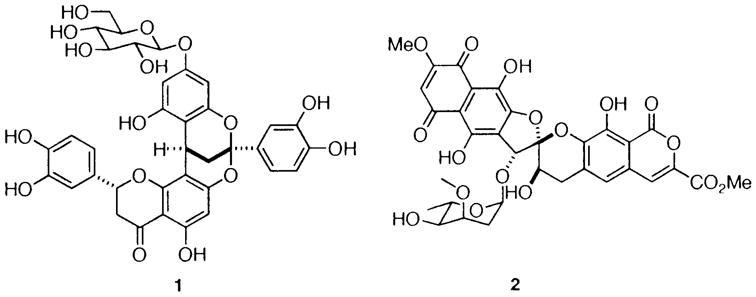
The benzopyran “chroman” nucleus is found in a variety of therapeutic agents.1 Our interest in structurally diverse benzopyrans such as the COX-2 inhibitor diinsininol 12 and the helicase inhibitor heliquinomycin 23 prompted us to consider methods for rapidly fashioning benzopyrans containing ample functionality. We recently described a new facile method for generating o-quinone methides in situ from o-OBOC salicylic aldehydes and alcohols and examined their reactivity in 1,4-conjugate additions.4 We now report the extension of this procedure for the construction of a diverse range of benzopyrans.
o-Quinone methides (o-QMs) are extremely reactive, undergoing dimerization or trimerization5 in the absence of a nucleophile or electron-rich alkene. To account for this reactivity, the o-QM concentration must be kept low throughout the reaction. Therefore, various methods have been engineered to generate these species in the presence of the nucleophile necessary for the next reaction, allowing for the generation and immediate consumption of the o-QM. Naturally, the first applications involved intramolecular cyclization motifs.6 There are only a few accounts of successful intermolecular applications.7 Most are complicated by the preparation of the o-QM because they rely on high temperatures or powerful Lewis acids for their generation; therefore, the range of nucleophiles is severely limited as is the ultimate diastereoselectivity in [4+2] cycloadditions.
Building upon an account of McLoughlin,8 we reported that o-OBOC salicylaldehydes and salicyl alcohols could lead to β-substituted and β-unsubstituted o-QMs, respectively, at low temperature upon the addition of an organometallic reagent (Scheme 1).4a,b Herein, we report our observations regarding the generation of 3 and 4 and their performance in subsequent inverse electron demand [4+2] cycloaddition reactions with various styrenes, enol ethers, enamines, imines, and heteroaromatics. As can be seen, the mild low-temperature procedure allows three components to be combined in a single pot, thereby producing a vast array of assorted benzopyrans.
Scheme 1.
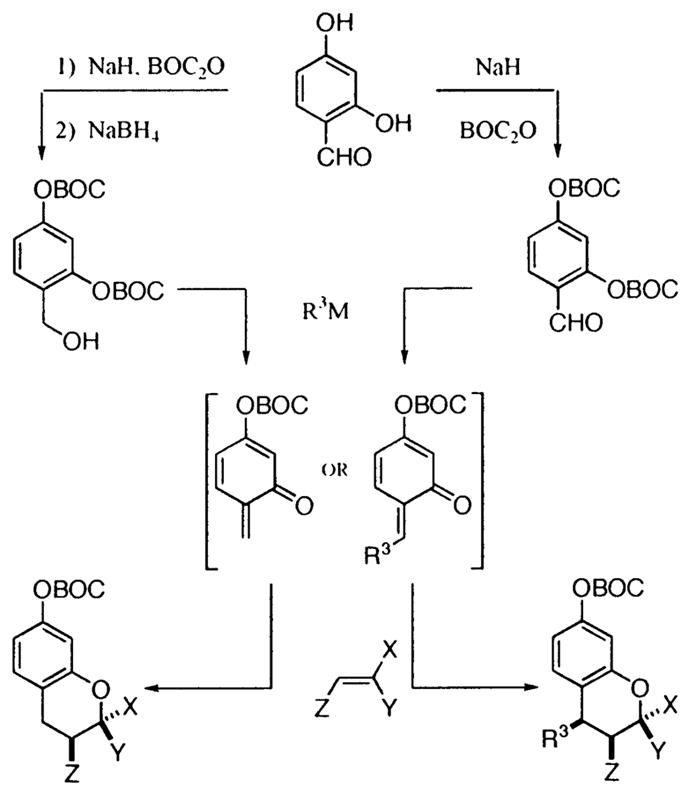
Cycloadditions of Salicylic Aldehydes and Alcohols
To generate the o-QM from alcohol 3 or aldehyde 4, an organomagnesium reagent is added. For more precise control, an organolithium reagent is added, followed by the addition of MgBr2·OEt2. In both cases, the addition results in a metal alkoxide intermediate that attacks the neighboring BOC residue. A migration ensues resulting in a phenoxide that, if M = Mg, undergoes β-elimination of the benzylic OBOC residue to form an o-QM. When M = Li, β-elimination does not occur until addition of MgX2 or LiX, which both act as Lewis acid catalysts. When the o-QM is generated in the presence of an electron-rich alkene, a diastereoselective [4+2] cycloaddition ensues to afford primarily a cis 1,3-substituted benzopyran.
Results and Conclusions
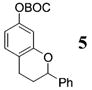
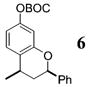
As shown in Table 1, the process proves quite general and far more convenient than previous methods involving o-QMs. Since the cycloaddition occurs between −78 and −10 °C it is much more sensitive to electronic effects and proceeds with better stereoselectivity than previous methods. Moreover, the pliable nature of the procedure allows many substituents to be introduced at the benzylic carbon of the benzopyran nucleus through variations in the organometallic reagent used to initiate formation of the o-QM. Although non-β-substituted o-QMs appear to be more reactive than their β-substituted counterparts, β-substituted o-QMs generated in this manner still undergo reaction with both activated and inactivated alkenes in respectable yields. For example, styrene (17) adds to the o-QM generated from alcohol 3 by the addition of t-BuMgCl (26) to produce 5 in 50%. On the other hand, addition of 17 to the o-QM generated by the addition of MeMgCl (31) to aldehyde 4 proceeds in only 27%. In both of these examples, styrene (17) was used as the solvent to maximize the percent yield.
TABLE 1.
One-Pot Diastereoselective Benzopyran Synthesis
| SM | R′M | 2π | Cycloadduct | cis/transa | % | |
|---|---|---|---|---|---|---|
| 1b | 3 | t-BuMgCl 26 |
|
|
NA | 50 |
| 2b | 4 | MeLi 27 + MgBr2 |
|
|
~6:1 | 27 |
| 3b | 4 | PhMgBr 28 |
|
|
>50:1c | 73 |
| 4b | 4 |

|
|
|
>50:1c | 70 |
| 5b | 4 |
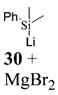
|
|
|
>50:1c | 86 |
| 6b | 4 | MeMgCl 31 |
|
|
~24:1 | 66 |
| 7d | 4 | MeMgCl 31 |
|
|
~4:1 | 55 |
| 8e | 4 | MeLi 27 + MgBr2 |
|
|
NA | 57 |
| 9e | 4 | MeLi 27 + MgBr2 |
|
|
NA | 58 |
| 10d | 4 |
|
|
|
>50:1c | 70 |
| 11e | 4 | MeMgCl 31 |
|
|
<1:50c | 94 |
| 12b | 4 | MeMgCl 31 |
|
|
>50:1c | 76 |
Cis/trans ratio determined by 1H NMR analysis of crude material.
2π component used as the solvent for the reaction.
>50:1 signifies that no other isomers could be found in the 400-MHz 1H NMR spectra.
5–10 equiv of 2π component used.
2–5 equiv of 2π component used.
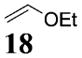
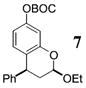
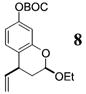
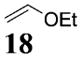
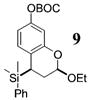
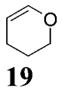
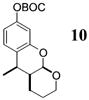
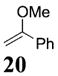
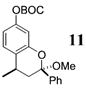
More reactive alkenes proceed with better stereoselectivity and require fewer molar equivalents relative to the o-QM. For example, ethyl vinyl ether (10 equiv) adds to the various β-substituted o-QMs generated from 4 (entries 3–5, Table 1) with a greater than 50:1 preference in the corresponding cis:trans ratio. The cis-2-ethoxy-4-phenylbenzopyran (7), the cis-2-ethoxy-4-vinylbenzopyran (8), and the cis-2-ethoxy-4-(dimethylphenylsilyl)-benzopyran (9) are assembled from three components: an aldehyde, an enol ether, and the necessary organometallic reagents (cf. 28, 29, and 309). Although more congested enol ethers, such as those disubstituted in a vicinal or geminal fashion, prove to undergo less stereo-selective cycloadditions, the results are still quite satisfactory. Addition of Grignard 31 to aldehyde 4 in the presence of dihydropyran (19, 10 equiv) proceeds to 10 with a 24:1 ratio of cis:trans isomers. The enol ether 20,10 which presents two groups (−OMe and −Ph) capable of secondary orbital interactions, affords 11 with 4:1 selectivity, apparently favoring the endo approach of the phenyl residue.
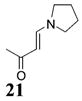
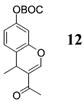
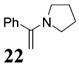
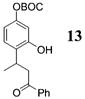
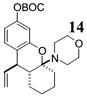
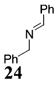
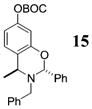
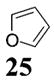
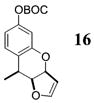
Other noteworthy adducts include the combination of o-QMs with enamines 21–23 and imine 24 (entries 8–11, Table 1). The reactions proceed quite rapidly in good overall yield. For example, the E-configured vinylogous amide 2111 smoothly undergoes cycloaddition with the o-QM that is generated by the addition of MeLi (32) to aldehyde 4 followed by the addition of MgBr2·OEt2. However, the initial cycloadduct proves to be unstable and the pyrrolidine is eliminated upon chromatography to yield the chromene 12. In a somewhat analogous fashion, the cycloadduct that emerges from addition of enamine 2212 to the same o-QM undergoes pyrrolidine hydrolysis and hydrate collapse upon chromatography to produce the ketone 13. On the other hand, cycloaddition of trisubstituted enamine 23 with the β-vinyl substituted o-QM, generated by addition of vinyl Grignard 29 to aldehyde 4, proceeds to the apparently more stable cycloadduct 14. However, stirring 14 with acid does lead to the corresponding chromene by elimination of the morpholine residue. The reaction between imine 24 and the o-QM generated by addition of 31 to aldehyde 4 proceeds to 15 in the highest yield of all 2π donors examined. Interestingly, the stereochemistry is trans, suggesting an exo-oriented combination of reactants. Most likely, however, the cis isomer can undergo an intramolecular equilibration to the more thermodynamically stable trans conformation, a process facilitated by the Lewis acid MgBr2 (Figure 2). Generation of an o-QM in the presence of a heterocyclic aromatic also yields benzopyrans in high regioselectivity and diastereoselectivity. The addition of methyl Grignard 31 to aldehyde 4 in the presence of furan affords adduct 16 (entry 12, Table 1) in 76% yield. The regiochemistry is explained by analysis of the HOMO of furan, in which the 2-position has the highest orbital coefficient. This predicts the furan oxygen will be at the 3-position in the cycloadduct.
FIGURE 2.
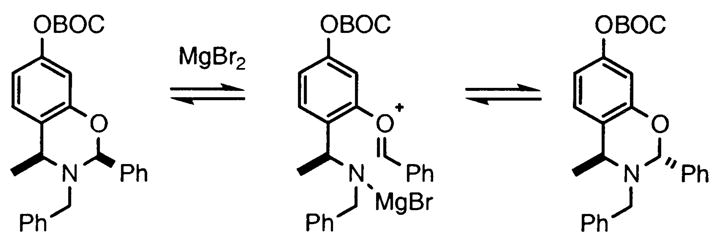
Equilibration of the stereochemistry in 15.
A wide assortment of benzopyrans are produced with excellent diastereoselectivity via inverse electron demand [4+2] cycloadditions with use of this low-temperature method for generating o-QMs from o-OBOC salicylaldehydes and alcohols. The 2π donors tested include the following in the order of reactivity: imines > enamines > enols > furans > styrenes. Future goals will include the application of this procedure in syntheses of diverse benzopyrans and other natural products, as well as adaptation of the cycloaddition to an asymmetric format. Developments will be reported in due course.
Experimental Section
General Information
These reactions required reagents of the highest quality. All reagents were newly purchased or freshly prepared. Starting aryl aldehydes that were not scrupulously dried often led to lower than expected yields. All column chromatography was conducted with silica gel, eluting with the indicated solvent system. The stereochemistry is established via 1H NMR by analysis of 1H-couplings and NOE interactions.
5: To a flame-dried 5-mL vial was added alcohol 3 (24.0 mg, 0.071 mmol). Styrene (17, 0.71 mL) was added, and the vial was cooled to −78 °C. To this mix was added t-BuMgCl (26, 53 μL, 0.106 mmol, 2 M in Et2O) dropwise. The reaction was allowed to slowly warm to rt and was monitored by TLC. Upon completion, the reaction was quenched with 0.1 M HCl and extracted with Et2O. After the ether layer was washed with brine, the combined aqueous layers were saturated with NaCl and extracted with Et2O. The combined Et2O layers were then dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel eluting with 99:1 petroleum ether/ethyl acetate, yielding 5 as a white solid (11.5 mg, 50% yield). 1H NMR [CDCl3, 400 MHz] δ 7.44–7.31 (m, 5H), 7.07 (d, 1H, J = 8.24 Hz), 6.75 (d, 1H, J = 2.38 Hz), 6.70 (dd, 1H, J1 = 8.24 Hz, J2 = 2.38 Hz), 5.06 (dd, 1H, J1 = 10.15 Hz, J2 = 2.47 Hz), 3.01–2.93 (m, 1H), 2.82–2.76 (m, 1H), 2.25–2.19 (m, 1H), 2.13–2.03 (m, 1H), 1.56 (s, 9H); 13C NMR [CDCl3, 100 MHz] δ 155.7, 152.2, 150.3, 141.6, 130.0, 128.7, 128.1, 126.2, 119.6, 113.6, 110.2, 94.6, 83.6, 30.0, 27.9, 24.9; IR [CH2Cl2, νmax cm−1] 3627, 2936, 1756, 1733; MS (EI) m/z 226 (78), 104 (24), 57 (100); HRMS (EI) m/z calcd for C20H22O4 326.1518, found 326.1528.
6: To a flame-dried 5-mL vial was added aldehyde 4 (39.6 mg, 0.12 mmol). Styrene (17, 0.59 mL), and Et2O (0.59 mL) were added, and this mixture was cooled to −20 °C. MeLi (27, 88 μL, 0.123 mmol, 1.4 M in Et2O) was added dropwise, and the resulting solution was stirred for 15 min at −20 °C. After MgBr2·OEt2 (37.0 mg, 0.14 mmol) was added to the solution, the reaction was slowly warmed to room temperature. Upon completion, as noted by TLC, the reaction was quenched with 1 M NaHCO3 and extracted with Et2O. The Et2O layer was washed with brine, and the combined aqueous layers were saturated with NaCl. The saturated aqueous layer was washed with Et2O; the combined Et2O extracts were dried over Na2-SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel eluting with petroleum ether, yielding 6 as a white solid (10.7 mg, yield 27%). 1H NMR [CDCl3, 400 MHz] δ 7.46–7.34 (m, 5H), 7.28–7.25 (m, 1H), 6.76–6.73 (m, 2H), 5.10–5.07 (m, 1H), 3.20–3.14 (m, 1H), 2.24–2.19 (m, 1H), 1.81 (q, 1H, J = 11.7 Hz), 1.56 (s, 9H), 1.36 (d, 3H, J = 6.78 Hz); 13C NMR [CDCl3, 100 MHz] δ 155.5, 152.2, 150.3, 141.6, 128.8, 128.2, 127.8, 126.3, 125.0, 113.7, 110.1, 94.6, 83.7, 40.0, 30.1, 27.9, 20.3; IR [CH2Cl2, νmax cm−1] 3054, 2930, 1757; MS (EI) m/z 240 (57), 225 (59), 57 (100); HRMS (EI) m/z calcd for C21H24O4 340.1675, found 340.1682.
7: To a flame-dried 5-mL vial was added aldehyde 4 (51.9 mg, 0.15 mmol). Ethyl vinyl ether (18, 1 mL) was added and the vial was cooled to −78 °C. To this mixture was added PhMgBr (28, 192 μL, 0.18 mmol, 0.96 M in THF) dropwise. The reaction was allowed to slowly warm to rt and was monitored by TLC. Upon completion, the reaction was quenched with 1 M NaHCO3 and extracted with Et2O. The ether layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with 98:2 petroleum ether/ethyl acetate, yielding 7 as a white solid (41.3 mg, yield 73%). 1H NMR [CDCl3, 400 MHz] δ 7.35–7.30 (m, 2H), 7.28–7.24 (m, 1H), 7.21–7.19 (m, 2H), 6.75 (d, 1H, J = 2.38 Hz), 6.69 (dd, 1H, J1 = 8.42 Hz, J2 = 0.92 Hz), 6.60 (dd, 1H, J1 = 8.42 Hz, J2 = 2.38 Hz), 5.27 (dd, 1H, J1 = 8.61 Hz, J2 = 2.38 Hz), 4.18–4.14 (m, 1H), 4.11–4.03 (m, 1H), 3.71–3.63 (m, 1H), 2.41–2.36 (m, 1H), 2.23–2.15 (m, 1H), 1.56 (s, 9H), 1.27 (t, 3H, J = 7.05 Hz); 13C NMR [CDCl3, 100 MHz] δ 154.2, 152.1, 150.6, 144.0, 130.1, 128.8, 127.0, 123.6, 114.0, 110.2, 99.9, 94.6, 83.7, 64.7, 41.2, 37.3, 27.9, 15.4; IR [CH2Cl2, νmax cm−1] 2982, 1757; MS (EI) m/z 311 (34), 224 (56), 57 (100); HRMS (EI) m/z calcd for C22H26O5 370.1780, found 370.1783.
8: To a flame-dried 5-mL vial was added aldehyde 4 (78.8 mg, 0.23 mmol). Ethyl vinyl ether (18, 2.3 mL) was added and the vial was cooled to −78 °C. To this mixture was added vinylmagnesium bromide (29, 270 μL, 0.25 mmol, 0.955 M in THF) dropwise. The reaction was slowly warmed to room tempearture and monitored by TLC. Upon completion, the reaction was quenched with 1 M NaHCO3, and extracted with Et2O. The Et2O layer was washed with brine; the combined aqueous layers were saturated with NaCl and extracted with Et2O. The combined Et2O layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel, eluting with 98:2 petroleum ether/ethyl acetate, yielding 8 as a white solid (52.0 mg, yield 70%). 1H NMR [CDCl3, 400 MHz] δ 7.09 (dt, 1H, J1 = 8.06 Hz, J2 = 0.92 Hz), 6.72–6.69 (m, 2H), 5.95–5.86 (m, 1H), 5.22–5.09 (m, 3H), 4.03–3.95 (m, 1H), 3.67–3.59 (m, 1H), 3.53–3.47 (m, 1H), 2.23–2.17 (m, 1H), 1.97–1.89 (m, 1H), 1.56 (s, 9H), 1.25 (t, 3H, J = 7.05 Hz); 13C NMR [CDCl3, 100 MHz] δ 153.1, 152.1, 150.7, 141.3, 129.8, 121.8, 115.9, 113.9, 110.3, 98.8, 83.7, 64.6, 39.2, 34.2, 27.9, 15.4; IR [CH2Cl2, νmax cm−1] 2980, 1757, 1614; MS FAB m/z 261 (24), 174 (54), 57 (100); HRMS (EI) m/z calcd for C18H24O5 320.1624, found 320.1616.
9: To a flame-dried 5-mL vial was added aldehyde 4 (30.1 mg, 0.089 mmol). Ethyl vinyl ether (18, 0.89 mL) was added and the vial was cooled to −78 °C. To this solution was added dimethylphenylsilyllithium (30, 740 μL, 0.27 mmol, 0.36 M in THF) dropwise. The reaction was stirred at −78 °C for 30 min. After MgBr2·OEt2 (32.2 mg, 0.125 mmol) was added to the solution, the reaction was slowly warmed to room temperature. Upon completion, as noted by TLC, the reaction was quenched with 1 M NaHCO3 and extracted with Et2O. The Et2O layer was washed with brine, and the combined aqueous layers were saturated with NaCl. After the saturated aqueous layer was washed with Et2O, the combined Et2O extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel eluting with 98:2 petroleum ether/ethyl acetate, yielding 9 as a white solid (32.8 mg, yield 86%). 1H NMR [CDCl3, 400 MHz] δ 7.54–7.52 (m, 2H), 7.38–7.36 (m, 3H), 6.75 (dd, 1H, J1 = 8.42 Hz, J2 = 0.73 Hz), 6.66 (d, 1H, J = 2.38 Hz), 6.57 (dd, 1H, J1 = 8.42 Hz, J2 = 2.56 Hz), 5.09–5.07 (m, 1H), 4.00–3.92 (m, 1H), 3.66–3.58 (m, 1H), 2.64–2.61 (m, 1H), 2.15–2.09 (m, 1H), 2.06–2.00 (m, 1H), 1.55 (s, 9H), 1.25 (t, 3H, J = 7.05 Hz), 0.32 (s, 3H), 0.26 (s, 3H); 13C NMR [CDCl3, 100 MHz] δ 152.7, 152.1, 149.2, 139.5, 134.3, 129.1, 128.5, 128.0, 122.6, 113.6, 110.4, 98.3, 83.5, 64.4, 29.2, 27.9, 22.3, 15.3, −2.8, −3.9; IR [CH2Cl2, νmax cm−1] 2983, 1757, 1613; MS (EI) m/z 148 (67), 135 (77), 57 (100); HRMS (EI) m/z calcd for C24H32O5Si 428.2019, found 428.2018.
10: To a flame-dried 5-mL vial was added aldehyde 4 (32.0 mg, 0.095 mmol). Dihydropyran (19, 0.95 mL) was added and the vial was cooled to −78 °C. To this mixture was added MeMgCl (31, 37 μL, 0.104 mmol, 2.85 M in THF) dropwise. The reaction was slowly warmed to rt and monitored by TLC. Upon completion, the reaction was quenched with 1 M NaHCO3 and extracted with Et2O. The Et2O layer was washed with brine; the combined aqueous layers were saturated with NaCl and extracted with Et2O. The combined Et2O layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel, eluting with 98:2 petroleum ether/ethyl acetate, yielding 10 as a white solid (20 mg, yield 66%). 1H NMR [CDCl3, 400 MHz] δ 7.11 (dt, 1H, J1 = 7.69 Hz, J2 = 1.28 Hz), 6.73–6.70 (m, 2H), 5.48 (d, 1H, J = 2.56 Hz), 4.00–3.94 (m, 1H), 3.77–3.73 (m, 1H), 3.14 (quintet, 1H, J = 6.41 Hz), 2.04–2.00 (m, 1H), 1.77–1.65 (m, 1H), 1.64–1.58 (m, 3H), 1.56 (s, 9H), 1.30 (d, 3H, J = 7.14 Hz); 13C NMR [CDCl3, 100 MHz] δ 154.3, 152.2, 150.6, 127.2, 122.3, 113.8, 109.2, 97.5, 83.7, 61.1, 38.0, 32.0, 27.9, 24.7, 17.6, 15.4; IR [CH2Cl2, νmax cm−1] 2978, 1758, 1613; MS (EI) m/z 220 (53), 205 (75), 57 (100); HRMS (EI) m/z calcd for C18H24O5 320.1624, found 320.1617.
11: To a flame-dried 5-mL vial was added aldehyde 4 (29.2 mg, 0.086 mmol). Enol ether 20 (116 mg, 0.86 mmol) and toluene (0.86 mL) were added and the vial was cooled to 0 °C. To this mixture was added MeMgCl (31, 46 μL, 0.14 mmol, 3 M in THF) dropwise. The reaction was slowly warmed to rt and monitored by TLC. Upon completion, the reaction was quenched with 1 M NaHCO3 and extracted with Et2O. The Et2O layer was washed with brine, then dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel, eluting with 99:1 petroleum ether/ethyl acetate, yielding 11 as white solid (17.5 mg, yield 55%, inseparable mix of diastereomers). Major isomer: 1H NMR [CDCl3, 400 MHz] δ 7.61–7.28 (m, 5H), 7.17 (dd, 1H, J1 = 8.42 Hz, J2 = 0.92 Hz), 6.89 (d, 1H, J = 2.38 Hz), 6.79 (dd, 1H, J1 = 8.42 Hz, J2 = 2.38 Hz), 3.13 (s, 3H), 2.73 (sextet, 1H, J = 6.78 Hz), 2.15 (qd, 2H, J1 = 42.3 Hz, J2 = 13.73 Hz, J3 = 6.23 Hz), 1.58 (s, 9H), 1.43 (d, 3H, J = 7.14 Hz); 13C NMR [CDCl3, 100 MHz] δ 153.0, 152.2, 150.3, 141.2, 128.6, 127.5, 126.5, 125.6, 114.2, 110.2, 102.3, 101.0, 83.6, 50.3, 41.8, 27.9, 27.5, 22.2; IR [CH2Cl2, νmax cm−1] 2967, 2935, 1758; MS (EI) m/z 255 (29), 223 (24), 57 (100); HRMS (EI) m/z calcd for C22H26O5 370.1780, found 370.1790.
General Procedure for Compounds 12 and 13
A flame-dried flask containing aldehyde 4 (0.1 M in Et2O) was cooled to −78 °C. The appropriate organolithium reagent was added in dropwise fashion. After the mixture was stirred for 15 min, the dienophile (5 equiv) was added, followed 5 min later by MgBr2·OEt2 (1 equiv), and the reaction was permitted to slowly warm to rt. Upon completion by TLC, 400 mg of silica gel was added to the solution suspended in 2 mL of Et2O; the mixture was then stirred for 3 h. The reaction was then quenched with 1 M NaHCO3 and extracted with an equal volume of Et2O. The ether layer was washed with an equal volume of brine, and the organic layer was then dried over Na2SO4, filtered, and concentrated in vacuo.
12: White solid (NMR yield 52%). 1H NMR [CDCl3, 400 MHz] δ 7.61 (s, 1H), 7.18 (d, 1H, J = 8.29 Hz), 6.94 (dd, 1H, J1 = 8.29 Hz, J2 = 2.3 Hz), 6.87 (d, 1H, J = 2.3 Hz), 3.93 (q, 1H, J = 6.76 Hz), 2.32 (s, 3H), 1.57 (s, 9H), 1.27 (d, 3H, J = 6.91 Hz); 13C NMR [CDCl3, 100 MHz] δ 195.9, 152.0, 150.1, 129.7, 124.3, 122.0, 118.2, 109.9, 84.1, 77.4, 29.9, 27.9, 27.3, 25.6, 25.5; IR [CH2Cl2, νmax cm−1] 2928, 1759, 1644, 1587, 1501; MS (EI) m/z 204 (21), 189 (90), 57 (100); HRMS (EI) m/z calcd for C17H20O5 304.1311, found 304.1315.
13: Brown oil (NMR yield 56%). 1H NMR [CDCl3, 400 MHz] δ 7.95 (d, 2H, J = 7.37 Hz), 7.58 (t, 1H, J = 7.37 Hz), 7.45 (t, 2H, J = 7.7 Hz), 7.18 (d, 1H, J = 8.29 Hz), 6.76–6.72 (m, 2H), 3.78–3.73 (m, 1H), 3.47 (dd, 1H, J1 = 18.77 Hz, J2 = 9.52 Hz), 3.29 (dd, 1H, J1 = 18.58 Hz, J2 = 3.22 Hz), 1.54 (s, 9H), 1.41 (d, 3H, J = 7.06 Hz); 13C NMR [CDCl3, 100 MHz] δ 201.8, 154.8, 152.2, 150.3, 136.2, 134.0, 130.8, 128.9, 128.6, 127.1, 114.1, 111.2, 83.6, 48.7, 27.9, 26.2, 21.7; IR [CH2Cl2, νmax cm−1] 3686, 3053, 2960, 2928, 2855, 1755, 1733, 1671, 1599; MS (EI) m/z 281 (60), 57 (74), 43 (100); HRMS (EI) m/z (M + Na)+ calcd for C21H24O5Na+ 379.1521, found 379.1527.
14: To a flame-dried 5-mL vial was added aldehyde 4 (32.5 mg, 0.096 mmol). Diethyl ether (0.96 mL) and enamine 23 (161 μL, 0.96 mmol) were added and the vial was cooled to −78 °C. To this mixture was added vinylmagnesium chloride (29, 110 μL, 0.106 mmol, 0.955 M in THF) dropwise. The reaction was slowly warmed to rt and monitored by TLC. Upon completion, the reaction was quenched with 1 M NaHCO3 and extracted with Et2O. The Et2O layer was washed with brine; the combined aqueous layers were saturated with NaCl and extracted with Et2O. The combined Et2O layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel, eluting with 98:2 petroleum ether/ethyl acetate, yielding 14 as a white solid (28.0 mg, yield 70%). 1H NMR [CDCl3, 400 MHz] δ 7.06 (dt, 1H, J1 = 8.06 Hz, J2 ) 0.916 Hz), 6.67–6.64 (m, 2H), 5.87–5.78 (m, 1H), 5.22–5.14 (m, 2H), 3.70–3.60 (m, 4H), 3.36–3.32 (m, 1H), 2.82–2.71 (m, 4H), 2.15–2.10 (m, 1H), 1.84–1.72 (m, 3H), 1.66–1.56 (m, 12H), 1.49 (s, 1H), 1.42–1.36 (m, 1H); 13C NMR [CDCl3, 100 MHz] δ 153.4, 152.2, 150.9, 142.1, 130.3, 120.8, 116.3, 113.1, 109.8, 91.6, 83.4, 67.6, 45.0, 44.0, 38.0, 29.9, 28.0, 27.8, 22.6, 22.1; IR [CH2Cl2 solution, νmax cm−1] 2936, 2858, 1757, 1610, 1591, 1498; MS (EI) m/z 167 (100), 57 (14), 43 (15); HRMS (EI) m/z calcd for C24H33NO5 415.2359, found 415.2348.
15: To a flame-dried 5-mL vial was added aldehyde 4 (33.3 mg, 0.098 mmol). Ether (1 mL) and imine 24 (100 μL, 0.49 mmol) were added and the vial was cooled to −78 °C. To this mixture was added MeMgCl (31, 41 μL, 0.108 mmol, 2.65 M in THF) dropwise. The reaction was slowly warmed to rt and monitored by TLC. Upon completion, the reaction was quenched with 1 M NaHCO3 and extracted with Et2O. The Et2O layer was washed with brine; the combined aqueous layers were saturated with NaCl and extracted with Et2O. The combined Et2O layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel, eluting with petroleum ether, yielding 15 as a white solid (40.0 mg, yield 94%). 1H NMR [CDCl3, 400 MHz] δ 7.77–7.75 (m, 2H), 7.45–7.41 (m, 2H), 7.35–7.29 (m, 5H), 7.25–7.22 (m, 1H), 6.99 (d, 1H, J = 8.42 Hz), 6.89 (d, 1H, J = 2.38 Hz), 6.75 (dd, 1H, J1 = 8.42 Hz, J2 = 2.38 Hz), 6.09 (s, 1H), 3.86–3.79 (m, 2H), 3.34 (d, 1H, J = 14.83 Hz), 1.59 (s, 9H), 1.55 (d, 1H, J = 6.96 Hz); 13C NMR [CDCl3, 100 MHz] δ 154.8, 152.3, 150.3, 139.6, 138.2, 129.3, 128.5, 128.4, 128.2, 128.1, 127.0, 126.7, 123.0, 114.1, 110.2, 86.0, 83.8, 52.4, 49.8, 27.9, 24.1; IR [CH2Cl2, νmax cm−1] 2975, 2935, 1758, 1617; MS FAB m/z 136 (27), 91 (100) 57 (82); HRMS (EI) m/z calcd for C27H29-NO4 431.2097, found 430.2113.
16: To a flame-dried 5-mL vial was added aldehyde 4 (36.2 mg, 0.107 mmol). Furan (25, 1.07 mL) was added and the vial was cooled to −78 °C. To this mixture was added MeMgCl (31, 48 μL, 0.118 mmol, 2.45 M in THF) dropwise. The reaction was slowly warmed to rt and monitored by TLC. Upon completion, the reaction was quenched with 1 M NaHCO3 and extracted with Et2O. The Et2O layer was washed with brine; the combined aqueous layers were saturated with NaCl and extracted with Et2O. The combined Et2O layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was chromatographed through silica gel, eluting with 98:2 petroleum ether/ethyl acetate, yielding 16 as a white solid (24.7 mg, yield 76%). 1H NMR [CDCl3, 400 MHz] δ 7.15 (dd, 1H, J1 = 8.24 Hz, J2 = 1.28 Hz), 6.83 (dd, 1H, J1 = 8.24 Hz J2 = 2.38 Hz), 6.71 (d, 1H, J = 2.38 Hz), 6.39–6.37 (m, 1H), 5.5 (ddd, 1H, J1 = 8.24 Hz, J2 = 2.56 Hz, J3 = 0.92 Hz), 5.07 (td, 1H, J1 = 2.56 Hz, J2 = 0.37 Hz), 4.92 (dd, 1H, J1 = 8.24 Hz, J2 = 2.93 Hz), 3.08 (qd, 1H, J1 = 6.96 Hz, J2 = 2.93 Hz), 1.58–1.53 (m, 12H); 13C NMR [CDCl3, 100 MHz] δ 155.2, 152.0, 151.8, 150.4, 126.6, 126.2, 115.1, 111.9, 101.5, 85.2, 83.6, 82.3, 31.1, 27.9, 13.7; IR [CH2Cl2, νmax cm−1] 2975, 2935, 1758, 1612; MS (EI) m/z 189 (46), 136 (27), 57 (100); HRMS (EI) m/z calcd for C17H20O5 304.1311, found 304.1316.
FIGURE 1.
Examples of biologically active benzopyrans.
Acknowledgments
Research support from the University of California Coordinating Committee on Cancer Research in the form of two awards (19990641 and SB010064) is greatly appreciated along with additional support from NSF (CHE-9971211), PRF (PRF34986-G1), NIH (GM-64831), the UC-AIDS Initiative (K00-SB-039), and Research Corporation (R10296). We are also thankful for the support of our M.S. facility funded in part by DAAD19-00-1-0026.
Footnotes
Supporting Information Available: Spectral data for compounds 5–16. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nicolaou KC, Pfefferkorn JA, Barluenga S, Mitchell HJ, Roecker AJ, Cao GQ. J Am Chem Soc. 2000;122:9968–9976. [Google Scholar]; Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. J Am Chem Soc. 2000;122:9939–9953. [Google Scholar]; Nicolaou KC, Pfefferkorn JA, Mitchell HJ, Roecker AJ, Barluenga S, Cao GQ, Affleck RL, Lillig JE. J Am Chem Soc. 2000;122:9954–9967. [Google Scholar]
- 2.Ogundaini A, Farah M, Perera P. J Nat Prod. 1996;59:587–590. doi: 10.1021/np960386k. [DOI] [PubMed] [Google Scholar]
- 3.Chino M, Nishikawa K, Yamada A, Ohsono M, Sawa T, Hanaoka F, Ishizuka M, Takeuchi T. J Antibiot. 1998;51:480–486. doi: 10.7164/antibiotics.51.480. [DOI] [PubMed] [Google Scholar]
- 4.(a) Van De Water RW, Magdziak DJ, Chau JN, Pettus TRR. J Am Chem Soc. 2000;122:6502–6503. [Google Scholar]; (b) Jones RM, Van De Water RW, Lindsey CC, Hoarau C, Ung T, Pettus TRR. J Org Chem. 2001;66:3435–3441. doi: 10.1021/jo001752e. [DOI] [PubMed] [Google Scholar]; (c) Hoarau C, Pettus TRR. Molecules. 2002 in press. [Google Scholar]
- 5.Turner AB. Q Rev. 1964;18:347–360. [Google Scholar]
- 6.Talley JJ. J Org Chem. 1985;50:1695–1699. [Google Scholar]
- 7.Chambers JD, Crawford J, Williams HWR, Dufresne C, Scheigetz J, Bernstein MA, Lau CK. Can J Chem. 1992;70:1717–1732. [Google Scholar]; Inoue T, Inoue S, Sato K. Bull Chem Soc Jpn. 1990;63:1062–1068. [Google Scholar]
- 8.McLoughlin BJ. J Chem Soc, Chem Commun. 1969:540–541. [Google Scholar]
- 9.Lipshutz BH, Sclafani JA, Takanami T. J Am Chem Soc. 1998;120:4021–4022. [Google Scholar]
- 10.Gassman PG, Burns SJ, Pfister KB. J Org Chem. 1993;58:1449–1457. [Google Scholar]
- 11.Kozmin SA, Janey JM, Rawal VH. J Org Chem. 1999;64:3039–3052. doi: 10.1021/jo981563k. [DOI] [PubMed] [Google Scholar]; Kanner CB, Pandit UK. Tetrahedron. 1982;38:3597–3604. [Google Scholar]
- 12.Pine S, Pettit RJ, Gieb GD, Cruz SG, Gallego CH, Tijerina T, Pine RD. J Org Chem. 1985;50:1212–1216. [Google Scholar]; Petasis N, Lu SP. Tetrahedron Lett. 1995;36:2393–2396. [Google Scholar]