

Abstract
The striatum contains a high density of histamine H3 receptors, but their role in striatal function is poorly understood. Previous studies have demonstrated antagonistic interactions between striatal H3 and dopamine D1 receptors at the biochemical level, while contradictory results have been reported about interactions between striatal H3 and dopamine D2 receptors. In the present study, by using reserpinized mice, we demonstrate the existence of behaviorally significant antagonistic postsynaptic interactions between H3 and D1 and also between H3 and dopamine D2 receptors. The selective H3 receptor agonist imetit inhibited, while the H3 receptor antagonist thioperamide potentiated locomotor activation induced by either the D1 receptor agonist SKF 38393 or the D2 receptor agonist quinpirole. High scores of locomotor activity were obtained with H3 receptor blockade plus D1 and D2 receptor co-activation, i.e., when thioperamide was co-administered with both SKF 38393 and quinpirole. Radioligand binding experiments in striatal membrane preparations showed the existence of a strong and selective H3-D2 receptor interaction at the membrane level. In agonist/antagonist competition experiments stimulation of H3 receptors with several H3 receptor agonists significantly decreased the affinity of D2 receptors for the agonist. This kind of intramembrane receptor-receptor interactions are a common biochemical property of receptor heteromers. In fact, by using Bioluminescence Resonance Energy Transfer techniques in co-transfected HEK-293 cells, H3 (but not H4) receptors were found to form heteromers with D2 receptors. The present study demonstrates an important role of postsynaptic H3 receptors in the modulation of dopaminergic transmission by means of a negative modulation of D2 receptor function.
Keywords: Histamine H3 receptor, dopamine D2 receptor, receptor heteromer, striatum
1. Introduction
The histaminergic system consists of a group of about 64,000 neurons whose cellular bodies are localized in one single hypothalamic nucleus, the tuberomamillary nucleus and which project extensively to all major parts of the forebrain, brainstem, cerebellum and spinal cord (Schwartz et al., 1991; Brown et al., 2001; Haas and Panula, 2003). The central actions of histamine are mediated by G protein-coupled receptors, mostly of the H1, H2 and H3 subtype. Another histamine receptor, the H4 subtype, has recently been cloned, but it is predominantly expressed in bone marrow, eosinophils and mast cells, with little expression in the brain (Oda et al., 2000; de Esch et al., 2005). The histaminergic system is involved in arousal, which is related to its innervation to the cortex, thalamus and basal forebrain (Brown et al., 2001), but the role of histamine in other parts of the forebrain are not well understood.
H3 receptors were first identified as autoreceptors, localized in histaminergic nerve terminals (Arrang et al., 1983), but they were later on found to act as heteroreceptors which modulate the release of various neurotransmitters (Leurs et al., 2005). The striatum contains one of the highest densities of H3 receptors in the brain (Pollard et al., 1993; Anichtchik et al., 2001; Pilot et al., 2002). In the striatum, presynaptic H3 receptors have also been identified in glutamatergic and dopaminergic terminals, where they seem to exert an inhibitory role of glutamate and dopamine release (Schlicker et al., 1993; Doreulee et al., 2001; Molina-Hernandez et al., 2000, 2001; Munzar et al., 2004). However, the major localization of striatal H3 receptors is postsynaptic, in the GABAergic striatal efferent neurons, where they are co-localized with D1 and D2 receptors in the D1 receptor-containing GABAergic dynorphinergic and the D2 receptor-containing GABAergic enkephalinergic neurons, respectively (Ryu et al., 1994; Pillot et al., 2002). This would give the rationale for the existence of functional postsynaptic H3-D1 and H3-D2 receptor interactions. In fact, antagonistic interactions between striatal H3 and D1 receptors have been observed at the biochemical level, by which stimulation of H3 receptor decreases cAMP accumulation and GABA release in striatal slices (Arias-Montano et al., 2001; Sanchez-Lemus and Arias-Montano, 2004). On the other hand, contradictory results about biochemical interactions between striatal H3 and D2 receptors have been reported (Hussain et al., 2002; Pillot et al., 2002).
In the present study, by analyzing the effects of H3 receptor agonists and antagonists on the locomotor activation induced by the D1 or D2 receptor agonists in reserpinized mice, we demonstrate the existence of behaviorally significant antagonistic postsynaptic H3-D1 and H3-D2 receptor interactions. We then show the existence of significant antagonistic H3-D2 receptor interactions at the membrane level in radioligand binding experiments in striatal membrane preparations. Finally, we demonstrate that H3 receptors can form heteromers with D2 receptors in co-transfected mammalian cells.
2. Materials and Methods
2.1. Locomotor activity in reserpinized mice
Male Swiss Webster mice (Charles River Laboratories, Inc., Wilmington, MA), experimentally naive at the start of the study and initially weighing 20 to 25 g, were housed four per cage with food and water available ad libitum. Mice were housed in a temperature- and humidity-controlled room and were maintained on a 12-h light/dark cycle (lights on from 6:00 to 18:00 h). Animals were used only once for locomotor activity experiments, which were conducted during the light phase one week after their arrival. Animals were maintained in accordance with guidelines of the Institutional Care and Use Committee of the Intramural Research Program, National Institute on Drug Abuse, NIH. Four locomotor activity chambers (Coulbourn Instruments, Allentown, PA) were used. Mouse open fields were 25.40 × 25.40 × 40.64 (high) cm. The beams were spaced 1.52 cm apart providing a 0.76 cm spatial resolution. Each chamber was equipped with one sensor ring used to accurately track the locomotion of each subject. Sessions programming and data recording made use of the TruScan Photobeam Activity System interface and the data acquisition TruScan 99 software package (Coulbourn Instruments) running on a PC compatible via the L18-16S/C Linc interface card (Coulbourn Instruments). Reserpine, the D1-like receptor agonist (+/−) – SKF 38393, the D2-like receptor agonist (−)-quinpirole and the H3 receptor antagonist thioperamide were purchased from Sigma (St. Louis, MO, USA). The H3–4 receptor agonist imetit dihydrobromide was purchased from Tocris (Ellisville, MO, USA). Reserpine was dissolved in a drop of glacial acetic acid which was made up to volume with 5.5% glucose and was administered (5 mg/kg) subcutaneously (s.c.) 20 h prior to the start of the locomotor activity recording. This dose of reserpine has been previously shown to produce pronounced striatal dopamine depletion in mice (more than 90% and 95% depletion 3 and 24 h after reserpine administration, respectively) (Starr et al., 1987), while histamine levels in the mouse brain are very little affected by reserpine (see Muroi et al., 1991). All the other drugs were dissolved in sterile saline and administered intraperitoneally (i.p.). The volume of injection was 10 ml/kg for all drugs. Imetit and thiperamide were administered 20 and 15 min prior to the locomotor activity recording, respectively, and SKF 38393 and quinpirole were administered immediately before the locomotor activity recording. Locomotor activity was recorded immediately after the animals, either reserpinized on non-reserpinized, were introduced in the open field without any acclimatization period. All values (in cm of ambulation) registered per 10 min were transformed (square root; Ferré et al., 1991, 1994) and the average of the results obtained during the 10-min periods of the first hour of recording were used for calculations. Differences between differently treated groups of animals were analyzed by ANOVA followed by post-hoc Newman-Keuls test.
2.2. Membrane preparation and protein determination
Membrane suspensions from sheep brain striatum were processed as described previously (Casadó et al., 1990). Tissue was disrupted with a Polytron homogenizer (PTA 20 TS rotor, setting 3; Kinematica, Basel, Switzerland) for three 5 s-periods in 10 volumes of 50 mM Tris-HCl buffer, pH 7.4. Membranes were obtained by centrifugation at 105,000 g (40 min, 4°C), and the pellet was resuspended and recentrifuged under the same conditions. The pellet was stored at −80°C and was washed once more as described above and resuspended in 50 mM Tris-HCl buffer for immediate use. Protein was quantified by the bicinchoninic acid method (Pierce Chemical Co., Rockford, IL, USA) using bovine serum albumin dilutions as standard.
2.3. Radioligand binding experiments
For competition experiments, membrane suspensions from sheep brain striatum (0.5 mg of protein/ml) were incubated for 2 h at 25ºC in 50 mM Tris-HCl buffer, pH 7.4, containing 10 mM MgCl2 with 1.4 nM of the D2 receptor antagonist [3H]YM-09151-2 (NEN Perkin Elmer) and increasing concentrations of the D2-like receptor agonist quinpirole (triplicates of 13 different competitor concentrations from 0.1 nM to 100 μM; Sigma) or the D2 receptor antagonist YM-09151-2 (triplicates of 8 different competitor concentrations from 0.01 nM to 1 μM; Sigma) in the absence or the presence of 20 nM of the H3 receptor agonist R-α-methylhistamine (RAMH, Sigma), imetit dihydrobromide (Tocris) or immepip dihydrobromide (Tocris) or 20 nM RAMH plus 1 μM of the H3 receptor antagonist thioperamide. Nonspecific binding was determined in the presence of 50 μM raclopride (Sigma) and confirmed that the value was the same as calculated by extrapolation of the displacement curves. To test the H3 receptor antagonist effect on D2 receptor ligand binding, membrane suspension from sheep brain striatum (0.5 mg of protein/ml) were incubated for 2 h at 25ºC in 50 mM Tris-HCl buffer, pH 7.4, containing 10 mM MgCl2 with 1.4 nM of the D2 receptor antagonist [3H]YM-09151-2 and 0, 0.1 or 1 μM of thioperamide. In all cases, free and membrane-bound ligand were separated by rapid filtration of 500 μl aliquots in a cell harvester (Brandel, Gaithersburg, MD, USA) through Whatman GF/C filters (Brandel) embedded in 0.3% polyethylenimine, which were subsequently washed for 5 s with 5 ml of ice-cold Tris-HCl buffer. The filters were incubated with 10 ml of Ecoscint H scintillation cocktail (National Diagnostics, Atlanta, GA, USA) overnight at room temperature and radioactivity counts were determined using a Tri-Carb 1600 scintillation counter (PerkinElmer, Boston, MA, USA) with an efficiency of 62% (Sarrió et al., 2000). Binding data from competition experiments were analyzed by nonlinear regression to the previously described equations (Casadó et al., 1992, Sarrió et al., 2000), using the commercial Grafit curve-fitting software (Erithacus Software, Surrey, UK). Goodness of fit was tested according to reduced χ2 value given by the nonlinear regression program and, in all cases, biphasic over monophasic competition curves were selected according to a p<0.01 using an F test (Ciruela et al., 2006). The equilibrium dissociation constants for the high-affinity (KDH) and low-affinity (KDL) binding sites were determined from IC50 values using the Cheng-Prusoff relation (Cheng and Prusoff, 1973). Results are given as parameter values ± S.E.M. from four independent experiments. Differences in KDH and KDL in the presence and absence of H3 agonists were tested for significance (two-tailed; p<0.05) using Student’s t test for unpaired samples.
2.4. Expression vectors
A plasmid encoding the cDNA of the human H3 receptor was provided by Johnson & Johnson Pharmaceutical Research & Development, L.L.C. (San Diego, California). The H3 receptor without its stop codon was amplified using sense and antisense primers harboring a unique EcoRI site. The fragment was then subcloned to be in-frame with Enhanced Yellow variant of green Fluorescent Protein (EYFP) into the EcoRI site of pEYFP-N1 (Clontech, Heidelberg, Germany) to provide the plasmid H3-YFP, which expresses EYFP on the C-terminal ends of the receptor. The D2-RLuc construct was generated by subcloning the cDNA of the human D2 receptor without its stop codon between the NheI and Xho I restriction sites of the Renilla luciferase-fusion protein expression vector (pRluc-N1, PerkinElmer). Constructs were verified by nucleotide sequencing. A pcDEF3 plasmid encoding the human cDNA of the H4 receptor fused to EYFP was used as negative control.
2.5. Cell culture and transfection
HEK-293 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamine (all from Invitrogen, Grand Island, NY, USA), at 37 °C in an atmosphere of 5% CO2. Cells were seeded in 35-mm diameter wells of 6-well plates and transient transfection with the corresponding fusion protein cDNAs was performed the following day using the calcium phosphate precipitation method (Jordan et al., 1996). Cells were harvested 48 h after transfection and used for BRET experiments. The empty vector pcDNA3.1 was used to equilibrate the total amount of transfected DNA.
2.6. cAMP determination
HEK-293 cells were grown in 25 cm2 flasks to 80% confluence and were transfected with 5 μg of cDNA corresponding to D2, H3 or H4 receptors or the fusion proteins D2-RLuc, H3-YFP or H4-YFP. Cells were incubated in serum-free medium for 16 h before the experiment and then were preincubated with 50 μM zardaverine (Tocris) as phosphodiesterase inhibitor for 10 min at 37ºC and treated for 10 min with 10 μM quinpirole, 100 nM RAMH, 10 μM imetit and/or 10 μM forskolin (Sigma). To stop the reaction cells were placed on ice and washed with ice-cold phosphate-buffered saline. Cells were incubated with 200 vL of HClO4 (4%) for 30 min and 1.5 M KOH was added to reach neutral pH and samples were centrifuged. The supernatant was frozen at −20ºC. Accumulation of cAMP was measured with Cyclic AMP (3H) Assay System (Amersham Biosciences, Uppsala, Sweden) as described in the manual from the manufacturer. Non-paired Student’s t test was used for statistics.
2.7. Bioluminescence Resonance Energy Transfer (BRET)
HEK-293 cells were transfected with 500 ng/well of the cDNA construct coding for D2-RLuc, acting as BRET donor, and increasing amounts (0.5–9 μg/well) of the cDNA construct coding for the BRET acceptor H3-YFP or the negative control H4-YFP. After 48 h of transfection cells were washed twice with Hanks’ balanced salt solution HBSS (137 mM NaCl, 5 mM KCl, 0.34 mM Na2HPO4×12H2O, 0.44 mM KH2PO4, 1.26 mM CaCl2×2H2O, 0.4 mM MgSO4×7H2O, 0.5 mM MgCl2, 10 mM HEPES, pH 7.4) supplemented with 0.1% glucose (w/v), detached by gently pipetting and resuspended in the same buffer. Sample protein concentration was determined to control cell number, using a Bradford assay kit (Bio-Rad, Munich, Germany) using bovine serum albumin dilutions as standards. Cell suspension (20 μg of protein) was dispensed in duplicates into 96-well black microplates with a transparent bottom (Porvair, King’s Lynn, UK) and the fluorescence was measured using a Mithras LB940 fluorescence-luminiscence detector (Berthold, Bad Wildbad, Germany) with an excitation filter of 485 nm and an emission filter of 535 nm. For BRET measurement, 20 μg of cell suspension were distributed in duplicates into 96-well white opaque microplates (Porvair) and coelenterazine H (Molecular Probes Europe, Leiden, The Netherlands) was added at a final concentration of 5 μM, after 1 min the readings were collected using sequential integration of signals detected at 440 to 500 nm and 510 to 590 nm. The same samples were incubated for 10 min and the luminescence was measured. Cells expressing BRET donors alone were used to determine background. The BRET ratio is defined as [(emission at 510–590)/(emission at 440–500)] – Cf where Cf corresponds to (emission at 510–590)/(emission at 440–500) for the -Rluc construct expressed alone in the same experiment. Curves were fitted using a nonlinear regression equation, assuming a single binding site with GraphPad Prism software (San Diego, CA, USA).
3. Results
3.1. H3-D1 and H3-D2 receptor interactions in reserpinized mice
Significant differences in locomotor activity were observed among groups of reserpinized mice that received different doses of the D1 receptor agonist SKF 38393 (0, 5 or 15 mg/kg, i.p.) with or without the previous administration of the H3 receptor antagonist thioperamide (3 or 10 mg/kg, i.p.) (one-way ANOVA: p<0.0001; n = 7–15/group) (Fig. 1a). Post-hoc Newman-Keuls tests demonstrated that the highest dose of SKF 38393 (15 mg/kg, i.p.) produced a modest but significant locomotor activation when administered alone (p<0.05). This effect was significantly potentiated by the previous administration of the H3 receptor antagonist thioperamide (3 and 10 mg/kg, i.p.; p<0.01 in both cases), which was not significantly effective when administered alone (Fig. 1a). Furthermore, thioperamide (3 and 10 mg/kg, i.p.) induced locomotor activation when administered previously to the low ineffective dose of SKF 38393 (5 mg/kg, i.p.; p<0.05 in both cases) (Fig. 1a). Significant differences in locomotor activity were also observed among groups of reserpinized mice that received different doses of the D2-like receptor agonist quinpirole (0, 0.15 or 0.5 mg/kg, i.p.) with or without the previous administration of thioperamide (3 or 10 mg/kg, i.p.) (one-way ANOVA; p<0.0001; n = 7–15/group) (Fig. 1b). Post-hoc Newman-Keuls tests demonstrated that the highest dose of quinpirole (0.5 mg/kg, i.p.) produced a modest but significant locomotor activation when administered alone (p<0.05), which was significantly potentiated by the previous administration of the H3 receptor antagonist thioperamide (3 and 10 mg/kg, i.p.; p<0.01 in both cases) (Fig. 1b). These results indicate that in reserpinized mice the already demonstrated high constitutive activity of H3 receptors (Morisset et al., 2000) or an endogenous histaminergic tone acting on H3 receptors is able to counteract the effects of both D1 and D2 receptor stimulation.
Figure 1.

Effects of the H3 receptor ligands on locomotor activity induced by dopamine receptor agonists in reserpinized mice. Results represent means + S.E.M. of the average of the transformed values (square root of cm of ambulation) obtained during the 10-min periods of the first hour of recording. a) Effect of saline or the H3 receptor antagonist thioperamide (Thiop; 3 and 10 mg/kg, i.p.) on the locomotor activation induced by saline or the D1 receptor agonist SKF 38393 (SKF; 5 and 15 mg/kg, i.p.; n = 8–16/group). b) Effect of saline or the H3 receptor antagonist thioperamide (Thiop; 3 and 10 mg/kg, i.p.) on the locomotor activation induced by saline or the D2 receptor agonist quinpirole (Quin; 0.15 and 0.5 mg/kg, i.p.; n = 9–15/group). * and **: significantly different compared to the control, saline-saline, group (ANOVA and post-hoc Newman-Keuls test: p<0.05 and p<0.01, respectively); ##: significantly different compared to the group treated with highest doses of SKF 38393 (15 mg/kg, i.p.) or quinpirole (0.5 mg/kg, i.p.) without thioperamide pretreatment (ANOVA and post-hoc Newman-Keuls test: p<0.01).
Significant differences in locomotor activity were also observed among groups of reserpinized mice that received SKF 38393 (15 mg/kg, i.p.) with or without the previous administration of the H3 receptor agonist imetit (10 mg/kg, i.p.) and with or without the previous administration of thioperamide (3 or 10 mg/kg, i.p.) (one-way ANOVA: p<0.01; n = 6–13/group) (Fig. 2). Post-hoc Newman-Keuls tests demonstrated that imetit significantly decreased locomotor activation induced by SKF 38393 (p<0.01), but not when thioperamide was also co-administered (Fig. 2). Similarly, significant differences in locomotor activity were observed among groups of reserpinized mice that received quinpirole (0.5 mg/kg, i.p.) with or without the previous administration of imetit (10 mg/kg, i.p.) and with or without the previous administration of thioperamide (3 or 10 mg/kg, i.p.) (one-way ANOVA: p<0.05; n = 8/group) (Fig. 2). Post-hoc Newman-Keuls tests demonstrated that imetit significantly decreased locomotor activation induced by quinpirole (p<0.05), but not when thioperamide was also co-administered (Fig. 2). In fact, when co-administered with imetit, thioperamide did not produce any potentiation of the motor effects induced by quinpirole or SKF 38393, strongly supporting an involvement of H3 receptors in the locomotor effects of thioperamide. The results indicate that stimulation of H3 receptors with exogenous agonists antagonize the locomotor activation induced by D1 or D2 receptor stimulation. Neither thioperamide (3 and 10 mg/kg, i.p.) or imetit (10 mg/kg, i.p.) significantly modified the exploratory locomotor activity in non-habituated, non-reserpinized mice (data not shown).
Figure 2.
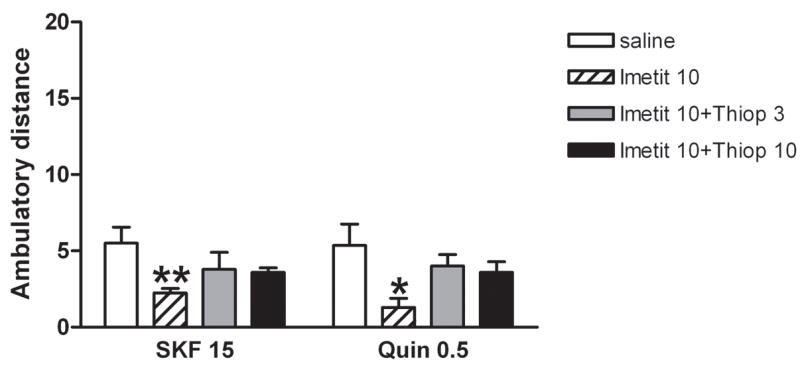
Effects of the H3 receptor agonist imetit (10 mg/kg, i.p.) on the locomotor activation induced by the D1 receptor agonist SKF 38393 (SKF; 15 mg/kg, i.p.) or the D2 receptor agonist quinpirole (Quin; 0.5 mg/kg, i.p.) with or without co-administration of the H3 receptor antagonist thioperamide (Thiop; 3 and 10 mg/kg, i.p.) in reserpinized mice (n = 8–12/group). Results represent means + S.E.M. of the average of the transformed values (square root of cm of ambulation) obtained during the 10-min periods of the first hour of recording. * and **: significantly different compared to the respective saline group (treated with SKF 38393 or quinpirole alone; ANOVA and post-hoc Newman-Keuls test: p<0.05 and p<0.01, respectively).
Finally, we analyzed the effects of H3 receptor blockade on the locomotor activation induced by the simultaneous activation of D1 and D2 receptors. Significant differences in locomotor activity were observed among groups of reserpinized mice that received different combinations of doses of SKF 38393 (5 or 15 mg/kg, i.p.) and quinpirole (0.15 or 0.5 mg/kg, i.p.) with or without the previous administration of thioperamide (3 mg/kg, i.p.) (one-way ANOVA: p<0.0001; n = 6–15/group) (Fig. 3). As previously reported (Rubinstein et al., 1988; Starr et al.. 1987; Ferré et al., 1991, 1994), co-administration of SKF 38393 and quinpirole in reserpinized mice produced a synergistic motor activating effect. Thus, post-hoc Newman-Keuls tests demonstrated that co-administration of low ineffective doses of SKF 38393 (5 mg/kg, i.p.) and quinpirole (0.15 mg/kg, i.p.) produced a significant locomotor activation (p<0.05), and co-administration of the effective doses of SKF 38393 (15 mg/kg, i.p.) and quinpirole (0.5 mg/kg, i.p.) produced a greater than additive activating effect (p<0.01) (Fig. 3). Importantly, the additional administration of thioperamide (3 mg/kg, i.p.) strongly potentiated the activating effects of both combinations of D1 and D2 receptor agonists (Fig. 3) (p<0.05 and p<0.01 for the combinations with low and high doses of dopamine agonists, respectively) (Fig. 3). In all the behavioral experiments, very similar effects were obtained with both doses of thioperamide used (3 and 10 mg/kg, i.p.). This dose range was similar to other studies in mice (see for instance, Brabant et al., 2006). Our results indicate that there is a ceiling effect at 3 mg/kg thioperamide, since pilot studies showed no significant effects with 1 mg/kg thioperamide (data not shown), indicating the existence of a small dose-dependent window for the H3 receptor-dependent effects of thioperamide.
Figure 3.
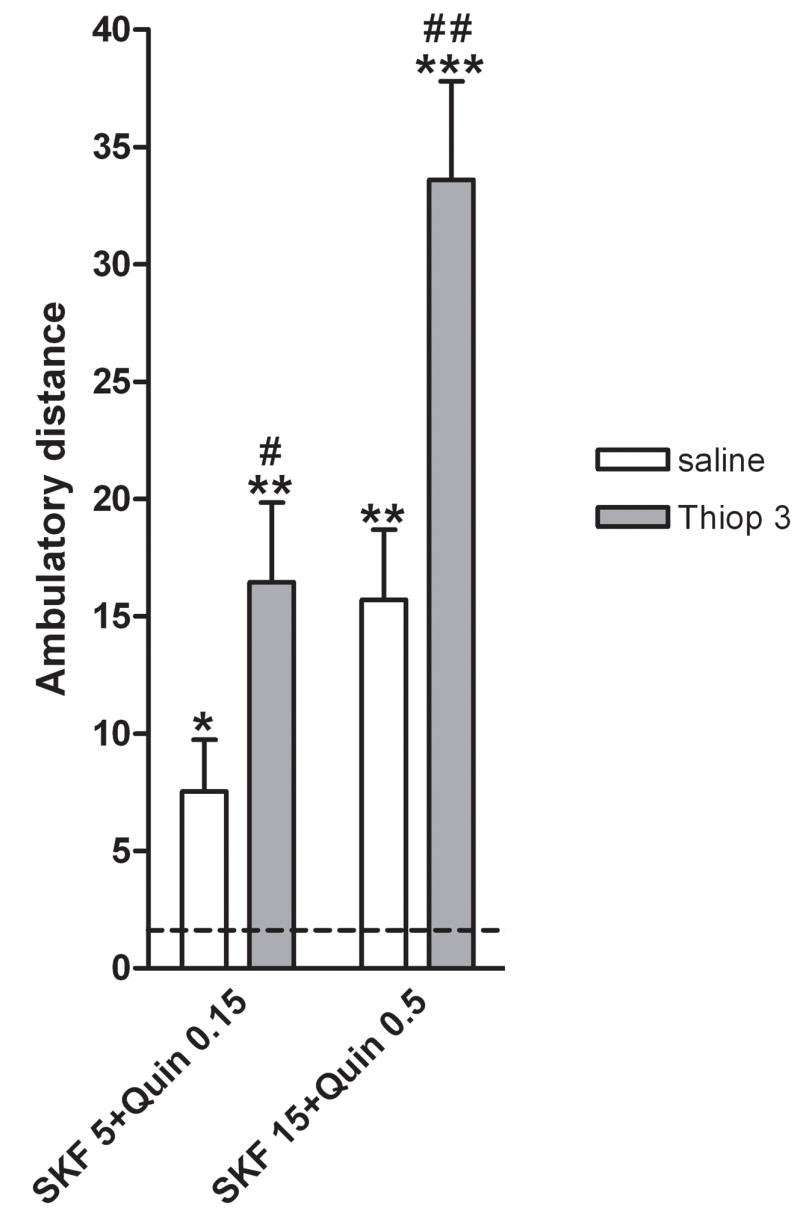
Effects of the H3 receptor antagonist thioperamide (Thiop; 3 mg/kg, i.p.) on the locomotor activation induced co-administration of the D1 receptor agonist SKF 38393 (SKF; 5 or 15 mg/kg, i.p.) and the D2 receptor agonist quinpirole (Quin; 0.15 or 0.5 mg/kg, i.p.) in reserpinized mice (n = 7–11/group). Results represent means + S.E.M. of the average of the transformed values (square root of cm of ambulation) obtained during the 10-min periods of the first hour of recording. The dashed line represents the mean of the control, saline-saline, group. * and **: significantly different compared to the control, saline-saline, group (represented by the dashed line) (ANOVA and post-hoc Newman-Keuls test: p<0.05 and p<0.01, respectively); # and ##: significantly different compared to the group treated with the same doses of the dopamine receptor agonists without thioperamide pretreatment (ANOVA and post-hoc Newman-Keuls test: p<0.05 and p<0.01, respectively).
3.2. Striatal H3-D2 intramembrane receptor interaction
To investigate the possible existence of a striatal H3-D2 intramembrane receptor interaction, competition experiments with the D2 receptor antagonist [3H] YM-09151-2 (1.4 nM) as radioligand and increasing concentrations of the D2 receptor agonist quinpirole as displacer were carried out in the presence or the absence of a high concentration (95% receptor saturation) of the selective H3 receptor agonist RAMH (20 nM). As shown in Fig. 4a, both competition curves are biphasic (significantly better fitting than monophasic; F test: p<0.01) and in the presence of the H3 receptor agonist a pronounced shift to the right in the competition curve was observed. From four independent experiments (different membrane preparations) in the absence of H3 receptor agonist the KDH and KDL values for quinpirole binding to D2 receptor were 0.9±0.2 nM and 1046.0±172.2 nM, respectively, whereas in the presence of RAMH the KDH and KDL values were 52.5±8.2 nM and 4030.0±75.5 nM, respectively (Student’s unpaired t test: p < 0.01 in both cases). Additional experiments using the same concentration than RAMH (20 nM; more than 95% receptor saturation) with the H3 receptor agonists imetit and immepip produced a similar effect than RAMH (one experiment with each agonist performed in triplicate). KDH values in the presence of imetit and immepip were 13.0 nM and 14.3 nM, respectively, and KDL values were 4600 nM and 5500 nM, respectively (Fig. 4a). These results indicate that H3 receptor activation induces a pronounced loss of affinity of both affinity states of the D2 receptor for agonists.
Figure 4.

H3 receptor ligand-mediated modulation of D2 receptor agonist binding in sheep striatal membranes. (A and C) Competition experiments of the D2 receptor antagonist [3H] YM-09151-2 (1.4 nM) versus increasing concentrations of the D2 receptor agonist quinpirole. In A, experiments were performed in the absence (■) or in the presence of the H3 receptor agonists RAMH (●), immepip (○) or imetit (▼) (20 nM in all cases). In C, experiments were performed in the presence of 20 nM RAMH (●) or 20 nM RAMH plus 1 μM thioperamide (△). Data are means ± S.D. from representative experiments performed in triplicate. . (B) Displacement of 1.4 nM [3H] YM-09151-2 binding by 0.1 μM (black bars) or 1 μM (white bars) of the H3 receptor agonists RAMH, immepip or imetit or the H3 receptor antagonist thioperamide. Values are expressed as specific binding percentage respect to the specific binding in absence of H3 receptor ligands. Data are means ± S.E.M. Significant differences were calculated by Student’s t test for unpaired samples (**: p<0.01).
The H3 receptor agonist-mediated loss of affinity was specific for the D2 agonist since no change in the maximum binding or the KD value for the YM-09151-2 binding to D2 receptor was observed in competition experiments using 1.5 nM [3H] YM-09151-2 and increasing concentrations of YM-09151-2 in the absence or the presence of 20 nM RAMH, imetit or immepip (Table 1), also indicating that H3 receptor agonists are not D2 receptor ligands. Accordingly, high concentrations of H3 receptor agonists did not change 1.5 nM [3H] YM-09151-2 binding and only very high thioperamide concentrations significativelly diminished the binding (Fig. 4b). Nevertheless, thioperamide was able to completely counteract the effect of RAMH on D2 receptor agonist binding (Fig. 4c).
Table 1.
Competition experiments of the D2 receptor antagonist [3H] YM-09151-2 with increasing concentrations of YM-09151-2 in the absence or the presence of H3 receptor agonists.
| Ligand | Parameters | |
|---|---|---|
| KD (nM) | Bmax (pmol/mg protein) | |
| YM-09151-2 | 5.4 ± 0.6 | 0.811 ± 0.092 |
| YM-09151-2 + RAMH | 4.4 ± 0.6 | 0.890 ± 0.101 |
| YM-09151-2 + Imetit | 4.2 ± 0.4 | 0.864 ± 0.097 |
| YM-09151-2 + Immepip | 4.4 ± 0.4 | 0.865 ± 0.097 |
Parameter values obtained by fitting data according to the one site model. Data are means ± SD values (of 3 experiments with triplicates). KD is the equilibrium dissociation constant and Bmax is the maximum binding.
3.3. H3-D2 receptor heteromerization
Since intramembrane receptor interactions are a functional property of some G protein-coupled receptor heteromers (Agnati et al., 2003; Ferré et al., 2007), we used the BRET approach to demonstrate the ability of H3 receptors to heteromerize with D2 receptors. Fusion of Rluc to D2 receptor or fusion of YFP to H3 or H4 receptors did not prevent receptor functionality, as determined by cAMP production in the presence of forskolin (Fig. 5A). The effect was significant and the degree of inhibition (around 30%) was similar to that observed with receptors without fusion proteins (Fig. 5A). BRET measurements were performed in transiently co-transfected HEK cells using a constant amount of D2-RLuc and increasing amounts of H3-YFP. A positive and saturable BRET signal for the transfer of energy between D2-RLuc and H3-YFP was also obtained (Fig. 5B). From the saturation curve a BRETmax of 0.052±0.004 and a BRET50 of 7.6±1.6 was calculated. The pair D2-RLuc and H4-YFP was used as a negative control, since the human H4 receptor is closely related to the human H3 receptor (31% sequence identity at the protein level, which increases to 54% in the transmembrane region) (de Esch et al., 2005). As shown in Fig. 5B this pair gave a linear non-specific BRET signal, indicating selectivity in the interaction between D2-RLuc and H3-YFP. HEK cells transfected with 0.5 μg cDNA corresponding to D2-RLuc expressed 1.5 pmol/mg protein of the receptor determined by radioligand binding. Analogously, cells transfected with increasing amounts of cDNA corresponding to the H3-YFP expressed 0.02 to 0.5 pmols/mg of the receptor protein. According to these values, in BRET50 conditions, the receptors expression was near the range of receptors expressed in sheep brain striatum (0.8 pmols/mg protein for D2 receptor and 0,1 pmols/mg protein for H3 receptor; values determined from saturation experiments using 0.02–10 nM [3H]YM 09151-2 and 0.02–10 nM [3H]RAMH, respectively).
Figure 5.
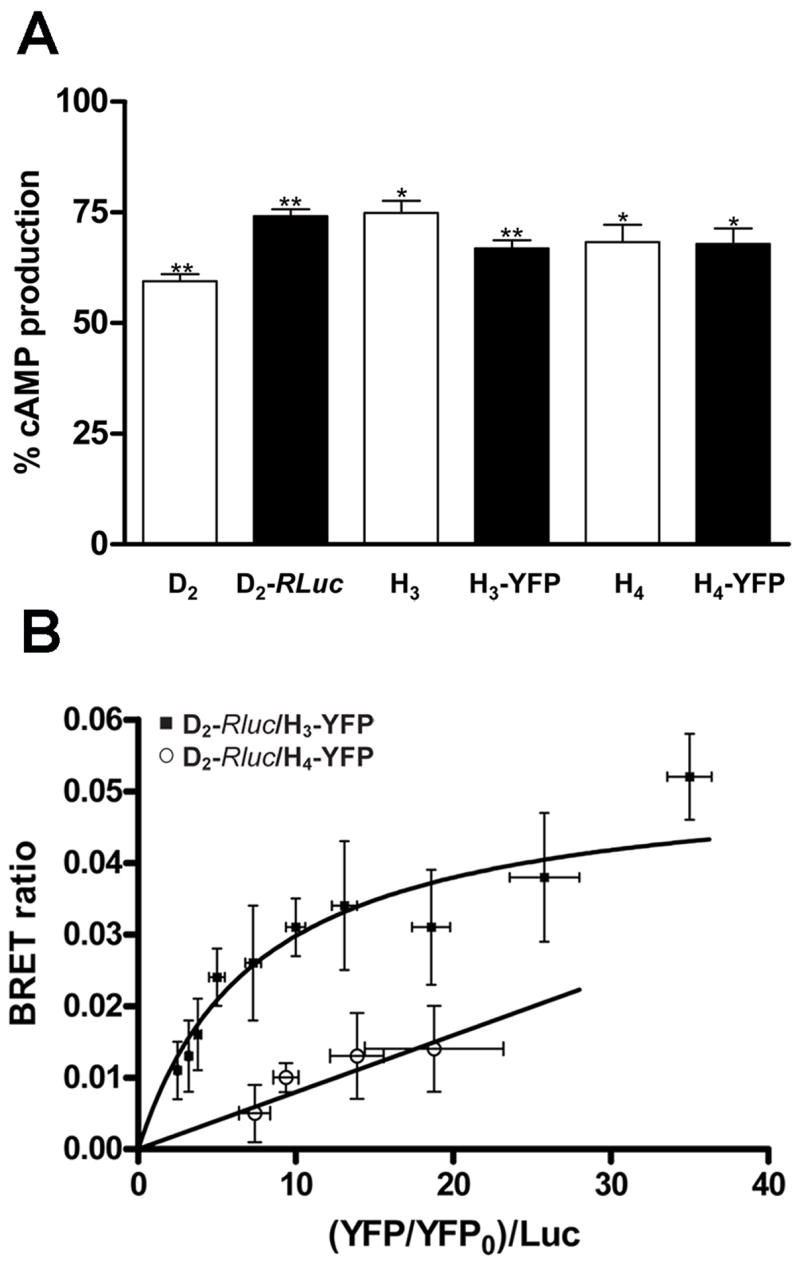
A. Production of cAMP by D2-RLuc, H3-YFP and H4-YFP constructs and by the same receptors without fusion proteins in HEK-293 cells. HEK-293 cells transfected with 5 μg of cDNA corresponding to D2-RLuc, H3-YFP or H4-YFP constructs were treated with 10 μM forskolin plus the D2 receptor agonist quinpirole (10 μM), the H3 receptor agonist RAMH (0.1 μM) or the H3–H4 receptor agonist imetit (10 μM). In the same conditions, cells transfected with the same amount of cDNA corresponding to the receptors not fused to bioluminescent or fluorescent proteins induced a 25–30% reduction of forskolin-induced cAMP. Results are expressed as percentage of the effect of forskolin alone (mean + SEM; n = 4); * and **: significantly different compared to forskolin alone (non-paired Student’s t test: p<0.05 and p<0.01, respectively). B. D2-H3 receptor heteromerization in HEK-293 cells. BRET saturation curve obtained from experiments with HEK-293 cells co-expressing D2-RLuc and H3-YFP (■) constructs or D2-RLuc and H4-YFP (○) constructs as a negative control. Co-transfections were performed with increasing amounts of plasmid for H3-YFP or H4-YFP whereas the D2-RLuc constructs were maintained constant. Both fluorescence and luminiscence of each sample were measured before every experiment to confirm equal expression of D2-RLuc while monitoring the increase of the acceptor. The relative amounts of BRET acceptor are expressed as the ratio between the fluorescence of the acceptor and the luciferase activity of the donor. YFP0 corresponds to the fluorescence value of cells expressing D2-RLuc alone. BRET data are expressed as means ± S.D. of three to thirteen different experiments grouped as a function of the amount of BRET acceptor.
4. Discussion
The evaluation of locomotor activity induced by dopamine receptor agonists in reserpinized mice is a very useful in vivo model to study the function of striatal postsynaptic D1 and D2 receptors, which are predominantly localized in GABAergic dynorphinergic and GABAergic enkephalinergic neurons, respectively, without the influence of endogenous dopamine (Rubinstein et al., 1988; Starr et al., 1987; Ferré et al., 1991, 1994b). By using the reserpinized mouse model, in the present study, we demonstrate the existence of H3 receptor-mediated negative modulation of D1 and D2 receptor function. In reserpinized mice, the selective H3 receptor agonist imetit inhibited, while the H3 receptor antagonist thioperamide potentiated the locomotor activation induced by either the D1 receptor agonist SKF 38393 or the D2 receptor agonist quinpirole.
GABAergic enkephalinergic and GABAergic dynorphinergic neurons give rise to two striatal efferent systems which connect the striatum with the output structures of the basal ganglia: the substantia nigra pars reticulata and the internal segment of the globus pallidus (entopeduncular nucleus in rodents) (Gerfen, 2004). These are called “direct” and “indirect” pathways. The direct pathway is made of GABAergic dynorphinergic neurons, which directly connect the striatum with the output structures. The indirect pathway consists of GABAergic enkephalinergic neurons, which connect the striatum with the external segment of the globus pallidus (globus pallidus in rodents), GABAergic neurons which connect the globus pallidus with the subthalamic nucleus and glutamatergic neurons which connect the subthalamic nucleus with the output structures (Gerfen, 2004). Stimulation of the direct pathway results in motor activation and stimulation of the indirect pathway produces motor inhibition. Dopamine and dopamine agonists induce motor activation by activating the direct pathway (acting on stimulatory D1 receptors localized in GABAergic dynorphinergic neurons) and by depressing the indirect pathway (acting on inhibitory D2 receptors localized in GABAergic enkephalinergic neurons) (Gerfen, 2004). The striatum contains a high density of postsynaptic H3 receptors, which are co-localized with both D1 and D2 receptors in the GABAergic dynorphinergic and enkephalinergic neurons, respectively (Ryu et al., 1994; Pillot et al., 2002). Previous studies have underscored the role of presynaptic H3 receptors (see introduction). The present study adds a new dimension to the role of histamine in basal ganglia physiology, by underscoring the role of postsynaptic H3 receptors in the modulation of D1 and D2 receptor function.
In the present study, we demonstrate the existence of an antagonistic intramembrane interaction between H3 and D2 receptors in striatal tissue, by which stimulation of H3 receptors significantly decreases the ability of an agonist, but not an antagonist, to bind to the D2 receptor. The antagonistic H3-D2 intramembrane interaction is particularly pronounced when compared with similar interactions involving striatal D2 receptors (Agnati et al., 2003). Thus, H3 receptor agonists produced a clear shift to the right in the competitive-inhibition curve of the D2 receptor antagonist [3H]YM-09151-2 versus the D2-like receptor agonist quinpirole, which was reversed by thioperamide. The analysis of the competition curves in the presence and absence of the H3 receptor agonist RAMH indicated that this effect was due to 50-fold and 4-fold H3 receptor-mediated decreases in the affinities of the high and low affinity states of the D2 receptor for the agonist binding, respectively. Therefore, this negative cross-talk could be the main mechanism responsible for the postsynaptic H3-D2 receptor negative interaction observed in our reserpinized mouse model and could also explain the biochemical results reported by Hussain et al. (2002), who found that an H3 receptor antagonist counteracts the increase in the striatal expression of the immediate-early gene c-fos induced by the D2 receptor antagonist haloperidol. In fact, it would be difficult to understand the H3-D2 functional cross-talk at the level of the intracellular signaling cascades interaction, because both D2 and H3 receptors couple to Gi/o proteins and their stimulation inhibits adenylyl cyclase (Lovenberg et al., 1999; Drutel et al., 2001; Neve et al., 2004; Leurs et al., 2005). Therefore, the pronounced antagonistic intramembrane H3-D2 receptor interaction most probably plays a key modulatory role of the function of the GABAergic enkephalinergic neuron.
Intramembrane receptor-receptor interaction implies a crosstalk that does not involve any signaling pathway, but an allosteric modification of one receptor secondary to stimulation of an adjacent receptor (Agnati et al., 2003; Franco et al., 2003; Ferré et al., 2007). An intramembrane receptor-receptor interaction constitutes a common biochemical characteristic of receptor heteromers (Ferré et al., 2007). Therefore, the existence of an antagonistic intramembrane H3-D2 receptor interaction in brain striatum strongly suggests the existence of a direct protein-protein interaction between both receptors. In fact, in the present study, we demonstrate the existence of H3-D2 receptor heteromerization in transfected cells by using BRET techniques. Thus, according to the recent UPHAR guidelines (Pin et al., 2007), the present study strongly suggests the existence of GPCR heteromers in the striatum.
Previous studies on neurotransmitter (GABA) release in striatal slices (Arias-Montano et al., 2001) and the present studies in reserpinized mice indicate the existence of antagonistic H3-D1 receptor interactions. Different from H3 receptors, D1 receptor couples to Gs/olf proteins and its main signaling pathway is the stimulation of the adenylyl cyclase-PKA cascade (Neve et al., 2004). The opposite effects of H3 and D1 receptors on adenylyl-cyclase predict the existence of antagonistic interactions of both receptors in the GABAergic dynorphynergic neuron. In fact, Sanchez-Lemus and Arias-Montano (2004) have recently demonstrated that H3 receptor activation efficiently inhibits D1 receptor-mediated adenylyl cyclase activation in rat striatal slices. The same authors could not find evidence for the existence of an H3-D1 intramembrane receptor interaction (Sanchez-Lemus and Arias-Montano, 2004). Therefore, an antagonistic H3-D1 receptor interaction at the second messenger level, with its consequent modulation of the function of the GABAergic dynorphinergic, might be mostly responsible for the antagonistic H3-D1 receptor interaction observed at the behavioral level.
In Parkinson’s disease a preferential degeneration of the nigrostriatal dopa-minergic system produces striatal dopamine depletion with the consequent impairment of the functioning of basal ganglia circuits, which is associated with hypokinesia. Reserpinized mice are used as a model to evaluate the possible antiparkinsonian activity of a drug. In the present study the H3 receptor antagonist thioperamide did not produce locomotor activation in reserpinized mice, but it potentiated the locomotor activation induced by the D1 receptor agonist SKF 38393 or by the D2 receptor quinpirole. Furthermore, very high scores of locomotor activity were obtained with H3 receptor blockade plus D1 and D2 receptor co-activation, i.e., when thioperamide was co-administered with both SKF 38393 and quinpirole. Although these results suggest that H3 receptor antagonists could be used as an adjuvant to dopamine receptor agonists in Parkinson’s disease, it must however be pointed out that thioperamide was not found to potentiate L-DOPA-mediated effects in another model of Parkinson’s disease, the unilateral 6-OH-dopamine lesioned rat (Huotari et al., 2000). Those negative results could be related to the neuroadaptations that develop upon chronic dopamine denervation. For instance, the upregulation of dopamine or H3 receptors observed after striatal dopamine denervation (Ryu et al., 1994, 1996; Anichtchik et al., 2000) could be accompanied by a reduction in H3-D1 or H3-D2 receptor interactions. Nevertheless, a possible antiparkinsonian role of drugs acting on striatal H3 receptors cannot be discarded and other disorders involving the cortico-striatal-thalamo-cortical circuits (Huntington’s disease, Tourette syndrome, obsessive compulsive disorder, schizophrenia, drug abuse) could benefit from a therapeutic approach based on the H3 receptor-mediated negative modulation of D1 and D2 receptor function.
Acknowledgments
This research was supported in part by grants from “Ministerio de Ciencia y Tecnología” (SAF2006-05481), “Fundació La Marató TV3” (grant 060110) and by the Intramural Research Program of the National Institutes of Health, National Institute on Drug Abuse, Department of Health and Human Services.
References
- Agnati LF, Ferré S, Lluis C, Franco R, Fuxe K. Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacological Reviews. 2003;55:509–550. doi: 10.1124/pr.55.3.2. [DOI] [PubMed] [Google Scholar]
- Anichtchik OV, Huotari M, Peitsaro N, Haycock JW, Mannisto PT, Panula P. Modulation of histamine H3 receptors in the brain of 6-hydroxydopamine-lesioned rats. European Journal of Neuroscience. 2000;12:3823–3832. doi: 10.1046/j.1460-9568.2000.00267.x. [DOI] [PubMed] [Google Scholar]
- Anichtchik OV, Peitsaro N, Rinne JO, Kalimo H, Panula P. Distribution and modulation of histamine H(3) receptors in basal ganglia and frontal cortex of healthy controls and patients with Parkinson’s disease. Neurobiology of Disease. 2001;8:707–716. doi: 10.1006/nbdi.2001.0413. [DOI] [PubMed] [Google Scholar]
- Arias-Montano JA, Floran B, Garcia M, Aceves J, Young JM. Histamine H(3) receptor-mediated inhibition of depolarization-induced, dopamine D(1) receptor-dependent release of [(3)H]-gamma-aminobutryic acid from rat striatal slices. British Journal of Pharmacology. 2001;133:165–171. doi: 10.1038/sj.bjp.0704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302:832–837. doi: 10.1038/302832a0. [DOI] [PubMed] [Google Scholar]
- Brabant C, Quertemont E, Tirelli E. Effects of the H3-receptor inverse agonist thioperamide on the psychomotor effects induced by acutely and repeatedly given cocaine in C57BL/6J mice. Pharmacology Biochemistry & Behavior. 2006;83:561–569. doi: 10.1016/j.pbb.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Progress in Neurobiology. 2001;63:637–672. doi: 10.1016/s0301-0082(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Casadó V, Cantí C, Mallol J, Canela EI, Lluís C, Franco R. Solubilization of A1 adenosine receptor from pig brain: characterization and evidence of the role of the cell membrane on the coexistence of high and low affinity status. Journal of Neuroscience Research. 1990;26:461–473. doi: 10.1002/jnr.490260409. [DOI] [PubMed] [Google Scholar]
- Casadó V, Casillas T, Mallol J, Canela EI, Lluis C, Franco R. The Adenosine Receptors Present on the Plasma Membrane of Chromaffin Cells Are of the A2b Subtype. Journal of Neurochemistry. 1992;59:425–431. doi: 10.1111/j.1471-4159.1992.tb09388.x. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical Pharmacology. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casado V, Rodrigues RJ, Lujan R, Burgueno J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortes A, Canela EI, Lopez-Gimenez JF, Milligan G, Lluis C, Cunha RA, Ferré S, Franco R. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. Journal of Neuroscience. 2006;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Esch IJ, Thurmond RL, Jongejan A, Leurs R. The histamine H4 receptor as a new therapeutic target for inflammation. Trends in Pharmacological Sciences. 2005;26:462–469. doi: 10.1016/j.tips.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Doreulee N, Yanovsky Y, Flagmeyer I, Stevens DR, Haas HL, Brown RE. Histamine H(3) receptors depress synaptic transmission in the corticostriatal pathway. Neuropharmacology. 2001;40:106–113. doi: 10.1016/s0028-3908(00)00101-5. [DOI] [PubMed] [Google Scholar]
- Drutel G, Peitsaro N, Karlstedt K, Wieland K, Smit MJ, Timmerman H, Panula P, Leurs R. Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Molecular Pharmacology. 2001;59:1–8. [PubMed] [Google Scholar]
- Ferré S, Herrera-Marschitz M, Grabowska-Anden M, Ungerstedt U, Casas M, Anden NE. Postsynaptic dopamine/adenosine interaction: I. Adenosine analogues inhibit dopamine D2-mediated behaviour in short-term reserpinized mice. European Journal of Pharmacology. 1991;192:25–30. doi: 10.1016/0014-2999(91)90064-w. [DOI] [PubMed] [Google Scholar]
- Ferré S, Gimenez-Llort L, Artigas F, Martinez E. Motor activation in short-and long-term reserpinized mice: role of N-methyl-D-aspartate, dopamine D1 and dopamine D2 receptors. European Journal of Pharmacology. 1994;255:203–213. doi: 10.1016/0014-2999(94)90099-x. [DOI] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Canals M, Marcellino D, Burgueno J, Casado V, Hillion J, Torvinen M, Fanelli F, Benedetti PdP, Goldberg SR, Bouvier M, Fuxe K, Agnati LF, Lluis C, Franco R, Woods AS. Adenosine A2A-dopamine D2 receptor-receptor heteromers. Targets for neuro-psychiatric disorders. Parkinsonism & Related Disorders. 2004;10:265–271. doi: 10.1016/j.parkreldis.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Woods AS, Lluis C, Franco R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends in Neurosciences. 2007;30:440–446. doi: 10.1016/j.tins.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Franco R, Canals M, Marcellino D, Ferré S, Agnati L, Mallol J, Casado V, Ciruela F, Fuxe K, Lluis C, Canela EI. Regulation of heptaspanning-membrane-receptor function by dimerization and clustering. Trends in Biochemical Sciences. 2003;28:238–243. doi: 10.1016/S0968-0004(03)00065-3. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. Basal Ganglia. In: Paxinos G, editor. The Rat Nervous System. Elsevier Academic Press; Amsterdam: 2004. pp. 445–508. [Google Scholar]
- Haas H, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nature Reviews in Neuroscience. 2003;4:121–130. doi: 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- Huotari M, Kukkonen K, Liikka N, Potasev T, Raasmaja A, Mannisto PT. Effects of histamine H(3)-ligands on the levodopa-induced turning behavior of hemiparkinsonian rats. Parkinsonism & Related Related Disorders. 2000;6:159–164. doi: 10.1016/s1353-8020(00)00007-9. [DOI] [PubMed] [Google Scholar]
- Hussain N, Flumerfelt BA, Rajakumar N. Muscarinic, adenosine A(2) and histamine H(3) receptor modulation of haloperidol-induced c-fos expression in the striatum and nucleus accumbens. Neuroscience. 2002;112:427–438. doi: 10.1016/s0306-4522(02)00069-6. [DOI] [PubMed] [Google Scholar]
- Jordan M, Schallhorn A, Wurm FM. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Research. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leurs R, Bakker RA, Timmerman H, de Esch IJ. The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nature Reviews in Drug Discovery. 2005;4:107–120. doi: 10.1038/nrd1631. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, Jackson MR, Erlander MG. Cloning and functional expression of the human histamine H3 receptor. Molecular Pharmacology. 1999;55:1101–1107. [PubMed] [Google Scholar]
- Molina-Hernandez A, Nunez A, Arias-Montano JA. Histamine H3-receptor activation inhibits dopamine synthesis in rat striatum. Neuroreport. 2000;11:163–166. doi: 10.1097/00001756-200001170-00032. [DOI] [PubMed] [Google Scholar]
- Molina-Hernandez A, Nunez A, Sierra JJ, Arias-Montano JA. Histamine H3 receptor activation inhibits glutamate release from rat striatal synaptosomes. Neuropharmacology. 2001;41:928–34. doi: 10.1016/s0028-3908(01)00144-7. [DOI] [PubMed] [Google Scholar]
- Morisset S, Rouleau A, Ligneau X, Gbahou F, Tardivel-Lacombe J, Stark H, Schunack W, Ganellin CR, Scwartz JC, Arrang JM. High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature. 2000;408:860–864. doi: 10.1038/35048583. [DOI] [PubMed] [Google Scholar]
- Munzar P, Tanda G, Justinova Z, Goldberg SR. Histamine h3 receptor antagonists potentiate methamphetamine self-administration and methamphetamine-induced accumbal dopamine release. Neuropsychopharmacology. 2004;29:705–717. doi: 10.1038/sj.npp.1300380. [DOI] [PubMed] [Google Scholar]
- Muroi N, Oishi R, Saeki K. Effect of reserpine on histamine metabolism in the mouse brain. Journal Of Pharmacology and Experimental Therapeutics. 1991;256:967–972. [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. Journal of Receptor Signal Transduction Research. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Oda T, Morikawa N, Saito Y, Masuho Y, Matsumoto S. Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. Journal of Biological Chemistry. 2000;275:36781–36786. doi: 10.1074/jbc.M006480200. [DOI] [PubMed] [Google Scholar]
- Pillot C, Heron A, Cochois V, Tardivel-Lacombe J, Ligneau X, Schwartz JC, Arrang JM. A detailed mapping of the histamine H(3) receptor and its gene transcripts in rat brain. Neuroscience. 2002;114:173–193. doi: 10.1016/s0306-4522(02)00135-5. [DOI] [PubMed] [Google Scholar]
- Pin JP, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, Lohse MJ, Milligan G, Palczewski K, Parmentier M, Spedding M. International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacological Reviews. 2007;59:5–13. doi: 10.1124/pr.59.1.5. [DOI] [PubMed] [Google Scholar]
- Pollard H, Moreau J, Arrang JM, Schwartz JC. A detailed autoradiographic mapping of histamine H3 receptors in rat brain areas. Neuroscience. 1993;52:169–189. doi: 10.1016/0306-4522(93)90191-h. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Gershanik O, Stefano FJ. Different roles of D-1 and D-2 dopamine receptors involved in locomotor activity of supersensitive mice. European Journal of Pharmacology. 1988;148:419–426. doi: 10.1016/0014-2999(88)90121-5. [DOI] [PubMed] [Google Scholar]
- Ryu JH, Yanai K, Iwata R, Ido T, Watanabe T. Heterogeneous distributions of histamine H3, dopamine D1 and D2 receptors in rat brain. Neuroreport. 1994;5:621–624. doi: 10.1097/00001756-199401000-00022. [DOI] [PubMed] [Google Scholar]
- Ryu JH, Yanai K, Zhao XL, Watanabe T. The effect of dopamine D1 receptor stimulation on the up-regulation of histamine H3-receptors following destruction of the ascending dopaminergic neurones. British Journal of Pharmacology. 1996;118:585–92. doi: 10.1111/j.1476-5381.1996.tb15441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Lemus E, Arias-Montano JA. Histamine H3 receptor activation inhibits dopamine D1 receptor-induced cAMP accumulation in rat striatal slices. Neuroscience Letters. 2004;364:179–184. doi: 10.1016/j.neulet.2004.04.045. [DOI] [PubMed] [Google Scholar]
- Sarrió S, Casadó V, Escriche M, Ciruela F, Mallol J, Canela EI, Lluis C, Franco R. The heat shock cognate protein hsc73 assembles with A(1) adenosine receptors to form functional modules in the cell membrane. Molecular and Cellular Biology. 2000;20:5164–5174. doi: 10.1128/mcb.20.14.5164-5174.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker E, Fink K, Detzner M, Gothert M. Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. Journal of Neural Transmission General Section. 1993;93:1–10. doi: 10.1007/BF01244933. [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M. Histaminergic transmission in the mammalian brain. Physiological Reviews. 1991;71:1–51. doi: 10.1152/physrev.1991.71.1.1. [DOI] [PubMed] [Google Scholar]
- Starr BS, Starr MS, Kilpatrick IC. Behavioural role of dopamine D1 receptors in the reserpine-treated mouse. Neuroscience. 1987;22:179–188. doi: 10.1016/0306-4522(87)90208-9. [DOI] [PubMed] [Google Scholar]