

Abstract
The oocyte-independent source for the generation of pluripotent stem cells is one of the ultimate goals in regenerative medicine. We report that upon exposure to mouse ES cell (ESC) extracts, reversibly permeabilized NIH3T3 cells undergo de-differentiation followed by stimulus-induced re-differentiation into multiple lineage cell types. Genome-wide expression profiling revealed significant differences between NIH3T3 control and ESC extract treated NIH3T3 cells including the re-activation of ESC specific transcripts. Epigenetically, ESC extracts induced CpG de-methylation of Oct4 promoter, hyper-acetylation of histones 3 and 4 and decreased lysine 9 (K-9) dimethylation of histone 3. In mouse models of surgically-induced hind limb ischemia (HLI) or acute myocardial infarction (AMI) transplantation of reprogrammed NIH3T3 cells significantly improved post-injury physiological functions and showed anatomical evidence of engraftment and trans-differentiation into skeletal muscle, endothelial cell and cardiomyocytes. These data provide evidence for the generation of functional multi-potent stem like cells from terminally differentiated somatic cells without the introduction of retroviral mediated trans-genes or ESC fusion.
Keywords: Somatic Cell Dedifferentiation, ES cells, Nuclear reprogramming, Epigenetics, Tissue Repair, Myocardial infarction
Introduction
The available evidence demonstrating improvement in myocardial function following transplantation of autologous bone marrow (BM) derived stem/progenitor cells, both in pre-clinical as well as in available clinical trials, remains a potent force driving discovery and clinical development simultaneously and has provided new hope for patients with debilitating heart diseases.1, 2 However, certain potential limitations of autologous BM or peripheral blood derived stem/progenitor cells have also been identified. Risk factors for coronary artery disease are reported to be associated with a reduced number and impaired functional activity of endothelial progenitor cells (EPC) in the peripheral blood of patients.3-6
De-differentiation of adult somatic cells into multi-potent cells might provide an attractive source of stem cells for regenerative medicine including post-infarct cardiac and other ischemic tissue repair. Recent experimental evidence has revealed that nuclear re-programming of terminally differentiated adult mammalian cells leading to their de-differentiation is possible.7-10 Recently, two breakthrough studies have reported the generation of embryonic stem (ES) cells from terminally differentiated human fibroblasts by the retroviral transduction of defined ESC-specific transcription factors.11, 12 The best example of somatic cell re-programming to totipotent stage comes from reproductive and therapeutic cloning experiments utilizing somatic nuclear transfer (SNT), wherein transplantation of somatic nuclei into enucleated oocyte cytoplasm can extensively reprogram somatic cell nuclei with new patterns of gene expression, new pathways of cell differentiation and successful generation of embryonic stem cells and birth of cloned animals.13-17 Therapeutic cloning by SNT for clinical application, though conceptually attractive is yet not practical given the technical difficulties, extremely low efficiency, oocyte-dependence, ethico-legal concerns and prohibitive cost associated with the process. It, therefore, becomes imperative to develop alternative strategies for somatic cell re-programming. One strategy would be to develop oocyte-independent systems, for instance, exposure of somatic cell nuclei to ESC-derived cell-free factors/proteins to drive somatic cell de-differentiation and nuclear re-programming. Indeed, alterations in the fate of one kind of differentiated somatic cells by cell-free extracts from other, leading to the acquirement of donor cell characteristics and functions by recipient cells, has been previously reported.18-24
In the present study we report that exposure to mouse ESC extracts induces marked epigenetic reprogramming in NIH3T3 fibroblasts including re-activation of ESC specific gene expression. These re-programmed cells possess multi-potent stem cell like characteristics including multi-lineage differentiation potential and more importantly therapeutic efficacy for improvement in physiological functions and anatomical tissue repair in mouse models of surgically induced hind limb ischemia and acute myocardial infarction.
Materials and Methods
Cell Culture
NIH3T3 Swiss-Albino fibroblasts (ATCC) were cultured in DMEM (Sigma-Aldrich) with 10% FCS, L-glutamine, and 0.1 mM β-mercaptoethanol. The D3-mESC were obtained from ATCC and cultured as described earlier.25
Cell Extracts
The mESC and 3T3 cell extracts were prepared as described previously.24 Briefly, the cells were washed in phosphate-buffered saline (PBS) and in cell lysis buffer (100 mM HEPES, pH 8.2, 50 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, and protease inhibitors), sedimented at 10,000 rpm, re-suspended in 1 volume of cold cell lysis buffer, and incubated for 30−45 min on ice. Cells were sonicated on ice in 200-μl aliquots using a sonicator fitted with a 3-mm-diameter probe until all cells and nuclei were lysed, as judged by microscopy. The lysate was sedimented at 12,000 rpm for 15 min at 4°C to pellet the coarse material. The supernatant was aliquoted, frozen in liquid nitrogen and stored at −80°C. Protein concentration of the mESC and NIH3T3 cell extracts were determined by Bradford assay.
Streptolysin –O (SLO)-mediated Permeabilization and Cell Extract Treatment
NIH3T3 cells were washed in cold PBS and in cold Ca2+- and Mg2+-free Hank's balanced salt solution (HBSS) (Invitrogen, Carlsbad, CA). Cells were re-suspended in aliquots of 100,000-cells/100 μl of HBSS, or multiples thereof; placed in 1.5-ml tubes; and centrifuged at 1500 rpm for 5 min at 4°C. After sedimentation, cells were suspended in 97.7 μl of cold HBSS, tubes were placed in a H2O bath at 37°C for 2 min, and 2.3 μl of SLO (Sigma-Aldrich) (100 μg/ml stock diluted 1:10 in cold HBSS) was added to a final SLO concentration of 230 ng/ml. Samples were incubated horizontally in a H2O bath for 50 min at 37°C with occasional agitation and set on ice. Samples were diluted with 200 μl of cold HBSS and cells were collected by sedimentation at 1500 rpm for 5 min at 4°C. Permeabilization efficiency of >80% was obtained as assessed by monitoring uptake of a 70,000-Mr Texas Red-conjugated dextran (50 μg/ml; Invitrogen). After permeabilization, NIH3T3 cells were suspended at 2000 cells/μl in 100 μl of mESC extract or control NIH3T3 cells extract containing an ATP-regenerating system (1 mM ATP, 10 mM creatine phosphate, and 25 μg/ml creatine kinase; Sigma-Aldrich), 100 μM GTP (Sigma-Aldrich), and 1 mM each nucleotide triphosphate (NTP; Roche Diagnostics, Indianapolis, IN). The cells were incubated for 1 hr at 37°C in a H2O bath with occasional agitation. To reseal plasma membranes, the cell suspension was diluted with complete DMEM medium containing 2 mM CaCl2, antibiotics and cells were seeded at 100,000 cells per well on a 48-well plate. After 2 hrs, floating cells were removed, and cells were cultured in D3 maintenance medium.
Determination of De-differentiation
De-differentiation of NIH3T3 following D3-cell extract treatment (hereto referred as 3T3/D3) was determined by the induction of ESC markers both at mRNA level by quantitative real time PCR and at protein level by immuno-staining. Total cellular RNA was harvested at various time points and the quantitative real-time RT-PCR was performed to determine mRNA expression of selected embryonic stem cell markers in self extract (hereto referred as 3T3/3T3) and 3T3/D3 cells, as described previously.25, 26 Relative mRNA expression of target genes was normalized to the endogenous 18S control gene (Applied Biosystems). Induction of embryonic stem cell specific mRNAs was further corroborated by immunofluorescence protein staining of induced specific stem cell markers in 3T3/D3 cells. For immuno-staining, 3T3/3T3 and 3T3/D3 cells were cultured in medium in the absence and presence of LIF for 10 days. Then the cells were harvested and cultured 1 × 104 cells per well in 4-well slides coated with 0.5% gelatin for another 2 days. The slides were stained with specific antibodies to stem cell markers, c-Kit, SSEA1 and Oct-4. De-differentiation was also determined by the lamin B and lamin A/C (markers of soma) protein expression.
Cardiomyocyte and Endothelial Cell Lineage Differentiation of re-programmed cells
To determine the re-differentiation potency of dedifferentiated 3T3/D3 cells into cardiomyocytes, cells were cultured in complete DMEM containing 5 ng/ml of LIF and 3 ng/ml of Bone morphogenic protein −2 (BMP2) in 6-well culture plates (1 × 106 cells per well) and 4-well chamber slides (1 × 104 cells per well) coated with 0.5% gelatin for 7 days.25 Total cellular RNA was harvested from 6-well culture plate and used to analyze quantitative mRNA expression of CMC specific markers, cardiotroponin I and T, connexin 43, GATA4, Mef2c, Nkx2.5 and Tbx5. The expression was normalized to that of 18S RNA. The protein expression of was determined by immunochemical staining. For EC lineage differentiation, cells were cultured in endothelial differentiation medium (10% FBS/EBM-2; Clonetics) medium containing supplements (SingleQuot Kit; Clonetics) for 7 days. mRNA expression for EC markers, CD31 and Flk1 was determined by real-time PCR and by incubation with DiI-acLDL (Biomedical Technologies) for one hour followed by Isolectin B4 staining. The dual stained cells were considered endothelial cells.
Induction of Neuronal and Adipogenic Differentiation
The neuronal differentiation was performed as described earlier.27 Briefly, cells were seeded in complete DMEM medium at 5 × 105 cells per 90-mm sterile culture dish. Suspension cultures were maintained for 24 hrs before adding 10 μM all-trans-retinoic acid (Sigma-Aldrich). Cells were cultured for 3 wks in retinoic acid, replacing the medium every 2−3 days. Subsequently, cell aggregates were washed in complete DMEM medium and plated onto poly-l-lysine (10 μg/ml; Sigma-Aldrich)-coated plates in complete DMEM medium containing the mitotic inhibitors fluorodeoxyuridine (10 μM; Sigma-Aldrich), cytosine arabinosine (1 μM; Sigma-Aldrich), and uridine (10 μM; Sigma-Aldrich). The culture dishes were stained for neuronal markers nestin and β-tubulin-III. The adipogenic differentiation was performed as described elsewhere.28 Briefly, the cells were cultured for 21 days in complete DMEM/Ham's F-12 medium containing dexamethasone, insulin and indomethacin. Cells were fixed with 4% paraformaldehyde, washed in 5% isopropanol, and stained for 15 min with Oil-Red-O (Sigma Aldrich).
Immunochemical Staining
For immunochemical staining, cells under different culture conditions were cultured on 4 well slides for indicated time, rinsed once with PBS and fixed with 4% paraformaldehyde (Sigma) in PBS for 30 min. The slides were again rinsed three times with PBS and then permeabilized with 0.3% of Triton X-100 (Sigma) in PBS for 5 min. After 2 washes with PBS, specific primary antibodies diluted in PBS containing 1% FBS were added and incubated overnight at 4°C. After 3 washes with PBS, the slides were incubated with respective secondary antibodies for 1 hr at 37°C. The excess secondary Abs on the slides were rinsed off with PBS three times. Finally, to visualize nuclei, slides were stained with DAPI for 5 min, washed 3 times with PBS, allowed to dry for 5 min and then mounted on Vectashield mounting medium for fluorescence imaging. The photographs were taken in a Nikon TE200 Digital Imaging system.
Determination of Oct4 Promoter methylation, bisulfite genomic sequencing and chromatin immunoprecipitation (ChIP)
Genomic DNA prepared from mES cells (D3), 3T3/D3 and 3T3/3T3 cells was amplified for the Oct4 promoter and the PCR product was digested with HpyCH4IV restriction enzymes that only cleave at methylated CpG sites. The digested products were analyzed on agarose gels. For genomic bisulphite sequencing, genomic DNA from cells was digested with EcoR1 and was used for bisulphite treatment using an EZ DNA methylation-Gold kit essentially following the manufacturer's instructions. The treated DNA was ethanol-precipitated and resuspended in water and then amplified by PCR using mouse methylation specific Oct4 primers PCR products were digested with the HpyCH4IV (New England Biolabs) restriction enzyme. Because only unmethylated cytosine residues were changed to thymines by the sodium bisulphite reaction, PCR fragments from non-methylated genomic DNA were resistant to HpyCH4IV, and those from methylated DNA were digested by the enzymes. The resultant products of restriction mapping were assessed by agarose gel electrophoresis. The Remaining PCR products were purified through the Wizard DNA Clean-Up system (Promega, Madison, WI), and were directly sequenced to determine the methylation status of all 10 CpG residues present in the amplified promoter region. Chromatin Immunoprecipitation (ChIP) assays were performed essentially as described in our recent publication.29 Anti-Acetyl H3, anti-acetyl H4 and anti-dimethyl K9H3 antibodies were purchased from Upstate Biotech and Santa Cruz.
Genome-wide Expression Profiling and Gene Expression Analyses
Affymetrix mouse genome A2 GeneChips were used for hybridization. Using a poly-dT primer with a incorporated T7 promoter, double-stranded cDNA was synthesized from 5 μg total RNA using a double-stranded cDNA synthesis kit (Invitrogen, Carlsbad, CA). Double-stranded cDNA was purified with the Affymetrix sample cleanup module (Affymetrix, Santa Clara, CA). Biotin–labeled cRNA was generated from the double-stranded cDNA template though in-vitro transcription with T7 polymerase, and a nucleotide mix containing biotinylated UTP (3’-Amplification Reagents for IVT Labeling Kit; Affymetrix). The biotinylated cRNA was purified using the Affymetrix sample cleanup module. For each sample, 15 μg of IVT product was digested with fragmentation buffer (Affymetrix, Santa Clara, CA) for 35 minutes at 94°C, to an average size of 35 to 200 bases. 10 μg of the fragmented, biotinylated cRNA, along with hybridization controls (Affymetrix), was hybridized to a Mouse 430A 2.0 GeneChip for 16 hours at 45°C and 60 rpm. Arrays were washed and stained according to the standard Antibody Amplification for Eukaryotic Targets protocol (Affymetrix). The stained arrays were scanned at 532 nm using an Affymetrix GeneChip Scanner 3000.
During analysis and for quality control, GeneChip® arrays were first inspected using a series of quality control steps. Present call rates were consistent across the arrays, ranging from 56% to 63%. The hybridization controls (BioB, BioC, Cre) were found to be present 100% of the time. Images of all arrays were examined, and no obvious scratches or spatial variation was observed. A visual inspection of the distribution of raw PM probe values for the twelve arrays showed no outlying arrays. Similarly, digestion curves describing trends in RNA degradation between the 5’ end and the 3’ end of each probe set were generated, and all twelve proved comparable. Probe sets with no present calls across the twelve arrays as well as Affymetrix control probe sets were excluded from further analyses. Raw intensity values for the remaining 17,213 probe sets were processed first by RMA (Robust Multi-Array Average) using the R package affy.26 Specifically, expression values were computed from raw CEL files by first applying the RMA model of probe-specific correction of PM (perfect match) probes. These corrected probe values were then normalized via quantile normalization, and a median polish was applied to compute one expression measure from all probe values. Resulting RMA expression values were log2-transformed. (Please see the affy manual at www.bioconductor.org/repository/devel/vignette/affy.pdf for details). Distributions of expression values processed via RMA of all arrays were very similar with no apparent outlying arrays. Pearson correlation coefficients and Spearman rank coefficients were computed on the RMA expression values (log2-transformed) for each set of biological triplicates. Spearman coefficients ranged from .990 to .996; Pearson coefficients ranged between 0.991 and 0.997. Differential expression of genes was determined by one-way ANOVA on the RMA expression values of each probe set, using the R package limma.30 A multiple testing adjustment31 was performed on the resulting statistics to adjust the false discovery rate. Differentially expressed probes with adjusted p-values < 0.01 and a fold-change of greater than two (absolute log fold change of greater than 1) were extracted for further inspection. Hierarchical clustering was obtained by using the Pearson correlation coefficient and an average agglomeration method, and the heatmaps were generated using z-scored probes, in which z-scores (subtraction of mean and division by standard deviation of normalized values) were computed for each probe across all arrays.
GFP- Transduction and DiI Labeling of Cells for Transplantation
For tracking of transplanted cells in AMI model, cells were transduced with a lentivirus-GFP construct.25 For tracking of transplanted cells in hind limb ischemic tissues, the cells were labeled with DiI before the transplantation.27
Hind Limb Ischemia, Cell Transplantation, and Laser Doppler Imaging and Histology
All procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee. The hind limb ischemia was established by the excision of femoral artery in the left hind limb in 10 male 8-week- old FVB mice (Jackson Laboratory, Bar Harbor, ME) essentially as described in our prior publication.29 The animals were grouped into 2 (n=15/group), each receiving either a total of DiI-labeled- 3T3/3T3 cells or 3T3/D3 cells (1 × 105) at multiple sites into the ischemic muscle. Laser Doppler imaging to determine blood flow was performed immediately after surgery (day 0) and at days 7, 10 and 14 after cell injections. Fourteen days after cell transplantation, the tissues were harvested and assayed by histochemical/immuno-fluorescence staining for isolectin B4, CD31 (EC identity), Desmin, alpha-SMA (muscle), and DiI followed by fluorescence microscopy. In some experiments, animals were perfused with FITC–BS-1 lectin to identify capillaries before sacrifice and tissue retrieval.
Establishment of Acute Myocardial Infarction
The study involved 8-week-old male C57BL/6J mice (n=30; Jackson Laboratories). Mice underwent surgery to induce acute myocardial infarction by ligation of the left anterior descending coronary artery, as described before.32,33 Animals were sub-divided into 3 groups and received intramyocardial injection of 5×104 lentiviral-GFP transduced D3-extract treated cells, 3T3 fibroblast control cells and saline, respectively, in total volume of 10μl at 5 sites (basal anterior, mid anterior, mid lateral, apical anterior and apical lateral) in the peri-infarct area.
Physiological Assessments of LV Function and Histology
Mice underwent echocardiography just before MI (base level) and one, two and four weeks after AMI as described before.31,33. Briefly, transthoracic echocardiography was performed with a 6 to 15 MHz transducer (SONOS 5500, Hewlett Packard). Two-dimensional images were obtained in the parasternal long and short axis and apical 4-chamber views. M-mode images of the left ventricular short axis were taken just below the level of the mid-papillary muscles. Left ventricular end-diastolic and end-systolic dimensions were measured and functional shorting was determined according to the modified American Society of Echocardiography-recommended guidelines. A mean value of 3 measurements was determined for each sample. On day 28 post-AMI, Mice were euthanized and the aortas were perfused with saline. The hearts were sliced into 4 transverse sections from apex to base and fixed with 4% paraformaldehyde, methanol or frozen in OCT compound and sectioned into 5-μm thickness. Immunoflurorescence staining was performed to determine CMC and EC differentiation of transplanted cells. For the measurement of fibrosis, tissues sections were frozen in OCT compound and sectioned for elastic tissue/trichrome to measure the average ratio of the external circumference of fibrosis area to LV area.
Statistical analyses
All experiments were carried out at least 3 times with similar results. Results are presented as mean ± SEM. Comparisons were done by ANOVA (GB-STAT; Dynamic Microsystems Inc.) or χ2 test for percentages. All tests were 2-sided, and a P value of less than 0.05 was considered statistically significant.
Results
Exposure of NIH3T3 Fibroblasts to mESC Extracts Leads to the Induction of ESC Specific Genes
Reversibly permeabilized (by Sreptolysin O) NIH3T3 cells were exposed to either whole cell extracts from NIH3T3 cells (‘self’ as control) or D3 cells in an ATP-regenerating reaction (see Methods). Plasma membranes were sealed by including 2mM calcium chloride in medium and cells were further cultured in D3 maintenance medium (complete DMEM + 10 ng/mL of LIF). NIH3T3 cells treated with the self extracts (hereto referred as 3T3/3T3) did not show any morphological changes up to day 10 post-treatment (Fig. 1A[a]), whereas NIH3T3 cells treated with D3 extracts (hereto referred as 3T3/D3) showed noticeable changes in cell morphology as early as day 3 post-treatment forming colonies resembling ESC morphology by day 10 (Fig. 1A [b-d]). Quantitative mRNA expression of mESC specific transcripts Oct4, Nanog, SSEA1, SCF and c-Kit was induced in 3T3/D3 cells while 3T3/3T3 cells did not express measurable mRNA for any of these stem cell markers (Fig. 1B). Enhanced mRNA expression of stem cell specific genes was further corroborated by immuno-fluorescence staining for selected proteins Oct4 and SSEA1(Fig. 1C-D) Further corroboration of NIH3T3 de-differentiation was evident by the loss of lamin-A protein expression, a specific marker of somatic cells, in 3T3/D3 cells (Fig. 2A-B). Taken together, these data suggest that as an alternative to ESC-somatic cell fusion and somatic nuclear transfer, cell-free ESC extracts could provide the necessary regulatory components required to induce somatic nuclear reprogramming and alter the differentiation status of non-embryonic cells. We further confirmed whether the D3 proteins and not the contaminating nucleic acids drove 3T3/D3 de-differentiation. Inactivation of proteins from D3 extracts prior to reaction either by temperature, trypsin or proteinase K digestion failed to induce any morphological changes or to the activation of Oct4 mRNA (Fig. 2C), whereas inactivation of either DNA or RNA had no such effect.
Figure 1. mES cell free extracts induce reactivation of ES-specific genes in NIH3T3 cells.
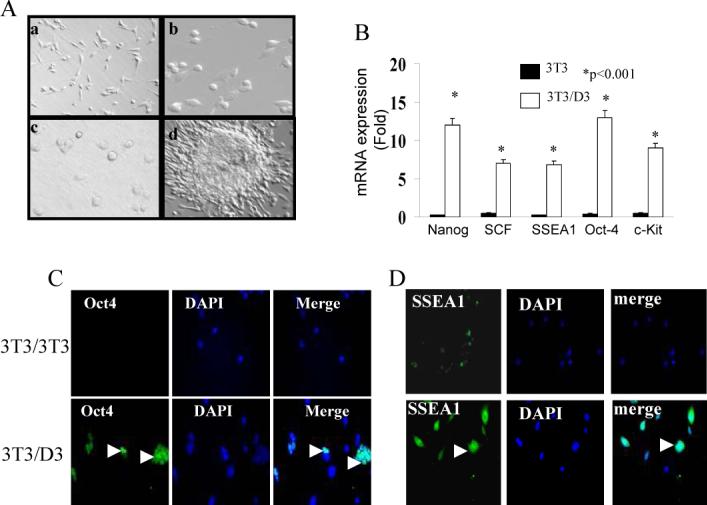
NIH3T3 fibroblasts were reversibly permeabilized with SLO and exposed to D3-mESC whole cell extracts or to control self (NIH3T3) extracts. Cells were cultured in DMEM supplemented with LIF (10ng/ml) and monitored daily for morphological changes. (A) Representative phase contrast image of self-extract treated (3T3/3T3) on day 10 (a) and D3-extract treated (3T3/D3) on days 3 (b), 5 (c) and 10 (d). (B) Total RNA from 3T3/D3, 4 weeks after initial treatment, was isolated and analyzed for the quantitative mRNA expression by real-time RT-PCR for indicated ESC markers. Data is plotted as fold mRNA expression compared to the mRNA levels in self-extract treated 3T3 cells averaged from 3 similar experiments. 3T3/D3 cells (4 weeks after initial treatment) were cultured on 4 well slides and protein expression of Oct4 (C) and SSEA1 (D) was determined by immuno-fluorescence staining. Representative micrographs are shown.
Figure 2. Re-programmed 3T3/D3 exhibit loss of somatic cell marker.

Protein expression of somatic cell marker lamin A was determined in 3T3/3T3 and 3T3/D3 cells by immuno-fluorescence cyto-chemistry (A) and by western blotting (B). Expression of lamin A was exclusively reduced in 3T3/D3 cells. (C) Prior to re-programming reaction, D3-extracts were pre-treated with RNases, DNase, Proteinase K, Trypsin or were exposed to 70 °C temperature. Induction of Oct4 mRNA was determined in 3T3 cells exposed to variously pre-treated D3-extracts. Only protein inactivation treatments resulted in the loss of mES extracts ability to reactivate Oct4 mRNA induction.
D3-Extract Induced Epigenetic Changes in 3T3/D3 Cells Involve Oct4 Promoter Demethylation and Post-translational Histone modifications
DNA methylation of CpG residues leading to the silencing of pluripotent embryonic genes, including that of Oct4, is known as an integral step governing differentiation and development. Since our data indicated that D3-extract exposure leads to the induction of Oct4 mRNA and protein expression in 3T3/D3 cells, we performed CpG methylation status analysis of Oct4 promoter. As depicted in Fig. 3A, upon bisulphite treatment (see methods) all 10 CpG sites in D3 cells were unmethylated (open circles). All 10 sites were methylated in 3T3/3T3 cells (closed circles), however, treatment of 3T3 cells with D3 extracts led to de-methylation at 8/10 CpG residues in 3T3/D3 cells. The D3-extract induced Oct4 promoter demethylation in the 3T3/D3 cells was independently corroborated by restriction enzyme digestion. In the Oct4 promoter region, there is one HpyCH4IV (methylated CpG specific restriction enzyme) site at −202. We analyzed the DNA methylation status of the −202 site by HpyCH4IV restriction digestion analysis of PCR amplified Oct4 promoter in D3, 3T3/3T3 and 3T3/D3 cells. As shown in Fig 3B, the PCR product was not digested with HpyCH4IV in D3 cells, indicating that the genomic DNA of D3 cells was unmethylated at this particular Oct4 promoter site. In contrast, the PCR product was readily digested in 3T3/3T3 cells indicating methylation of the Oct4 −202 CpG site. Interestingly, the PCR product from 3T3/D3 cells was resistant to digestion by HypCH4IV, suggesting that treatment of 3T3 cells with D3 extracts induced de-methylation of CpG sites thereby reversing the repression of Oct4 mRNA expression, observed in 3T3/3T3 cells.
Figure 3. D3 extract treatment induces epigenetic changes in 3T3/D3 cells.

(A) Genomic DNA from indicated cells was digested with EcoRI and was treated with sodium meta-bisulphite. Oct4 promoter was amplified from modified DNA using specific primers by PCR and PCR products were sequenced for the evidence of cytosine conversion to thymine at unmethylated CpG. Filled circle represents methylated CpG and open circles represent unmethylated CpG in Oct4 promoter. (B) Oct4 promoter fragment was amplified from bisulphite treated genomic DNA of indicated cells and was subjected to digestion with HypCH4IV restriction enzyme that specifically cleaves methylated CpG. Post-digestion DNA was resolved on 2% ethidium bromide stained gels and photographed. The promoter of Oct4 was analyzed by ChIP for histone H3 and H4 acetylation (C) and for dimethylation status of lysine 9 of histone H3 (D). Gels from 3 separate experiments were quantified by NIH image analysis and average values were plotted against levels observed in D3 cells (arbitrarily given a numerical value of 1).
DNA methylation/demethylation dependent gene suppression/activation is coupled with post-translational modifications to the histone proteins, which together lead to chromatin remodeling and new patterns of gene expression.30, 31 Therefore, we further confirmed the D3-extract induced epigenetic changes by assessing the acetylation of histones (H) 3 and 4 and methylation status of lysine 9 (K9) residue in histone 3 protein within Oct4 promoter by Chromatin Immunoprecipitation (ChIP) assays29 using specific antibodies. The Oct4 promoter was amplified from immunoprecipitated chromatin DNA by PCR. ChIP analyses showed that the promoter of Oct4 had increased acetylation of H3 and H4 (Fig. 3C) and decreased dimethylation of K9-histone 3 (Fig. 3D) in 3T3/D3 cells compared to 3T3/3T3 cells. Together these data suggest that D3-extract induced de-differentiation and nuclear re-programming of 3T3/D3 cells is mediated, at least in part, by chromatin remodeling leading to the activation of Oct4. Considering that DNA methylation and histone modifications are involved in various biological phenomena, such as tissue-specific gene expression, cell differentiation, X-chromosome inactivation, genomic imprinting, changes in chromatin structure, and tumorogenesis,35-43 it is conceivable that the changes in Oct4 promoter CpG methylation and histone modifications by exposure of 3T3 cells to ESC extracts, may be one of the principal epigenetic events underlying de-differentiation and activation of ESC specific genes in 3T3 cells.
D3-Extract Treatment Induces Genome-wide Gene Expression Profile Changes in Reprogrammed 3T3/D3 cells
To gain further insights into the changes in gene expression patterns in reprogrammed 3T3/D3 cells, we performed global gene-expression profiles of D3, 3T3/3T3 and 3T3/D3 using Affymetrix mouse genome 2A gene chips.44-45 Differentially expressed probes with adjusted p-value < 0.001 and fold-change of greater than 2 (absolute log fold change of > than 1) were extracted for further inspection. This resulted in 3,286 probes with statistically significant differential expression between cell types 3T3/3T3 and 3T3/D3 including the significant up-regulation of ESC specific genes and down-regulation of somatic genes. The heatmap of z-scored probes illustrating this clustering, and the expression pattern of a subset of 99 significantly up-regulated genes in 3T3/D3 and D3 cells, including Oct4 and nanog, is shown in supplementary figures S1A, B. and functional grouping of all 3,286 genes found to be differentially expressed between 3T3 and 3T3/D3 cell types is shown in Fig. S1C. The expression levels of representative up-regulated and down-regulated genes observed in gene chip experiments were independently confirmed by real-time RT-PCR which confirmed the expression pattern observed in gene profiling experiments (Fig. 4). A list of selected genes, their relative expression level and functional description is depicted in Table S1
Figure 4.

Selected genes from expression profiling experiments (figure S1) that were either up or down-regulated in 3t3/D3 cells, were confirmed for their mRNA expression pattern quantitative real-time PCR.
Re-programmed 3T3/D3 Cells Re-differentiate into Cells of Multiple Lineages
We next determined the re-differentiation potential of de-differentiated 3T3/D3 cells into multiple cell types, in vitro. Under culture conditions conducive for cell-type specific differentiation, 3T3/D3 cells acquired morphology (Fig. S2), mRNA and protein expression of cardiomyocyte specific (Figs. 5A, B) and endothelial cell specific (Figs. 5C, D) genes. Furthermore, under specific culture conditions (see methods) 3T3/D3 cells differentiated into neuronal and adipocyte phenotypes (Fig. S3).
Figure 5. Cardiomyocyte and Endothelial differentiation of reprogrammed 3T3/D3 cells.
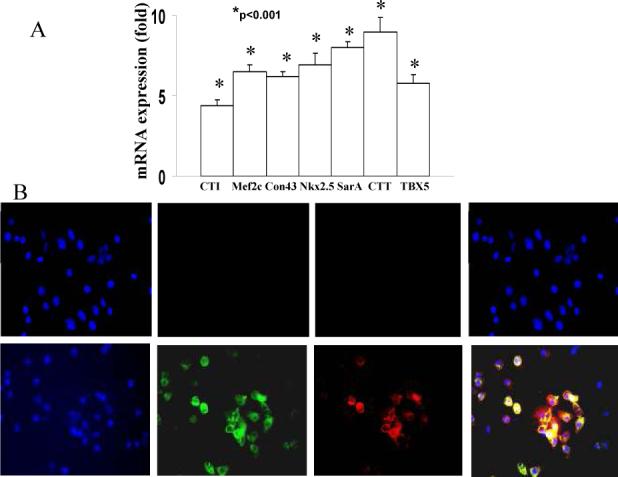
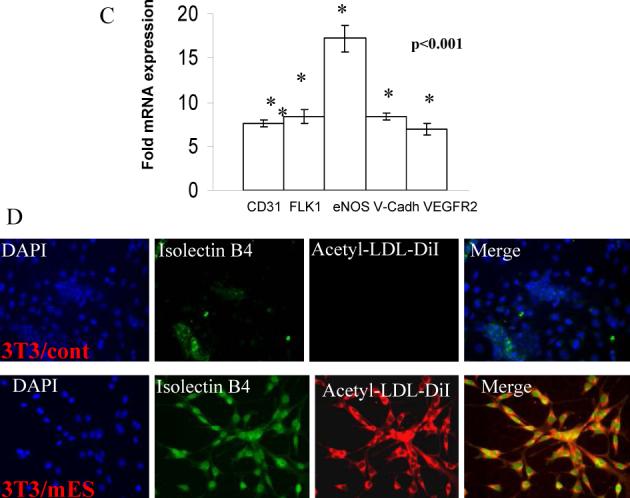
(A) 3T3/D3 and 3T3/3T3 cells were cultured under CMC differentiation conditions and total RNA extracted and was analyzed for the mRNA expression of CMA-specific transcripts by real-time PCR. Data is shown as fold up-regulation in 3T3/D3 cells over 3T3/3T3 cells (A). (B) Under CMC-differentiation culture conditions 3T3/D3 cells showed the protein expression of CMC -specific proteins, connexin43 (green) and cardiotroponin I (red). Merged image depicts cells expressing both of these CMC markers (yellow fluorescence). (C) Total RNA extracted from 3T3/3T3 and 3T3/D3 cells cultured under EC differentiation conditions was analyzed for the mRNA expression of EC-specific transcripts by real-time PCR. Data is shown as fold up-regulation in 3T3/D3 cells over 3T3/3T3 cells. (D) Under culture conditions conducive to endothelial cell differentiation, 3T3/D3 cells show expression of EC-specific proteins (merged image depicts DiI-AceLDL + Isolectin B4 double positive cells).
Transplantation of Re-programmed 3T3 Cells into Surgically Induced Mouse Hind Limb Ischemia Model Improves Functional and Anatomical Repair
To ascertain the functional efficacy of reprogrammed cells in ischemic tissue repair, we conducted cell transplantation studies in a well-established mouse hind limb ischemia model described in our prior publication.28 Immediately following the surgery, mice were assigned to two groups (n=15 each) and 3T3/D3 or 3T3/3T3 cells (1×105), labeled with DiI for tracing purposes, were injected into the ischemic muscles at 3 different sites. Physiological blood flow recovery was assessed by Laser Doppler Perfusion Imaging (LDPI) on days 7, 10 and 14 (n=5 each time point) post- surgery, in both groups of mice. As shown in representative perfusion images in Fig. 6A and quantified as the ratio of blood flow in ischemic to non-ischemic limb, in Fig. 6B, mice transplanted with 3T3/D3 cells displayed significantly improved perfusion in the ischemic limb at all time points compared to mice treated with 3T3/3T3 cells (p<0.01). Enhanced blood flow recovery in mice transplanted with 3T3/D3 cells was further corroborated by increased capillary density in the ischemic hind limbs (Fig. 6C). Histological analyses on retrieved tissues indicated both EC and skeletal muscle differentiation of transplanted 3T3/D3 cells (Fig. S4)
Figure 6. Transplantation of 3T3/D3 cells improves recovery of ischemic hind limb perfusion.
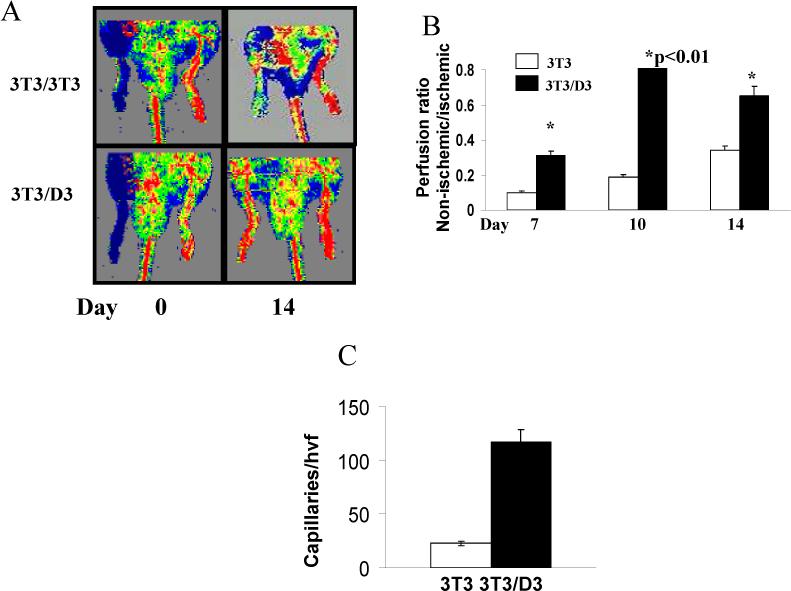
(A) Representative perfusion imaging of ischemic hind limbs immediately after HLI (left panels) and 14 days post transplantation of either 3T3/D3 (top right panel) or 3T3/D3 (bottom right panel) cells. (B) Cumulative perfusion of ischemic hind limbs on various days plotted as the ratio of blood flow in ischemic: non-ischemic limbs. (C) Capillaries in the ischemic limbs were identified as fluorescent structures, stained with in vivo perfusion with FITC-BS-1 lectin. The number of capillaries/per high visual field in different sections was quantified and averaged.
Intra-cardiac Transplantation of 3T3/D3 Cells leads to Cardiomyocyte Differentiation and Improves Left Ventricular Functions in mouse AMI model
Functional efficacy of 3T3/D3 cells in tissue repair was also investigated in a model of acute myocardial infarction (AMI). Mice underwent surgery to induce AMI by ligation of the left anterior descending coronary artery, as described.32, 34 Animals were sub-divided into 3 groups (10 each), and received intramyocardial injection of 5×104 lentiviral-GFP transduced 3T3/D3, 3T3/3T3 cells or saline, respectively. Echocardiography data revealed that left ventricular end-diastolic areas (LVEDA) were similar in the 3T3/3T3 cell and saline groups before and at all time points after AMI (Fig. 7A, red and grey lines, respectively). In contrast, however, mice treated with the 3T3/D3 cells revealed less ventricular dilation (Fig. 7A blue line, p< 0.01 in 3T3/D3 vs. control groups). Additionally, fractional shortening (FS), an indicator of contractile function, was consistently depressed in mice receiving saline and 3T3/3T3 cells (Fig. 7B). However, treatment with 3T3/D3 cells significantly improved FS at all time points tested (at 4 weeks post-surgery, p<0.05 vs. 3T3/3T3 cell treated group). Gains in post-AMI physiological functions in mice transplanted with 3T3/D3 cells were further corroborated by histological evaluation of hearts from each group of mice. As shown in Fig. 7C, the percent fibrosis areas in mouse hearts receiving either saline or 3T3/3T3 cells were significantly larger than in mice that received 3T3/D3 cells (p<0.001). Tissue sections were also stained with BS1 lectin to determine the capillary density at the border zone of the infarcted myocardium. Significantly higher capillary density was observed in the mice receiving 3T3/D3 cells than in mice receiving 3T3/3T3 cells or saline (Fig. 7D, p<0.01). Immuno-fluorescence staining on myocardial sections was performed to determine CMC and EC differentiation of the transplanted (GFP+) cells. EC differentiation of transplanted cells was investigated by co-expression of GFP+CD31 by transplanted cells. As shown in Fig. 8A, GFP+CD31 double positive cells were observed in myocardial sections obtained from mice transplanted with 3T3/D3 cells, while sections obtained from 3T3/3T3 transplanted hearts did not show the evidence for EC differentiation. CMC differentiation of the transplanted cells in the myocardium, was determined by the co-expression of GFP (green) and cardiomyocyte specific cardiotroponin I (CTI, red). As shown in Fig. 8B, GFP+CTI double positive cells (yellow fluorescence in merged image) were observed in mice treated with 3T3/D3 cells, suggesting that some of the transplanted cells differentiated into CMC lineage in vivo, while no evidence of CMC differentiation was observed for transplanted 3T3/3T3 cells. Transplanted 3T3/D3 cells also showed the evidence of proliferation in vivo (day 28), determined by co-localization of GFP+cells with nuclear proliferation antigen, ki67 (Fig. S5), while no GFP+Ki67 double positive cells were observed in 3T3/3T3 cell transplanted myocardial sections.
Figure 7. Transplantation of 3T3/D3 cells improves Left Ventricular functions in a mouse model of acute myocardial infarction.

Transplantation of 3T3/D3 cells significantly improved left ventricular end-diastolic areas (A) and left ventricular fractional shortening (B) as compared to mice treated with 3T3/3T3 cells and/or saline. (C) Quantification of % fibrosis area in 3 groups of mice. (D) Quantification of capillary density. Mice were perfused with FITC-BS1 lectin and fluorescently labeled capillaries were counted in 6 randomly selected tissue sections at the border zone from each animal.
Figure 8. Transplanted 3T3/D3 cells differentiate into EC and CMCs, in vivo.

(A) Representative merged images showing co-localization of GFP+ (green) transplanted 3T3/D3 and 3T3/3T3 cells and EC specific marker, CD31+ (red) cells. Double positive cells are identified by yellow fluorescence in the merged images. Higher magnification of area in inset is shown on right. (B) Representative merged micrographs showing co-localization of GFP+ (green) transplanted 3T3/D3 and 3T3/3T3 cells and CMC specific marker, cardiotroponin I+ (red) cells. Double positive cells are identified by yellow fluorescence in the merged images. Higher magnification of area in inset is shown on right.
Discussion
The goal of therapeutic cloning is to produce pluripotent stem cells with the nuclear genome of the patient and induce the cells to differentiate into replacement cells, for example, CMCs for repairing damaged heart tissue. Reports on the generation of pluripotent stem cells14-16 or histocompatible tissues41 by nuclear transplantation, and on the correction of a genetic defect in cloned ESCs,47 suggest that therapeutic cloning could, in theory, provide a source of cells for regenerative therapy. Recent evidence on the efficacy of human therapeutic cloning, however, underscores the difficulties associated with the generation of human ESC lines for therapeutic purposes. Moreover, a number of limitations may hinder the strategy of therapeutic cloning for future clinical applications. Extremely low efficiency of somatic nuclear transfer is a major concern.48 Analysis of the literature on mouse SNT derived ESC lines raises concerns about the feasibility and relevance of therapeutic cloning, in its current embodiment, for human clinical practice.48-50 This limitation might be alleviated with oocytes from other species41 but mitochondrial genome differences between species are likely to pose a problem. It is therefore desirable to develop alternative strategies to oocyte-dependent autologous stem cell generation. Our data indicating mESC extract-mediated reverse de-differentiation of terminally differentiated murine fibroblasts and multi-lineage re-differentiation of these reprogrammed cells not only support the feasibility of such an approach but more importantly, provide evidence that the stem-like cells obtained using this methodology are functionally competent for tissue repair.
Oocyte-independent epigenetic re-programming of somatic cell by somatic-ES cell fusion reported by several recent studies has generated a lot of enthusiasm9, 10 However, this strategy, although excellent for mechanistic studies, retains certain drawbacks that are associated with oocyte-dependent therapeutic cloning. Firstly, the 2 cells used to generate hybrid cells are not derived from autologous source, secondly the efficiency of fusion remains low (1−3/1000), thirdly the genetic stability of heterokaryon hybrids remains to be established:- one will have to devise the technological innovations to delete additional set of chromosomes, and finally the efficacy of reprogrammed cell to retain the ES like properties if ES cell derived nucleus is removed remains to be elucidated. Recently, a major breakthrough was reported whereby forced expression of transcription factors Oct4, Sox2, c-Myc and Klf4 was shown to induce pluripotency in primary mouse fibroblasts51 and in human fibroblasts using either Oct4, Sox2, c-myc and Klf4 combination11 or Oct4, nanog, Sox2, lin28 combination.12 While these studies did provide the evidence that over-expression of these ES-specific genes leads to the derivation of ES-like cells from primary human fibroblasts, several critical questions were not answered by these studies. First, it appears, that the requirement of these factors or their combination is varied. These studies utilized different combinations of transcription factors in similar human cells and achieved similar results indicating that some of these factors may be dispensable. Secondly, these studies did not answer the question that how these transcription factors re-activated epigenetically silent endogenous pluripotent genes (e.g. that of Oct4) since chromatin changes would be required to make transcriptionally silent promoters of these pluripotent genes in order to bind these transcription factors. Finally four transcription factors were transduced by constitutively expressed retroviral vectors it is unclear why the cells could be induced to differentiate and whether continuous vector expression was required for the maintenance of the pluripotent state. Despite these drawbacks, these studies did provide the identification of certain transcription factors which in combination drive the de-differentiation of somatic cells leading to reactivation of pluripotent genes and ES like phenotype. mES cell-free extracts express all of the above proteins besides other putative chromatin modifying factors. Our data suggests that similar changes in somatic cell fate reported in these studies can be achieved without the introduction of transgenes or viral vectors.
Our observations demonstrating that mESC protein extracts can reprogram somatic cells towards multipotency, would argue that multipotent epigenome could be activated in somatic cells without fusion and forced expression of nucleic acids. More importantly, our's is the first study to demonstrate that transplantation of de-differentiated somatic cells can repair ischemic tissue and mediate gain in physiological functions in relevant models of tissue injury. We, however, recognize certain limitations that our data does not sufficiently clarify. First, in vitro differentiation of reprogrammed 3T3/D3 cells did not reveal any “beating cell”. This may either reflect incomplete in vitro differentiation or more likely a reflection on in vitro culture conditions employed for CMC differentiation. We only used 2 factors (LIF+BMP2 for 4−7 days) for CMC-differentiation, conditions that we have previously demonstrated lead to CMC pre-commitment of mES cells.25 Inclusion of additional cytokine as well as co-culture with rat neonatal CMCs may enhance the CMC-differentiation potential of 3T3/D3 cells. Second only 2−3% of transplanted 3T3/D3 cells were retained in the myocardium by day 28, although 3T3/D3 cells were retained in higher number than control 3T3/3T3 cells. Thus it appears that our re-programmed cells share the similar low retention as has been observed with the transplantation of other adult stem cells. Finally, limited CMC and EC differentiation of transplanted 3T3/D3 cells may not be solely responsible for gains in the physiological functions. Indeed it is quite possible that these reprogram cells may also participate in the ischemic tissue repair by secreting paracrine growth factors resulting in enhanced neovascularization and/or resident cardiomyocyte protection/salvage. Limited in vivo differentiation may also reflect the fact that the cells used in this study represented pooled cells many of which were not completely reprogrammed. We have recently obtained clonal derivatives from reprogrammed cells which display long-term expression of mES specific genes. Future studies utilizing these cells will help clarify these and other issues.
Taken together our biochemical, molecular and functional data provide an oocyte-independent approach for the generation of functional autologous multipotent cells from terminally differentiated somatic cells. The refinement of techniques and additional experimental data to elucidate applicability of this approach in primary somatic cells of different lineages and derivation of single cell clones displaying stable, long-term reprogramming may hold significant promise for future use of such generated cells in regenerative medicine, including cardiac repair and regeneration.
Table 1.
Changes in expression level (fold) of selected transcripts in 3T3/D3 cells (4 weeks after reprogramming)
| Name | Description | 3T3/D3 | Pattern |
|---|---|---|---|
| Somatic cell markers | |||
| LMNA | Nuclear lamin A | 0.045 | down |
| LMNB1 | Nuclear lamin B1 | 1.67 | NA |
| LMNB2 | Nuclear lamin B2 | 1.1 | NA |
| NPR3 | Atrionatriuretic peptide receptor | 0.009 | down |
| PRC1 | Protein regulator of cytokinesis | 0.004 | down |
| CKAP2 | Cytoskeleton assosciated protein2 | 0.001 | down |
| FIN15 | FGF inducible15 | 0.018 | down |
| Embryonic stem cell/pluripotency markers | |||
| Pou5f1 | POU domain class 5 (Oct4) | 14.3 | up |
| Nanog | Nanog | 43.4 | up |
| Sox2 | Sex determining region Y box2 | 3.4 | up |
| Dppa3 | Developmental pluripotency associated | 5.1 | up |
| Dppa5 | Developmental pluripotency associated | 3.8 | up |
| FOXD3 | Forkhead box D3 | 34.0 | up |
| Kit | kit oncogene | 9.6 | up |
| DAD1 | Defender against cell death | 54.0 | up |
| Gdf3 | Netrin G1 | 2.0 | up |
| S1c2a3 | Solute carrier family 2 | 3.2 | up |
| Chromatin structure/function | |||
| JARID1B | Jumonji interactive domain | 6.38 | up |
| Hist1H2bc | Histonel, H2bc | 32.3 | up |
| HBP1 | High mobility group box trans. Factor | 4.8 | up |
| HDAC11 | Histone deacetylase 11 | 4.2 | up |
| HDAC1 | Histone deacetylase 1 | 6.7 | down |
| SOAT1 | Sterol O acetyltransferase 1 | 3.2 | up |
| Atp6v0d1 | ATPase H+ transporting | 43.0 | up |
| CREB | cAMP response element binding protein | 3.8 | up |
| LSD1 | Lysine specific histone demethylase | 11.2 | down |
| Acaa1a | acetyl transferase | 3.8 | up |
| Dnmt1 | DNA methyltransferase | 5.6 | down |
| Suv39h2 | Histone demethylase | 6.9 | up |
| Smarcad1 | SWI/SNF-related regulator of chromatin | 17.6 | up |
| Smc2 | Structural maintenance of chromatin | 12.6 | down |
| Nusap1 | Nucleolar and spindle associated protein | 13.3 | down |
Sources of Funding
Work described in this manuscript was in part supported by American Heart Association grant 0530350N and National Institute of Health grant AA014575 to R.K. and National Institute of Health grants HL63414 to D.W.L.
Footnotes
Publisher's Disclaimer: This is an un-copyedited author manuscript accepted for publication in Circulation Research, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at http://circres.ahajournals.org/. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
Disclosures
None
Supplementary Material
References
- 1.Losordo DW, Dimmeler S. Therapeutic angiogenesis and vasculogenesis for ischemic disease: part II: cell-based therapies. Circulation. 2004;109:2692–2697. doi: 10.1161/01.CIR.0000128596.49339.05. [DOI] [PubMed] [Google Scholar]
- 2.Dimmeler S. ATVB in focus: novel mediators and mechanisms in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol. 2005;11:2245. doi: 10.1161/01.ATV.0000187471.06942.17. [DOI] [PubMed] [Google Scholar]
- 3.Walter DH, Haendeler J, Reinhold J, Rochwalsky U, Seeger F, Honold J, Hoffmann J, Urbich C, Lehmann R, Arenzana-Seisdesdos F, Aicher A, Heeschen C, Fichtlscherer S, Zeiher AM, Dimmeler S. Impaired CXCR4 signaling contributes to the reduced neovascularization capacity of endothelial progenitor cells from patients with coronary artery disease. Circ Res. 2005;97:1142–1151. doi: 10.1161/01.RES.0000193596.94936.2c. [DOI] [PubMed] [Google Scholar]
- 4.Urbich C, Dimmeler S. Risk factors for coronary artery disease, circulating endothelial progenitor cells, and the role of HMG-CoA reductase inhibitors. Kidney Int. 2005;67:1672–1676. doi: 10.1111/j.1523-1755.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- 5.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 6.Ii M, Takenaka H, Asai J, Ibusuki K, Mizukami Y, Maruyama K, Yoon YS, Wecker A, Luedemann C, Eaton E, Silver M, Thorne T, Losordo DW. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ Res. 2006;98:697–704. doi: 10.1161/01.RES.0000209948.50943.ea. [DOI] [PubMed] [Google Scholar]
- 7.Byrne JA, Simonsson S, Western PS, Gurdon JB. Nuclei of adult mammalian somatic cells are directly reprogrammed to oct-4 stem cell gene expression by amphibian oocytes. Curr Biol. 2003;13:1206–1213. doi: 10.1016/s0960-9822(03)00462-7. [DOI] [PubMed] [Google Scholar]
- 8.Yamanaka S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell. 2007;1:39–49. doi: 10.1016/j.stem.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 9.Tada M, Takahama Y, Abe K, Nakastuji N, Tada T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr Biol. 2001;11:1553–1558. doi: 10.1016/s0960-9822(01)00459-6. [DOI] [PubMed] [Google Scholar]
- 10.Cowan CA, Atienza J, Melton DA, Eggan K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 2005;309:1369–1373. doi: 10.1126/science.1116447. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 12.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 13.Gurdon JB, Byrne JA. The first half-century of nuclear transplantation. Proc Natl Acad Sci USA. 2003;100:8048–8052. doi: 10.1073/pnas.1337135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cibelli JB, Stice SL, Golueke PJ, Kane JJ, Jerry J, Blackwell C, Ponce de Leon FA, Robl JM. Transgenic bovine chimeric offspring produced from somatic cell-derived stem-like cells. Nat Biotechnol. 1998;16:642–646. doi: 10.1038/nbt0798-642. [DOI] [PubMed] [Google Scholar]
- 15.Munsie MJ, Michalska AE, O'Brien CM, Trounson AO, Pera MF, Mountford PS. Isolation of pluripotent embryonic stem cells from reprogrammed adult mouse somatic cell nuclei. Curr Biol. 2000;10:989–992. doi: 10.1016/s0960-9822(00)00648-5. [DOI] [PubMed] [Google Scholar]
- 16.Wakayama T, Tabar V, Rodriguez I, Perry AC, Studer L, Mombaerts P. Differentiation of embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Science. 2001;292:740–743. doi: 10.1126/science.1059399. [DOI] [PubMed] [Google Scholar]
- 17.Hwang WS, Lee BC, Lee CK, Kang SK. Human embryonic stem cells and therapeutic cloning. J Vet Sci. 2005;6:87–96. [PubMed] [Google Scholar]
- 18.Gaustad KG, Boquest AC, Anderson BE, Gerdes AM, Collas P. Differentiation of human adipose tissue stem cells using extracts of rat cardiomyocytes. Biochem Biophys Res Commun. 2004;314:420–427. doi: 10.1016/j.bbrc.2003.12.109. [DOI] [PubMed] [Google Scholar]
- 19.Hakelien AM, Gaustad KG, Taranger CK, Skalhegg BS, Kuntziger T, Collas P. Long-term in vitro, cell-type-specific genome-wide reprogramming of gene expression. Exp Cell Res. 2005;309:32–47. doi: 10.1016/j.yexcr.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Landsverk HB, Hakelien AM, Kuntziger T, Robl JM, Skalhegg BS, Collas P. Reprogrammed gene expression in a somatic cell-free extract. EMBO Rep. 2002;3:384–389. doi: 10.1093/embo-reports/kvf064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGann CJ, Odelberg SJ, Keating MT. Mammalian myotube dedifferentiation induced by newt regeneration extract. Proc Natl Acad Sci USA. 2001;98:13699–13704. doi: 10.1073/pnas.221297398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansis C, Barreto G, Maltry N, Niehrs C. Nuclear reprogramming of human somatic cells by xenopus egg extract requires BRG1. Curr Biol. 2004;14:1475–1480. doi: 10.1016/j.cub.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 23.Qin M, Tai G, Collas P, Polak JM, Bishop AE. Cell extract-derived differentiation of embryonic stem cells. Stem Cells. 2005;23:712–718. doi: 10.1634/stemcells.2004-0195. [DOI] [PubMed] [Google Scholar]
- 24.Taranger CK, Noer A, Sorensen AL, Hakelien AM, Boquest AC, Collas P. Induction of Dedifferentiation, Genomewide Transcriptional Programming, and Epigenetic Reprogramming by Extracts of Carcinoma and Embryonic Stem Cells. Mol Biol Cell. 2005;16:5719–5735. doi: 10.1091/mbc.E05-06-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajasingh J, Bord E, Hamada H, Lambers E, Qin G, Losordo DW, Kishore R. STAT3-Dependent Mouse Embryonic Stem Cell Differentiation Into Cardiomyocytes. Analysis of Molecular Signaling and Therapeutic Efficacy of Cardiomyocyte Precommitted mES Transplantation in a Mouse Model of Myocardial Infarction. Circ Res. 2007;101:910–918. doi: 10.1161/CIRCRESAHA.107.156786. [DOI] [PubMed] [Google Scholar]
- 26.Irizarry RA, Hobbs R, Collin R, Beazer-Barclay YD. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 27.Stewart R, Christie VB, Przyborski SA. Manipulation of human pluripotent embryonal carcinoma stem cells and the development of neural subtypes. Stem Cells. 2003;21:248–256. doi: 10.1634/stemcells.21-3-248. [DOI] [PubMed] [Google Scholar]
- 28.Boquest AC, Shahdadfar A, Fronsdal K, Sigurjonsson O, Tunheim SH, Collas P, Brinchmann JE. Isolation and transcription profiling of purified uncultured human stromal stem cells: alteration of gene expression after in vitro cell culture. Mol Biol Cell. 2005;3:1131–1141. doi: 10.1091/mbc.E04-10-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kishore R, Qin G, Luedemann C, Bord E, Hanley A, Silver M, Gavin M, Yoon YS, Goukassian D, Losordo DW. The cytoskeletal protein ezrin regulates EC proliferation and angiogenesis via TNF-alpha-induced transcriptional repression of cyclin A. J Clin Invest. 2005;115:1785–1796. doi: 10.1172/JCI22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trojer P, Reinberg D. Histone Lysine Demethylase and their impact on epigenetics. Cell. 2006;125:213–217. doi: 10.1016/j.cell.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 32.Iwakura A, Shastry S, Luedemann C, Hamada H, Kawamoto A, Kishore R, Zhu Y, Qin G, Silver M, Thorne T, Eaton E, Masuda H, Asahara T, Losordo DW. Estradiol enhances recovery after myocardial infarction by augmenting incorporation of bone marrow-derived endothelial progenitor cells into sites of ischemia-induced neovascularization via endothelial nitric oxide synthase-mediated activation of matrix metalloproteinase-9. Circulation. 2006;113:1605–1614. doi: 10.1161/CIRCULATIONAHA.105.553925. [DOI] [PubMed] [Google Scholar]
- 33.Kusano KF, Pola R, Murayama T, Curry C, Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, Thorne T, Takenaka H, Aikawa R, Goukassian D, von Samson P, Hamada H, Yoon YS, Silver M, Eaton E, Ma H, Heyd L, Kearney M, Munger W, Porter JA, Kishore R, Losordo DW. Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat Med. 2005;11:1197–1204. doi: 10.1038/nm1313. [DOI] [PubMed] [Google Scholar]
- 34.Yoon YS, Murayama T, Gravereaux E, Tkebuchava T, Silver M, Curry C, Wecker A, Kirchmair R, Hu CS, Kearney M, Ashare A, Jackson DG, Kubo H, Isner JM, Losordo DW. VEGF-C gene therapy augments postnatal lymphangiogenesis and ameliorates secondary lymphedema. J Clin Invest. 2003;111:717–725. doi: 10.1172/JCI15830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;2:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 36.Cho JH, Kimura H, Minami T, Ohgane J, Hattori N, Tanaka S, Shiota K. DNA methylation regulates placental lactogen I gene expression. Endocrinology. 2001;142:3389–3396. doi: 10.1210/endo.142.8.8347. [DOI] [PubMed] [Google Scholar]
- 37.Okamoto K, Okazawa H, Okuda A, Sakai M, Muramatsu M, Hamada H. A novel octamer binding transcription factor is differentially expressed in mouse embryonic cells. Cell. 1990;60:461–472. doi: 10.1016/0092-8674(90)90597-8. [DOI] [PubMed] [Google Scholar]
- 38.Rosner MH, Vigano MA, Ozato K, Timmons PM, Poirier F, Rigby PW, Staudt LM. A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature. 1990;345:686–692. doi: 10.1038/345686a0. [DOI] [PubMed] [Google Scholar]
- 39.Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Scholer H, Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 40.Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 41.Deb-Rinker P, Ly D, Jezierski A, Sikorska M, Walker PR. Sequential DNA methylation of the Nanog and Oct-4 upstream regions in human NT2 cells during neuronal differentiation. J Biol Chem. 2005;280:6257–6260. doi: 10.1074/jbc.C400479200. [DOI] [PubMed] [Google Scholar]
- 42.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 43.Ng HH, Jeppesen P, Bird A. Active repression of methylated genes by the chromosomal protein MBD1. Mol Cell Biol. 2000;4:1394–1406. doi: 10.1128/mcb.20.4.1394-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smyth GK, Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Limma: linear models for microarray data. Bioinformatics and Computational Biology Solutions using R and Bioconductor. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 45.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:133–140. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 46.Lanza RP, Cibelli JB, West MD. Prospects for the use of nuclear transfer in human transplantation. Nat Biotechnol. 1999;17:1171–1174. doi: 10.1038/70709. [DOI] [PubMed] [Google Scholar]
- 47.Rideout WM, 3rd, Hochedlinger K, Kyba M, Daley GQ, Jaenisch R. Correction of a genetic defect by nuclear transplantation and combined cell and gene therapy. Cell. 2002;109:17–27. doi: 10.1016/s0092-8674(02)00681-5. [DOI] [PubMed] [Google Scholar]
- 48.Mombaerts P. Therapeutic cloning in the mouse. Proc Natl Acad Sci USA. 2003;100:11924–11925. doi: 10.1073/pnas.1934141100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cibelli JB, Grant KA, Chapman KB, Cunniff K, Worst T, Green HL, Walker SJ, Gutin PH, Vilner L, Tabar V, Dominko T, Kane J, Wettstein PJ, Lanza RP, Studer L, Vrana KE, West MD. Parthenogenetic stem cells in nonhuman primates. Science. 2002;295:819. doi: 10.1126/science.1065637. 2002. [DOI] [PubMed] [Google Scholar]
- 50.Humpherys D, EgganK, Akutsu H, Friedman A, Hochedlinger K, Yanagimachi R, Lander ES, Golub TR, Jaenisch R. Abnormal gene expression in cloned mice derived from embryonic stem cell and cumulus cell nuclei. Proc Natl Acad Sci USA. 2002;99:12889–12894. doi: 10.1073/pnas.192433399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.