

Abstract
Secondary active transport of substrate across the cell membrane is crucial to many cellular and physiological processes. The crystal structure of one member of the secondary active transporter family, the sn-glycerol-3-phosphate (G3P) transporter (GlpT) of the inner membrane of Escherichia coli, suggests a mechanism for substrate translocation across the membrane that involves a rocker-switch-type movement of the protein. This rocker-switch mechanism makes two specific predictions with respect to kinetic behavior: the transport rate increases with the temperature, whereas the binding affinity of the transporter to a substrate is temperature-independent. In this work, we directly tested these two predictions by transport kinetics and substrate-binding experiments, integrating the data on this single system into a coherent set of observations. The transport kinetics of the physiologically relevant G3P–phosphate antiport reaction were characterized at different temperatures using both E. coli whole cells and GlpT reconstituted into proteoliposomes. Substrate-binding affinity of the transporter was measured using tryptophan fluorescence quenching in detergent solution. Indeed, the substrate transport velocity of GlpT increased dramatically with temperature. In contrast, neither the apparent Michaelis constant (Km) nor the apparent substrate-binding dissociation constant (Kd) showed temperature dependence. Moreover, GlpT-catalyzed G3P translocation exhibited a completely linear Arrhenius function with an activation energy of 35.2 kJ mol−1 for the transporter reconstituted into proteoliposomes, suggesting that the substrate-loaded transporter is delicately poised between the inward- and outward-facing conformations. When these results are taken together, they are in agreement with a rocker-switch mechanism for GlpT.
Secondary active membrane transporter proteins use an electrochemical gradient generated across the membrane by primary active transport as the driving force for substrate translocation (1, 2). Over 100 families of secondary active membrane transporters have been identified to date, the largest of which is the major facilitator superfamily (MFS),1 with over 5000 known members (3–5). The MFS, members of which are ubiquitous in all three kingdoms of life, includes many medically and pharmaceutically important transporters, such as the efflux pumps that confer resistance to antibiotics in bacteria and chemotherapeutic drugs in humans (6, 7).
Over the years, extensive efforts have been made to understand the molecular mechanisms of these transporters by using an array of genetic, biochemical, and biophysical techniques. The advent of crystal structures of several MFS transporters (8–11) has now placed functional studies of this flavor of transporter on a much firmer footing and allowed for a more rigorous analysis. One of these transporters, the sn-glycerol-3-phosphate (G3P) transporter (GlpT) in the Escherichia coli inner membrane, transports G3P into the cell via an antiport mechanism that is energized by an inorganic phosphate (Pi) gradient (12). The crystal structure of GlpT (8) shows that it functions as a monomer composed of N- and C-terminal domains, each consisting of a compact 6 α-helical bundle and joined by a long, central, cytoplasmic loop region. The substrate translocation pore is located between the N- and C-terminal domains (Figure 1A). This structural information, in combination with an opus of previous biochemical, thermodynamic, and kinetic studies on both GlpT and LacY (12–17), and the closely related E. coli organic phosphate/inorganic phosphate antiporter, UhpT (18, 19), have allowed us to construct a model for substrate translocation by GlpT (8, 20) (parts B and C of Figure 1). This single-binding site, alternating-access mechanism, which involves a rocker-switch-type movement of the N- and C-terminal domains of the protein, is dependent upon binding-site reorientation between an inward (Ci) and outward (Co) facing conformation. The model allows the mechanism of GlpT-mediated antiport to be broken down into six discrete stages, with three stages dedicated to the binding, translocation, and release of one substrate (in this case, G3P), and the remaining three stages describing the transport of the countersubstrate, Pi (Figure 1B). Initially, the transporter exists in an unloaded, low-energy Co conformation, with the binding site accessible to the periplasmic side of the membrane. The first stage of the mechanism involves actual binding of substrate to yield the Co–S complex. This complex is only slightly higher in energy than the unloaded carrier, and its formation is paid for by intrinsic binding energy. From the Co–S state, the complex can move through the high-energy transition state and switch or flip into the Ci–S state (Figure 1C). During transition, the protein undergoes gross conformational change in which the N- and C-terminal domains participate in a rocker-switch type of movement that functions to translocate the bound substrate across the membrane (8, 20). Such global structural changes require considerable energy input, represented by the high transition-state energy level of the unloaded transporter (Figure 1C). The third stage of the transport process represents the release of the substrate to the cytoplasmic side of the membrane, thereby allowing access of the binding site to the countersubstrate. The process is then simply reversed for the translocation of countersubstrate from the cytoplasm to the periplasm. The whole process is enabled by the Pi gradient, because the cellular concentration of Pi is much greater than that in the extracellular environment (21).
Figure 1.
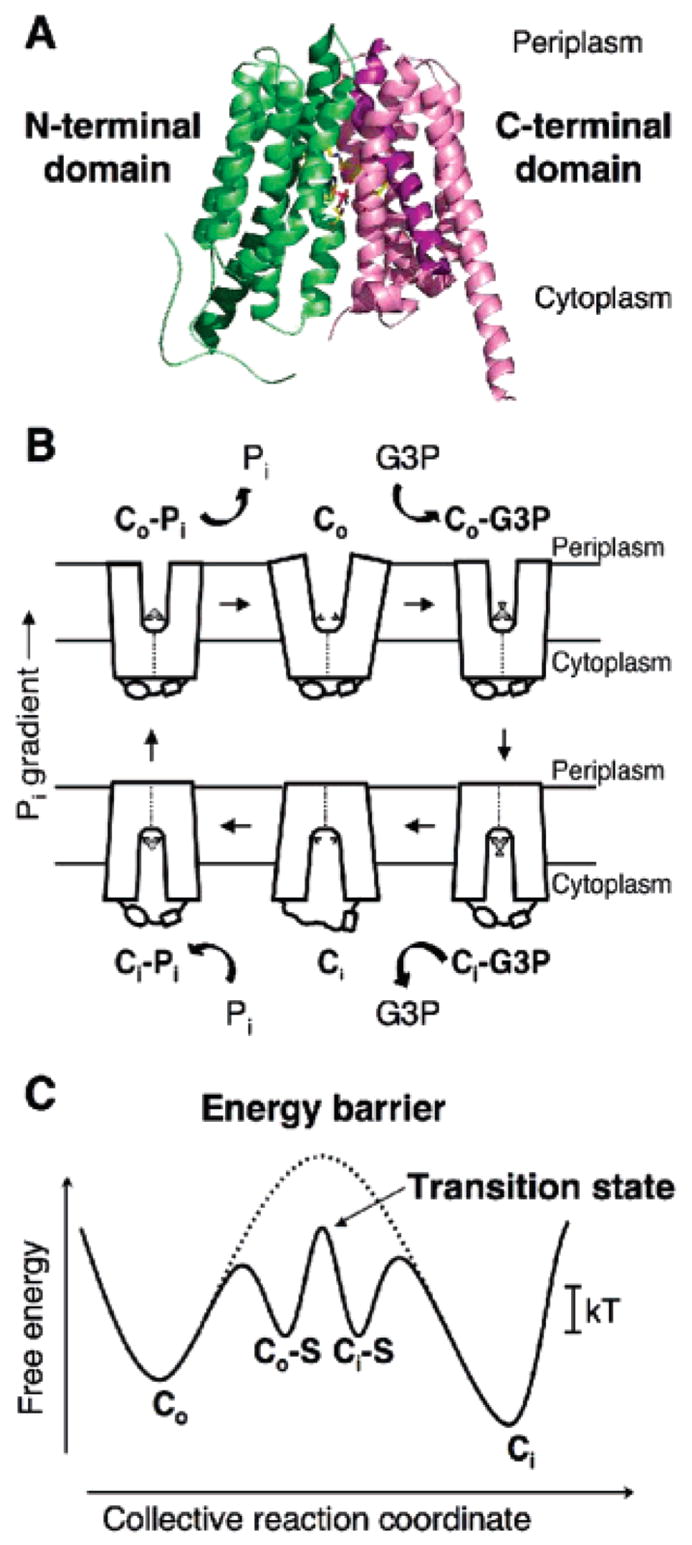
Crystal structure of GlpT and the rocker-switch mechanism. (A) Ribbon diagram of GlpT viewed parallel to the membrane. The molecule consists of 12 transmembrane α-helices. The N-terminal domain is colored green, and the C-terminal domain is colored pink. (B) Schematic diagram of the single-binding site, alternating the access mechanism with a rocker-switch type of movement for the GlpT-mediated G3P–Pi exchange reaction. The diagram describes the proposed conformational changes that the transporter undergoes during the reaction cycle. Co represents the protein in the outward-facing conformation, and Ci represents the inward-facing one. The G3P substrate is represented by a small disk and triangle, and Pi is represented by a small disk. (C) Schematic free-energy diagram illustrating the energy levels of the different conformations of GlpT that occur during the transport reaction cycle under physiological conditions. In the absence of substrate binding, the energy barrier (represented by a dotted line) prevents the conformational interconversion between the Co and Ci states of the transporter. Substrate binding lowers the energy barrier sufficiently to allow Brownian motion (kT) to drive the conformational interconversion. S denotes the substrate.
The rocker-switch model of the GlpT transporter immediately suggests two experimentally testable predictions with which its kinetic behavior should comply. (i) The activation energy barrier for its interconversion from the Co–S to the Ci–S state is surmounted by the input of thermal energy from the environment via Brownian motion. The actual interconversion is rate-limiting, and specifically, its Vmax is temperature-dependent. (ii) The substrate-binding affinity, Kd, in contrast, is independent of the temperature.
In the present work, we investigated the viability of the rocker-switch mechanism of the GlpT function by directly testing the above two predictions. We report on transport kinetics studies using E. coli whole cells and GlpT reconstituted into proteoliposomes, in combination with substrate-binding affinity studies of the transporter in detergent solution.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmids
For whole-cell transport assays, E. coli strain SH1200 (glpT phoR ugpA704::Tn10) (obtained from W. Boos, University of Konstanz, Germany) served as the host to the plasmids used in this part of the study (16). Strain SH1200 fails to transport G3P because of defects in GlpT and UgpT, the ATP-dependent G3P transporter. Plasmid pQ31 was constructed by introducing into pQE (Qiagen, Valencia, CA) a BamHI–HindIII fragment that encodes GlpT with a N-terminal polyhistidine (His6) extension (HisGlpT), placing GlpT expression under the control of the tac promoter and in the presence of lacIq. The E. coli strain and plasmid used for the overexpression of GlpT for use in reconstitution experiments and in substrate-binding affinity assays are described by Auer et al. (15).
Transport Assays Using E. coli Whole Cells
For whole-cell transport assays, E. coli strain SH1200 cells were grown overnight in M63 minimum medium (22) with 0.2% glucose as the carbon source, along with antibiotics and amino acids (each 50 μg/mL) (16). After overnight growth, cells were diluted 100-fold into the same medium and cultured at 35 °C to an OD660 of 0.4–0.8. After the cells were harvested by centrifugation, they were washed once with assay buffer (50 mM MOPS/K and 100 mM potassium chloride at pH 7.0) and then resuspended in the same buffer to an OD660 of 1.4. For kinetics experiments, after equilibration of the whole cells to the desired temperature, substrate transport was initiated by the addition of [14C]G3P (Sigma, St. Louis, MO), ranging from 6.25 to 400 μM in a final reaction volume of 110 μL (104.5 μL of cells and 5.5 μL of [14C]G3P). After 2 min, 100 μL was removed for filtration using Millipore filters (0.45 μm pore size); filters were washed twice with 5 mL of assay buffer. Assays were performed at temperatures of 8, 16, 26, and 33 °C. Triplicate measurements were made for each substrate concentration tested, and data shown are mean values from three independent experiments.
Purification of GlpT
GlpT was cloned and overexpressed as described by Auer et al. (15). The protein used in fluorescence binding assays was purified according to the same protocol. His-tagged GlpT for reconstitution into proteoliposomes was purified using modifications of the methods described by Auer et al. (15) and Fann et al. (16). In this case, cells were resuspended in 100 mM potassium phosphate at pH 7.0 containing 1 mM phenylmethanesulfonyl fluoride (PMSF) and protease inhibitor cocktail (Sigma) and broken by passage through a French pressure cell. After centrifugation to remove unbroken cells and large cell debris, the cell membrane was harvested by ultracentrifugation at 100000g for 2.5 h. The membrane pellet was solubilized in a buffer consisting of 100 mM potassium phosphate at pH 7.0, 400 mM NaCl, 10 mM G3P, 10 mM imidazole, 20% (v/v) glycerol, 0.2% (w/v) E. coli total lipid extract, and 1.5% (w/v) N-dodecyl-β-D-maltoside (DDM) (Anatrace, Maumee, OH). After centrifugation to remove any unsolubilized material, the His-tagged GlpT was purified by nickel–nitrilotriacetic acid (Ni–NTA) affinity chromatography as described in the published method (15), except that 100 mM potassium phosphate, 10 mM G3P, 0.2% (w/v) E. coli total lipid extract, and 1.5% (w/v) DDM was included in the wash and elution buffers. Purified protein was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and quantitated using a Micro BCA Protein Assay Kit (Pierce, Rockford, IL).
Reconstitution of GlpT
Reconstitution of GlpT into proteoliposomes was performed using modifications of the methods described by Rigaud et al. (23) and Knol et al. (24). E. coli total lipid extract (Avanti Polar Lipids, Alabaster, AL) and egg yolk L-α-phosphatidylcholine (Sigma) were mixed in a ratio of 3:1 (w/w), dissolved in chloroform, and then dried down on ice under a stream of nitrogen. Traces of any remaining solvent were removed under vacuum. The dried lipid (20 mg) was resuspended in 1 mL of ice-cold reconstitution loading buffer (100 mM potassium phosphate at pH 7.0) and vortexed vigorously to ensure that it was completely dissolved. Unilamellar vesicles were formed by sonication of the lipid sample on ice followed by seven rounds of freezing and thawing. To ensure a uniform size distribution, the liposomes were extruded 13 times through a 400 nm polycarbonate filter using a mini-extruder apparatus (Avanti Polar Lipids). The liposomes were then diluted to 4 mg of lipid/mL with loading buffer and destabilized by the addition of DDM to 4.4 mM. The destabilized liposomes were incubated, with gentle rocking, for 2 h at 4 °C and then mixed with freshly purified His-tagged GlpT at a lipid/protein ratio of 100:1 (w/w). The protein was allowed to incorporate into the liposomes by incubation for 30 min at room temperature, followed by a further hour at 4 °C, with nutation. Detergent removal was performed by the addition of Bio-Beads SM-2 (Bio-Rad, Richmond, CA), at a wet weight of 80 mg/mL of liposome suspension. After an initial 30 min of incubation at room temperature, the sample was incubated for a further 2 h at 4 °C. The Bio-Beads were removed by filtration through glass silk, and a fresh 80 mg aliquot of beads was added to the sample, followed by an overnight incubation at 4 °C. After another addition of fresh Bio-Beads and a final 2 h of incubation at 4 °C, the proteoliposomes were isolated by centrifugation for 1 h at 180000g. The protein content of the proteoliposomes was estimated by quantitating the amount of protein remaining in the supernatant using a Micro BCA Protein Assay Kit (Pierce). The surfaces of the proteoliposome pellets were washed twice in ice-cold transport assay buffer (100 mM K2SO4 and 50 mM MOPS/K at pH 7.0) prior to resuspension in the same buffer to a protein concentration of 25 μg/mL. The proteoliposomes were kept on ice until use in transport assays.
Transport Assays Using Proteoliposomes
Transport assays were performed at four different temperatures (10, 18, 28, and 37 °C). A total of 10 μL of freshly prepared, phosphate-loaded proteoliposomes were diluted 10-fold with assay buffer and allowed to equilibrate to the assay temperature in a thermostatically controlled water bath. The transport was initiated by the addition of [3H]G3P (American Radiolabeled Chemicals, St. Louis, MO), to concentrations covering a range from 5 to 400 μM (see the Results for exact concentrations used). The reaction was allowed to proceed for 60 s before it was terminated by harvesting the proteoliposomes on nitrocellulose filters (0.22 μm, Millipore GSWP 02500) mounted on a Hoeffer vacuum manifold and washing with two 5 mL aliquots of ice-cold assay buffer. The filters were placed in vials and incubated overnight in liquid scintillant (ScintiLene, Fisher Scientific) prior to measuring the incorporated radioactivity with a Wallac 1450 Microbeta Plus liquid scintillation counter. All experiments were performed in triplicate. After correction for background radiation, apparent kinetic constants (Km and Vmax) for G3P uptake were determined by fitting the data to the Michaelis–Menten equation (v = Vmax[S]/(Km + [S])) using a nonlinear regression analysis available in the Enzyme Kinetics Module of SigmaPlot 10 (Systat, Richmond, CA).
Tryptophan Fluorescence-Quenching and Binding Studies
Tryptophan fluorescence-quenching experiments were performed according to the published procedure (15), except that the emission wavelength was set to 334 nm and the GlpT protein was titrated with 0.1–15 μM sn-glycerol 3-phosphate bis(cyclohexylammonium) salt (G3P) (Sigma). Experiments were performed in triplicate at temperatures of 10, 18, 28, and 37 °C. After correction for dilution, the data underwent nonlinear regression analysis using the formula y = Bmax−[G3P]/(Kd + [G3P]).
RESULTS
Transport Activity in Whole Cells
To gain a preliminary overview of GlpT-mediated transport kinetics under physiological conditions, we first performed transport assays in E. coli whole cells. The initial rate of G3P uptake into E. coli was measured over a temperature range of 8–33 °C. The transport rate showed a clear temperature dependence, with the rate increasing incrementally with the temperature (Figure 2A). The maximal transport velocity, Vmax, at 8 °C was 11.1 ± 3.5 nmol min−1 (mg of whole-cell membrane protein)−1. The Vmax increased to 82.1 ± 6.4 nmol min−1 (mg of whole-cell membrane protein)−1 at 33 °C (Table 1). This represents a 7–8-fold increase in the transport rate over the temperature range studied. In contrast, the apparent Michaelis constant (Km) showed no temperature dependence, with values maintained between 42.4 ± 17.5 and 50.7 ± 3.1 μM in the temperature range of 8–33 °C (Table 1). The temperature dependence of the rate of the G3P transport was further analyzed using the Arrhenius plot (Figure 2B). The slope of the regression line (sArr) was used to calculate the activation energy (Ea) using the equation Ea = −sArr × R, where R = 8.314 J mol−1 K−1. Ea was determined to be 55.4 kJ mol−1. Such activation energy corresponds to a temperature coefficient (Q10 value) over the temperature range studied of 2.2, indicating the involvement of a large-scale conformational change in the transporter at some point during the reaction cycle.
Figure 2.
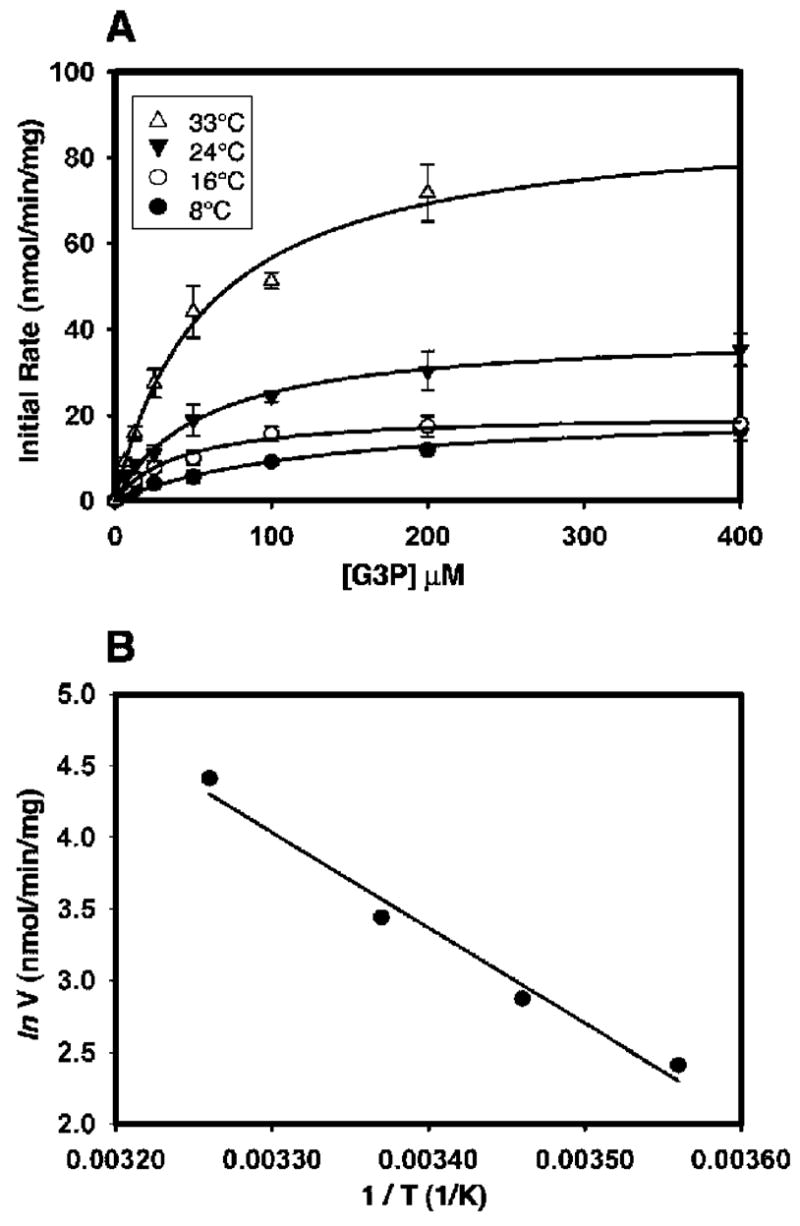
Kinetic measurements of the G3P transport by GlpT in whole cells. (A) Effect of temperature on the kinetic properties of the GlpT-mediated G3P transport in whole cells. The data represent the mean ± standard error (SE) of three separate measurements. Curves were fitted with the Michaelis–Menten equation and used to calculate the Km and Vmax values at each temperature studied. (B) Arrhenius plot of the rate of the G3P transport into whole cells of E. coli at various temperatures. The plot was constructed using the natural log of the Vmax values obtained for the rates of the G3P transport presented in Table 1. The filled circles represent the means of three individual measurements.
Table 1.
Kinetic Properties of the GlpT-Catalyzed G3P Transport into E. coli Whole Cells
| temperature (°C) | Vmax [nmol min−1 (mg of protein)−1] | Km (μM) |
|---|---|---|
| 8 | 11.1 ± 3.5 | 42.4 ± 17.5 |
| 16 | 17.6 ± 2.3 | 34.2 ± 0.8 |
| 24 | 31.1 ± 1.7 | 39.5 ± 15.4 |
| 33 | 82.1 ± 6.4 | 50.7 ± 3.1 |
Reconstitution of GlpT
To further investigate the effect of the temperature on the kinetics of GlpT-mediated G3P uptake, we decided to study a system that was free of interference from other protein. To this end, we reconstituted GlpT into proteoliposomes. Successful results were achieved using DDM-mediated reconstitution of partially purified (about 85–90% pure according to SDS–PAGE analysis) His-tagged GlpT into liposomes consisting of 3:1 E. coli total lipid/phosphatidylcholine. The presence of a His tag does not affect the activity of the transporter (16). Previous experiments have demonstrated that GlpT reconstitutes into liposomes in a random orientation (15). It is important to realize in this case, however, that the direction of transport is dictated by the driving gradient and not the orientation of the protein. Interestingly, activity was present only when the protein was purified in the presence of 100 mM KPi, 10 mM G3P, and 0.2% (w/v) E. coli lipid extract. Protein that had been stored at −20 or −80 °C, even briefly, showed a significant decrease in the transport activity compared to that of freshly prepared protein. Additionally, GlpT stored at 4 °C for more than 72 h also showed a lower activity (results not shown). Hence, all of our experiments were performed with freshly purified protein. The efficiency of GlpT incorporation into liposomes was estimated by the quantitation of protein remaining in the supernatant after the proteoliposomes were collected by ultracentrifugation. The efficiency of incorporation was about 60%, equivalent to about 0.25 μg of GlpT for each individual transport assay measurement.
Transport Activity of GlpT in Proteoliposomes
GlpT was reconstituted into liposomes preloaded with saturating concentrations of the physiological countersubstrate, KPi. This allowed for the measurement of the heterologous G3P–Pi antiport reaction driven by the Pi gradient and mediated via GlpT, i.e., the uptake of labeled G3P, mimicking the physiological role of the transporter. Control experiments confirmed that the proteoliposomes were tight and that no G3P was entering them via simple diffusion. To determine that G3P was indeed transported into the lumen of the proteoliposomes and not merely associated with the surface, additional control experiments were performed using liposomes without any GlpT incorporated. Only background radioactivity was associated with them (results not shown).
Initial rates of GlpT-mediated G3P transport (1 min) were plotted as a function of the external concentration of [3H]-G3P for each temperature studied (Figure 3A). The rates clearly show saturation kinetics, as well as temperature dependence (Figure 3A), mirroring the trend observed in whole cells (Figure 2A). At the lowest temperature, 10 °C, the Vmax was 2.66 μmol min−1 (mg GlpT)−1. The Vmax increased as the temperature was increased. The transport rate was greatest at 37 °C [Vmax = 9.73 μmol min−1 (mg of GlpT)−1], equivalent to a turnover number of about 8.6 mol of G3P (mol of GlpT)−1 s−1 (Table 2). This represents a 3.5-fold increase in transport velocity over the temperature range studied, about 50% of the increase that was observed in whole cells over a similar temperature range. While Vmax was clearly influenced by the temperature, the Km again proved to be independent of the temperatures employed in this study, with a mean value of about 200 μM (Table 2).
Figure 3.
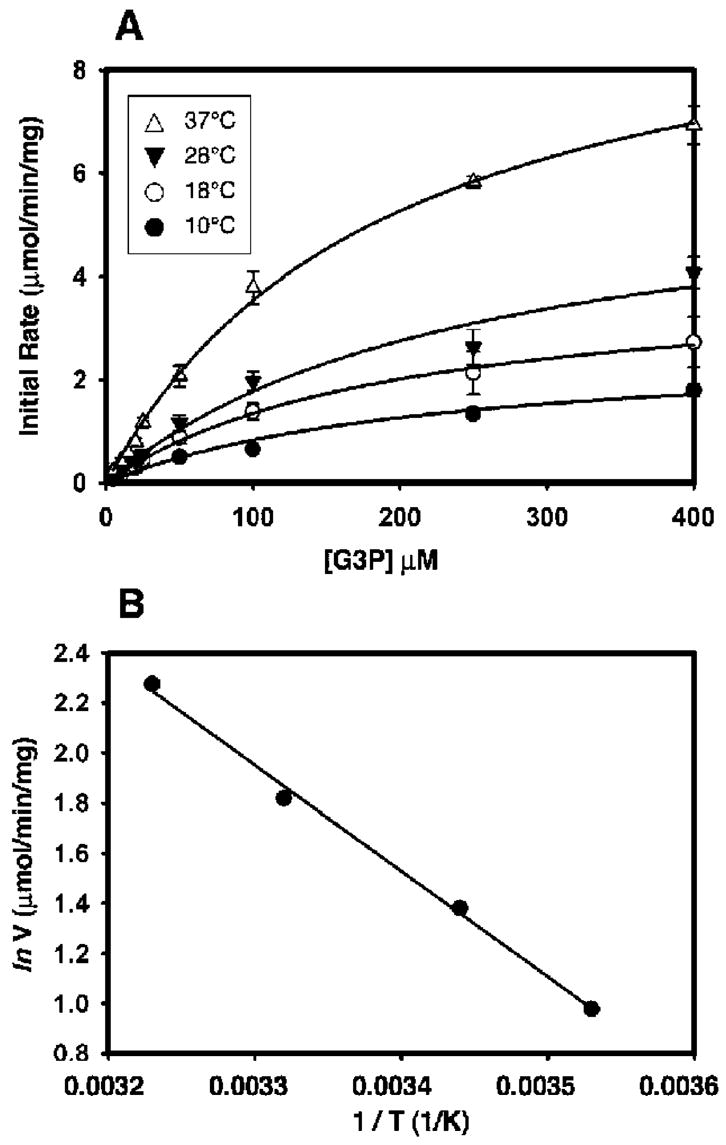
Kinetic measurements of the G3P transport by GlpT in reconstituted proteoliposomes. (A) Effect of temperature on the kinetic properties of the G3P transport mediated by GlpT reconstituted into proteoliposomes. The data represent the mean ± SE of three separate measurements. Curves were fitted with the Michaelis–Menten equation and used to calculate the Km and Vmax values at each temperature studied. The data clearly show the temperature dependence of the transport rate. (B) Arrhenius plot of the rate of the G3P transport into proteoliposomes between 10 and 37 °C. The plot was generated using the natural log of the Vmax values presented in Table 2.
Table 2.
Kinetic Properties of the G3P Transport into Proteoliposomes at Various Temperatures
| temperature (°C) | Vmax [μmol min−1 (mg of protein)−1] | Km (μM) | Kd (μM) |
|---|---|---|---|
| 10 | 2.66 ± 0.36 | 219.71 ± 60.28 | 0.71 ± 0.09 |
| 18 | 3.98 ± 0.61 | 195.30 ± 64.25 | 0.76 ± 0.10 |
| 28 | 6.16 ± 0.78 | 247.38 ± 61.80 | 0.68 ± 0.16 |
| 37 | 9.73 ± 1.11 | 163.89 ± 44.18 | 0.80 ± 0.17 |
An Arrhenius plot of the data again demonstrates the temperature dependence of the G3P transport (Figure 3B). The data are in general agreement with those calculated from whole-cell data and show no bi- or multiphasic behavior over the temperature range studied. Using this plot, an activation energy of 35.2 kJ mol−1 was calculated and a Q10 value of 1.6 was obtained for the temperature range between 10 and 37 °C. Although these activation energy and Q10 values are about 37 and 27% lower, respectively, than those observed for GlpT-catalyzed G3P translocation in whole cells, they do exhibit a temperature dependence that concurs with active transport, rather than simple diffusion (Q10 < 1.0) or flux through a channel (Q10 < 1.5). These data lend credence to our model of secondary active transport, in which a temperature-dependent conformational change is associated with the GlpT-mediated G3P–Pi antiport reaction. To test if substrate binding was equally affected by the temperature, an intrinsic tryptophan fluorescence-quenching assay was performed.
Binding Affinity to G3P at Various Temperatures
The kinetic analysis described above only allows Km and Vmax to be measured. These values, however, do not permit a distinction to be made between transport- or dissociation-limited models. Therefore, we determined the apparent dissociation constant (Kd) values of G3P binding to GlpT in detergent solution at each temperature under study by measuring the quenching of the protein intrinsic tryptophan fluorescence upon substrate binding (15). To minimize potential interference from contaminating protein, the GlpT used for fluorescence-quenching studies was purified to homogeneity using size-exclusion chromatography as described previously (15). Indeed, the dissociation constants for G3P binding to GlpT at neutral pH were not influenced by temperature (Figure 4). Between 10 and 37 °C, the Kd remained between 0.71 ± 0.09 and 0.80 ± 0.17 μM, respectively (Table 2).
Figure 4.
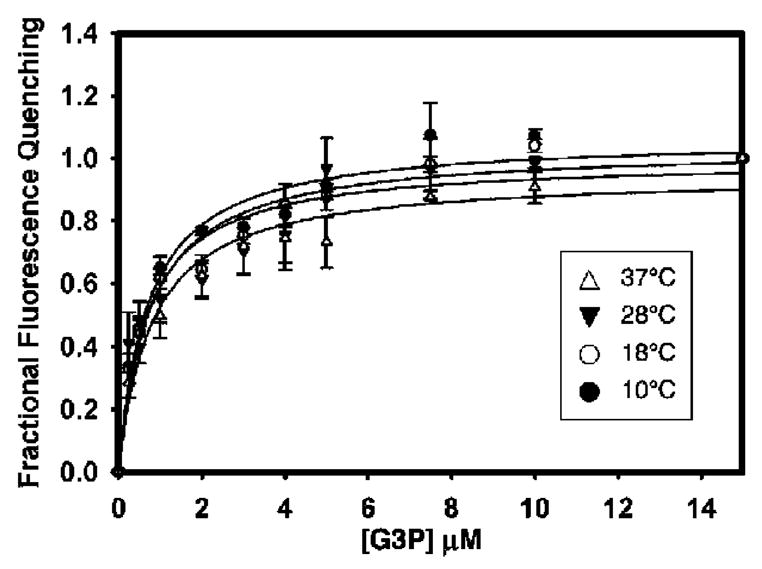
Measurement of G3P binding to GlpT at various temperatures using intrinsic tryptophan fluorescence quenching. G3P was titrated until fluorescence quenching was saturated (about 15 μM). The dissociation constant of G3P binding was found to be about 0.7 μM, and substrate binding did not show temperature dependence. Data points represent the mean ± SE of three separate measurements.
DISCUSSION
The results of our transport and binding experiments with E. coli whole cells and with GlpT in proteoliposomes and in solution are in accordance with a rocker-switch-type transport mechanism for GlpT. The GlpT-mediated transport rate not only displays substrate saturation, but also the Vmax increases markedly with the temperature. In complete contrast, both the apparent Km and the apparent substrate binding dissociation constant, Kd, are temperature-independent.
The temperature dependence of the GlpT transport process was reflected in the temperature coefficient and activation energy values obtained from Arrhenius plots of the data. Q10 and Ea values can be used to gain a mechanistic insight of the transport process. Data from whole-cell transport assays revealed a Q10 of 2.2 (Ea of 55.4 kJ mol−1) over the range of temperature investigated, and the Q10 derived from assays performed with GlpT reconstituted into proteoliposomes was 1.6 (Ea of 35.2 kJ mol−1). Published Q10 values for active transporters range from 1.5 to over 10 (25–27). Moreover, both Arrhenius plots are completely linear, suggesting that any conformational change is unaffected by the transition state of the lipid environment, as observed previously for equilibrium exchange catalyzed by a lactose permease mutant (28) and transport of glucose in E. coli (29). Although the calculated activation energy of 35.2 kJ mol−1 for GlpT reconstituted into proteoliposomes is very similar to that of the E325A lactose permease mutant that catalyzes lactose transport without proton translocation (29), significantly, it is only marginally greater than that observed for flow through membrane channels. The differences between the Ea and Q10 values calculated from whole-cell and proteoliposome data could be attributed to one or both of the following: (i) differences in the composition and asymmetry of the lipid environment experienced by the transporter [it has been shown previously that, for the LacY transporter, lipid composition affects coupling between lactose and proton symport (30)] and (ii) by the fact that in whole cells the transporter exists in a unique orientation within the membrane, whereas in proteoliposomes, it is distributed evenly between two different orientations (15). In the latter case, even though the Pi gradient provides the net driving force and sets the vectorial nature of the transport reaction, the two different orientations of the transporter with respect to the gradient may exhibit subtly different kinetics constants. Although such a kinetic bias has been reported for the Streptococcus thermophilus LacS (24) and E. coli PutP transporters (31), the two orientations of the E. coli glucose 6-phosphate transporter (UhpT), a close homologue of GlpT, exhibit functional symmetry and kinetics with respect to physiologically relevant substrates (33).
Taken at face value, the relatively low activation energies calculated for G3P transport in both whole cells and proteoliposomes would appear to argue against a substrate translocation mechanism that involves large, global conformational changes in the transporter. However, a relatively low activation energy for a substrate translocation event involving large conformational changes can be rationalized if the transporter exists in a delicately balanced or “poised” state when the substrate is bound, requiring only a small additional energy input to effect the conformational change and substrate translocation (Figure 1C). Thus, under conditions of substrate saturation, the transporter is available to cycle between inward- and outward-facing conformations, binding and releasing the substrate on opposite sides of the membrane (28).
Because conventional kinetic analysis alone (calculation of Km and Vmax values) cannot differentiate between a model in which conformational change is the rate-limiting step for transport and a model in which the dissociation of the substrate is rate-limiting, the ability to make a distinction between these models is crucial to verify the mechanism. To allow this distinction to be made, we measured the binding affinity of GlpT for its substrate, G3P. Although changing the temperature altered the Vmax of the GlpT-mediated transport in both whole cells and proteoliposomes, it did not affect either the Km or the apparent affinity, Kd, of GlpT for G3P. Our Kd value also shows binding to be much tighter than predicted by the Km (32). The fact that our calculated Km differs from the measured Kd by over 2 orders of magnitude (about 200 versus 0.7 μM) is consistent with an interconversion from the Co–S to the Ci–S state that is rate-limiting. In the absence of the substrate or even at nonsaturating concentrations, the transporter exists as a stable and segregated Ci or Co state. The experimental corroboration of the two predictions made from the rocker-switch model of the GlpT transport mechanism allow us to further refine our model of the GlpT transport mechanism.
Similar to other antiporters, GlpT operates via a kinetic mechanism that breaks down the complete antiport cycle into six discrete stages (Figure 1B). G3P binds to the unloaded transporter in the Co state, thereby decreasing entropy and enabling intrinsic binding energy to lower the energy barrier for substrate translocation. Formation of the Co–S complex is not temperature-sensitive, because neither substrate binding nor Km are influenced by temperature. The next stage of the mechanism represents the conformational interconversion of the protein from the Co to the Ci state, which is accompanied by the simultaneous translocation of substrate across the membrane (Figure 1B). This interconversion is the rate-limiting step of the whole transport process, as proposed for UhpT (33). Crucially, the transport step is temperature-dependent, with an enthalpic contribution actually driving the interconversion from one conformation to the other via Brownian motion (Figure 1C). Therefore, GlpT-mediated translocation of the substrate across the membrane is, in essence, a catalytic mechanism that serves to increase the rate of an otherwise improbable event. The final stage involves the release of bound substrate into the cytoplasmic compartment and presentation of the binding site to the countersubstrate to allow for the antiport reaction to proceed. Under physiological conditions, such a mechanism would avoid excessively high- or low-energy intermediates along the reaction pathway, because the former would present a barrier to biologically relevant turnover rates and the latter would effectively “lock” the transporter in an energy well.
The transport rates measured in proteoliposomes are over 2 orders of magnitude larger than those measured in whole cells. This apparent discrepancy can be addressed by the fact that the latter rates were measured on the basis of total membrane protein and not on the actual amount of active GlpT present in the whole-cell membrane. Because GlpT makes only a small contribution to the total membrane protein, the transport rates are vastly underestimated. The differences observed between the measured Km values for GlpT-catalyzed transport in whole cells and proteoliposomes (about 50 and 200 μM, respectively) can probably also be attributed to the intrinsic differences between the two systems under study.
Previous results for the GlpT-mediated transport of G3P in proteoliposomes published by our laboratory (15) reported transport rates that were 3-fold higher than those presented here. This inconsistency is probably due to the fact that the previous experiments used a commercially available assay kit (EnzChek, Invitrogen) that provides a spectrophotometric method for the quantitation of Pi (34). It was found subsequently that, in addition to inorganic phosphate, the kit also detected organic phosphates, such as G3P. This led to an overestimation, both of the amount of Pi released and, therefore, of the GlpT-mediated transport rate. Furthermore, the affinity for G3P binding in the present work (Kd of 0.7 μM) is 5-fold greater than that previously published (Kd of 3.54 μM) (15). In the current experiments, binding measurements were performed immediately using freshly purified protein, whereas the previous measurements were performed with protein that had been stored in detergent solution for several days, which may have led to a partial loss of the integrity of the transporter.
Temperature dependence of transport reaction rates has been observed before for other transporter proteins (26, 29, 34), although the significance in many cases was not explained in terms of the transport mechanism. Electrophysiology measurements of the voltage dependence of the L-asparagine transporter in endoderm cells from Xenopus also showed that Vmax increased with temperature but the Km value remained unaltered (25). The yeast mitochondrial citrate transporter, CTP, also showed a temperature dependence of transport rates (35). However, in contrast to our observations, the latter system also showed that Km was temperature-dependent. Unlike GlpT, CTP functions as a dimer, and this may explain the apparent temperature sensitivity of Km values. Similarly, the independence of the substrate-binding affinity to temperature has also been observed beforehand in thermodynamic studies of the LacY transporter of E. coli (36).
When these data are taken together, they are completely consistent with the rocker-switch mechanism for GlpT-mediated transport. This mechanism probably involves large, global conformational changes in the transporter during the reaction cycle. Our results are also consistent with the viewpoint that membrane transporters are, essentially, vectorial enzymes (37–39). Because the classical view of enzyme catalysis is based on a transition-state destabilization of the substrate (40), the transition state is therefore the essential step of the transport process, linking structural changes in the protein to the translocation event (38, 41). In the transport mechanism we propose, destabilization occurs as a global conformational change in the transporter, driven by both substrate binding (the entropic contribution, of which lowers the energy barrier) and Brownian motion (the enthalpic contribution, of which drives the conformational change). The transport event is also the rate-limiting step, because the magnitude of the protein movement is the major determinant for the rate. It must also be noted that the conformational changes that accompany translocation do not represent random free movements in a floppy structure but rather a series of programmed and precise events, probably involving mutual shifts of amino acid side chains, along with rotation, bending, and changes in inclination of transmembrane helices (8, 20). The rocker-switch mechanism that we describe here is probably applicable to substrate translocation by other antiporters of the MFS.
Acknowledgments
We thank Dr. Bill Rice (NYSBC) for advice on the use of the radioactive substrate.
Footnotes
This work was supported in part by NIH Grant DK-053973 (to D.-N.W.). The work of P.C.M. was supported by NIH Grant GM-24195.
Abbreviations: GlpT, glycerol-3-phosphate transporter; G3P, glycerol-3-phosphate; Pi, inorganic phosphate; MFS, major facilitator superfamily; Ea, activation energy; DDM, N-dodecyl-β-D-maltoside.
References
- 1.Mitchell P. Molecule, group and electron translocation through natural membranes. Biochem Soc Symp. 1963;22:142–168. [Google Scholar]
- 2.Tanford C. Mechanism of free-energy coupling in active transport. Ann Rev Biochem. 1983;52:379–409. doi: 10.1146/annurev.bi.52.070183.002115. [DOI] [PubMed] [Google Scholar]
- 3.Henderson PJF, Roberts PE, Martin GEM, Seamon KB, Walmsley AR, Rutherford NG, Varela MF, Griffith JK. Homologous sugar transport proteins in microbes and man. Biochem Soc Trans. 1993;21:1002–1006. doi: 10.1042/bst0211002. [DOI] [PubMed] [Google Scholar]
- 4.Pao SS, Paulsen IT, Saier MH. Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saier MH, Beatty JT, Goffeau A, Harley KT, Heijne WHM, Huang SH, Jack DL, Jahn PS, Lew K, Liu J, Pao SS, Paulsen IT, Tseng TT, Virk PS. The major facilitator superfamily. J Mol Microbiol Biotechnol. 2000;2:255–255. [PubMed] [Google Scholar]
- 6.Saidijam M, Benedetti G, Ren QH, Xu ZQ, Hoyle CJ, Palmer SL, Ward A, Bettaney KE, Szakonyi G, Meuller J, Morrison S, Pos MK, Butaye P, Walravens K, Langton K, Herbert RB, Skurray RA, Paulsen IT, O’Reilly J, Rutherford NG, Brown MH, Bill RM, Henderson PJF. Microbial drug efflux proteins of the major facilitator superfamily. Curr Drug Targets. 2006;7:793–811. doi: 10.2174/138945006777709575. [DOI] [PubMed] [Google Scholar]
- 7.Higgins CF. Multiple molecular mechanisms for multi-drug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 8.Huang YF, Lemieux MJ, Song JM, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301:616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 9.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 10.Hirai T, Heymann JA, Shi D, Sarker R, Maloney PC, Subramaniam S. Three-dimensional structure of a bacterial oxalate transporter. Nat Struct Biol. 2002;9:597–600. doi: 10.1038/nsb821. [DOI] [PubMed] [Google Scholar]
- 11.Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312:741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elvin CM, Hardy CM, Rosenberg H. Pi exchange mediated by the Glpt-dependent sn-glycerol-3-phosphate transport system in Escherichia coli. J Bacteriol. 1985;161:1054–1058. doi: 10.1128/jb.161.3.1054-1058.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abramson J, Smirnova I, Kasho V, Verner G, Iwata S, Kaback HR. The lactose permease of Escherichia coli: Overall structure, the sugar-binding site and the alternating access model for transport. FEBS Lett. 2003;555:96–101. doi: 10.1016/s0014-5793(03)01087-1. [DOI] [PubMed] [Google Scholar]
- 14.Ambudkar SV, Larson TJ, Maloney PC. Reconstitution of sugar phosphate transport systems of Escherichia coli. J Biol Chem. 1986;261:9083–9086. [PubMed] [Google Scholar]
- 15.Auer M, Kim MJ, Lemieux MJ, Villa A, Song JM, Li XD, Wang DN. High-yield expression and functional analysis of Escherichia coli glycerol-3-phosphate transporter. Biochemistry. 2001;40:6628–6635. doi: 10.1021/bi010138+. [DOI] [PubMed] [Google Scholar]
- 16.Fann MC, Busch A, Maloney PC. Functional characterization of cysteine residues in GlpT, the glycerol 3-phosphate transporter of Escherichia coli. J Bacteriol. 2003;185:3863–3870. doi: 10.1128/JB.185.13.3863-3870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maloney PC, Ambudkar SV, Anantharam V, Sonna LA, Varadhachary A. Anion exchange mechanisms in bacteria. Microbiol Rev. 1990;54:1–17. doi: 10.1128/mr.54.1.1-17.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ambudkar SV, Anantharam V, Maloney PC. UhpT, the sugar phosphate antiporter of Escherichia coli, functions as a monomer. J Biol Chem. 1990;265:12287–12292. [PubMed] [Google Scholar]
- 19.Fann M, Davies AH, Varadhachary A, Kuroda T, Sevier C, Tsuchiya T, Maloney PC. Identification of two essential arginine residues in UhpT, the sugar phosphate antiporter of Escherichia coli. J Memb Biol. 1998;164:187–195. doi: 10.1007/s002329900404. [DOI] [PubMed] [Google Scholar]
- 20.Lemieux MJ, Huang Y, Wang DN. Structural basis of substrate translocation by the Escherichia coli glycerol-3-phosphate transporter: A member of the major facilitator superfamily. Curr Opin Struct Biol. 2004;14:405–412. doi: 10.1016/j.sbi.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Vink R, Bendall MR, Simpson SJ, Rogers PJ. Estimation of H+ to adenosine 5′-triphosphate stoichiometry of Escherichia coli ATP synthase using 31P NMR. Biochemistry. 1984;23:3667–3675. doi: 10.1021/bi00311a015. [DOI] [PubMed] [Google Scholar]
- 22.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 23.Rigaud J-L, Pitard B, Levy D. Reconstitution of membrane proteins into liposomes: Application to energy-transducing membrane proteins. Biochim Biophys Acta. 1995;1231:223–246. doi: 10.1016/0005-2728(95)00091-v. [DOI] [PubMed] [Google Scholar]
- 24.Knol J, Veenhoff L, Liang WJ, Henderson PJF, Leblanc G, Poolman B. Unidirectional reconstitution into detergent-destabilized liposomes of the purified lactose transport system of Streptococcus thermophilus. J Biol Chem. 1996;271:15358–15366. doi: 10.1074/jbc.271.26.15358. [DOI] [PubMed] [Google Scholar]
- 25.Bergman C, Bergman J. Origin and voltage dependence of asparagine-induced depolarization in intestinal cells of Xenopus embryo. J Physiol (London) 1985;366:197–220. doi: 10.1113/jphysiol.1985.sp015792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chraibi A, Horisberger AD. Na self-inhibition of human epithelial Na channel: Temperature dependence and effect of extracellular proteases. J Gen Physiol. 2002;120:133–145. doi: 10.1085/jgp.20028612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klingenberg M, Grebe K, Appel M. Temperature dependence of ADP/ATP translocation in mitochondria. Eur J Biochem. 1982;126:263–269. doi: 10.1111/j.1432-1033.1982.tb06774.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, Kaback HR. Effect of the lipid phase transition on the lactose permease from Escherichia coli. Biochemistry. 2000;39:14538–14542. doi: 10.1021/bi001947g. [DOI] [PubMed] [Google Scholar]
- 29.Rottem S, Cirillo VP, Dekruyff B, Shinitzk M, Razin S. Cholesterol in Mycoplasma membranes–Correlation of enzymic and transport activities with physical state of lipids in membranes of Mycoplasma mycoides var. capri adapted to grow with low cholesterol concentrations. Biochim Biophys Acta. 1973;323:509–519. doi: 10.1016/0005-2736(73)90159-4. [DOI] [PubMed] [Google Scholar]
- 30.Chen CC, Wilson TH. Solubilization and functional reconstitution of the proline transport system of Escherichia coli. J Biol Chem. 1986;261:2599–2604. [PubMed] [Google Scholar]
- 31.Jung H, Tebbe S, Schmid R, Jung K. Unidirectional reconstitution and characterization of purified Na+/proline transporter of Escherichia coli. Biochemistry. 1998;37:11083–11088. doi: 10.1021/bi980684b. [DOI] [PubMed] [Google Scholar]
- 32.van Winkle LJ. Biomembrane Transport. Academic Press; New York: 1995. pp. 90–91. [Google Scholar]
- 33.Fann MC, Maloney PC. Functional symmetry of UhpT, the sugar phosphate transporter of Escherichia coli. J Biol Chem. 1998;273:33735–33740. doi: 10.1074/jbc.273.50.33735. [DOI] [PubMed] [Google Scholar]
- 34.Upson RH, Haugland RP, Malekzadeh MN, Haugland RP. A spectrophotometric method to measure enzymatic activity in reactions that generate inorganic pyrophosphate. Anal Biochem. 1996;243:41–45. doi: 10.1006/abio.1996.0479. [DOI] [PubMed] [Google Scholar]
- 35.Ma CL, Kotaria R, Mayor JA, Remani S, Walters DE, Kaplan RS. The yeast mitochondrial citrate transport protein–Characterization of transmembrane domain III residue involvement in substrate translocation. J Biol Chem. 2005;280:2331–2340. doi: 10.1074/jbc.M411474200. [DOI] [PubMed] [Google Scholar]
- 36.Nie YL, Smirnova I, Kasho V, Kaback HR. Energetics of ligand-induced conformational flexibility in the lactose permease of Escherichia coli. J Biol Chem. 2006;281:35779–35784. doi: 10.1074/jbc.M607232200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jencks WP. What Is a Coupled Vectorial Process? Curr Top Memb Trans. 1983;19:1–19. [Google Scholar]
- 38.Klingenberg M. Transport catalysis. Biochim Biophys Acta. 2006;1757:1229–1236. doi: 10.1016/j.bbabio.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 39.Tanford C. Simple model for the chemical-potential change of a transported ion in active transport. Proc Natl Acad Sci USA. 1982;79:2882–2884. doi: 10.1073/pnas.79.9.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jencks WP. Catalysis in Chemistry and Enzymology. Dover Publications, Inc; New York: 1987. pp. 599–614. [Google Scholar]
- 41.Krupka RM. Channelling free energy into work in biological processes. Exp Physiol. 1998;83:243–251. doi: 10.1113/expphysiol.1998.sp004109. [DOI] [PubMed] [Google Scholar]