

Abstract
Interferon (IFN)-λ, also known as IL-28A, IL-28B or IL-29, is a new type III IFN, which like type I IFN-(α/β), activates common elements of the JAK/STAT signaling pathway and exhibits antiproliferative activity. Currently, IFN-α is used in the treatment of certain forms of cancer, but its antitumor effects are limited and associated with high toxicity. In this study, we determined whether IFN-λ induced the same level of cell growth inhibition relative to IFN-α. To this effect HaCaT cells, which are typically growth inhibited by IFN-α, underwent apoptosis in response to IFN-λ. Next, in contrast to IFN-α stimulation, IFN-λ prolonged the duration of activated STAT1 and STAT2. Furthermore, the kinetics of IFN-stimulated genes was different as IFN-λ induced a delayed but stronger induction of IFN-responsive genes. Components of the JAK/STAT pathway remained essential for the antiproliferative effects of IFN-α and IFN-λ. IFN-λ-induced persistence of STAT activation required de novo protein synthesis and was in part due to a delay in STAT2 inactivation. Thus our data demonstrate that the duration of IFN-λ signaling is different from that of IFN-α, and that IFN-λ could be a suitable cytokine to evaluate for cancer therapy.
Keywords: Interferon-α, interferon-λ, STAT, antiproliferation, apoptosis
INTRODUCTION
Interferons (IFNs) are a family of pleiotropic cytokines that exhibit potent antiviral, antiproliferative, apoptotic, and immunoregulatory activities 1. These activities are controlled, in part, by a set of cellular genes that are rapidly induced upon binding of IFN to specific membrane-bound receptors 1-3. The type I IFN receptor consists of two chains: IFNAR1 and IFNAR2 1. Binding of type I IFNs (IFN-α/β) to their cognate receptor stimulates the auto-phosphorylation of members of the Janus family of tyrosine kinases, specifically JAK1 and TYK2 3. STAT1 and STAT2 are tyrosine-phosphorylated by the JAKs, and are key factors in mediating the biological effects of IFN 1.
Tyrosine phosphorylation of STAT1 and STAT2 causes these proteins to dimerize and translocate to the nucleus, where they associate with IFN-regulatory factor 9 (IRF9/p48) to form the IFN-stimulated gene factor-3 (ISGF3) complex 4. Following nuclear translocation, the ISGF3 complex binds to interferon-stimulated response elements (ISREs) in the DNA, and induces the transcription of IFN-stimulated genes (ISGs). Type I IFN also induces the formation of phosphorylated STAT1 homodimers, which translocate to the nucleus 5. In contrast to ISGF3, STAT1 homodimers bind to the gamma interferon-activated site (GAS), and induce the expression of a distinct set of ISGs. Collectively, these ISGs mediate the biological effects of IFN, such as inhibition of viral replication, cellular growth inhibition, and apoptosis.
Type III IFN, also known as IFN-λ, was recently identified as a member of the IL-10-related cytokine family 6. Human IFN-λ was discovered based on limited sequence homology to members of the class II cytokine receptor ligand family. Three distinct forms of IFN-λ were identified, IFN-λ1/IL-29, IFN-λ2/IL-28A, and IFN-λ3/IL-28B, with IFN-λ1 reported to be the most potent form of the molecule 6, 7. Unlike type I IFNs, human and mouse IFN-λ are cross reactive. The IFN-λ receptor complex (IFN-λR) consists of a unique ligand-binding chain, IFN-λR1 (also known as IL-28R), and an accessory chain, IL-10R2, that is shared with the IL-10 receptor and other IL-10-related cytokine receptors 6. Interestingly, while their receptor subunits display no detectable homology to type I IFNs, IFN-λ and type I IFNs (IFN-α and IFN-β) both activate the JAK/STAT pathway 6, 8. Signaling via the IFN-λR results in tyrosine phosphorylation of STAT1 and STAT2 (as well as STAT3 and STAT5) and induces assembly of the ISGF3 and STAT1 homodimer complexes 6, 9. Thus, many of the genes induced by type I IFN are also induced by IFN-λ 6, 7. These similarities led to the proposal that IFN-λ be deemed a new type of IFN: type III IFN 6.
Type I IFNs are FDA approved anticancer drugs, and disappointedly, the small subset of patients who benefit from this form of therapy must also endure the adverse effects of the treatment 10. Therefore, the timely identification of alternative treatment modalities that are less toxic and more effective in destroying tumor cells is a priority. The focus of our study was to compare the antiproliferative effects and kinetics of activation of the JAK/STAT pathway between IFN-α and the recently discovered IFN-λ. Our study demonstrates that in contrast to IFN-α, IFN-λ signals extend STAT1 and STAT2 tyrosine phosphorylation, prolong the duration of differentially-regulate ISG expression. Furthermore, IFN-λ can promote apoptosis in cell lines that previously shown only exhibit antiproliferative activity in response to IFN-α. These results reveal that IFN-λ could potentially be used, in addition to IFN-α in controlling tumor growth.
MATERIALS AND METHODS
Cell Culture and Cytokines
The human keratinocyte cell line HaCaT, the human fibrosarcoma 2fTGH cell line and mutants derived from this line deficient in STAT1 (U3A) and Jak1 (U4A) (a gift from G. Stark, Cleveland Clinic Foundation, OH) were maintained in DMEM, supplemented with 10% FBS, 0.4 mg/mL L-glutamine, 1% penicillin and 1% streptomycin. Cell culture reagents were obtained from Invitrogen (Carlsbad, CA), unless stated otherwise. Cell lines stably expressing transfected human IFN-λR1 were maintained in medium supplemented with 2 μg/mL puromycin (Sigma, St. Louis, MO). Human IFN-α-2a and human IFN-λ1 were obtained from Peprotech Inc. (Rocky Hill, NJ). Mouse IFN-α was purchased from PBL Biomedical Laboratories (Piscataway, NJ).
Cell Transfection
Parental, U3A and U4A 2fTGH cells were transfected with 5 μg of an expression plasmid encoding hemaggluttinin (HA)-tagged IFN-λR1 (kindly provided by Jean-Christophe Renauld, Ludwig Institute for Cancer Research) by using Metafectene reagent (Biontex,Germany) at a ratio of 1:3 (μg plasmid DNA: μL Metafectene). Following overnight transfection at 37°C, cells were switched to fresh medium containing 2 μg/mL puromycin. Two weeks later, surviving individual puromycin-resistant colonies were selected and stained with anti-HA antibody to detect surface expression of IFN-λR1 by flow cytometry.
Flow Cytometry
Surface IFN-λR1 expression was determined by staining cells with a mouse anti-HA mAb (Covance, Berkeley, CA) followed by incubation with a rabbit anti-mouse FITC-labeled immunoglobulin (R&D Systems, Minneapolis, MN). Stained cells were analyzed with a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Apoptosis Assay
Cells (5 × 105) were incubated with 100 μL of Annexin-V reagent (BD Biosciences, San Jose, CA) consisting of 1 μL Annexin-V-FITC, 10 μL propidium iodide; PI (50 μg/mL, Calbiochem, La Jolla, CA), 10 μL 10× binding buffer (100 mM Hepes pH 7.4, 1.4 M NaCl, 25 mM CaCl2) and 79 μL H2O for 15 min at room temperature in the dark. Following incubation, 500 μL of 1× binding buffer was added to each sample and the samples analyzed on a FACScan flow cytometer. Unstained cells and cells singly stained with either Annexin-V-FITC or PI only were used as controls. For inactivation of caspase activity, cells were pretreated for 30 min with 40 μM of the caspase inhibitor ZVAD-fmk (BD Biosciences, San Jose, CA), followed by stimulation with the various IFNs.
Proliferation Assay
Cellular viability was assessed by trypan blue dye exclusion (Invitrogen, Carlsbad, CA). Five-hundred cells in 100 μL volume were added to each well of a flat-bottom 96-well plate in triplicate. Cells were treated with or without IFN and incubated at 37°C for 72 h. Cell growth was determined by MTS,[3-(4,5-dimethythiazol-2-yl)]-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) salt assay (Promega, Madison, WI). Briefly, 20 μL of MTS reagent was added to each well and the plate incubated for 2-4 h at 37°C. Following color development, absorbance was measured at 490 nm on a Perkin Elmer (Wellesley, MA) Wallac Victor2 1420 multilabel counter.
Western Blotting
Cells were lysed in a buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, 2 mM EDTA pH 8.0, 0.5% Triton X-100, protease inhibitor cocktail (Roche Diagnostics Mannheim, Germany), 1 mM PMSF, and 1 μM Na3VO4. Proteins were resolved on pre-cast 4-12% SDS-polyacrylamide gels (Invitrogen, Carlsbad, CA) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA). Immunoblots were incubated with mouse anti-phosphotyrosine STAT1, pY-STAT1 (1:2000 dilution, BD Biosciences, Franklin Lakes, NJ), rabbit anti-phosphotyrosine STAT2, pY-STAT2 (1:2000 dilution, Upstate, Charlottesville, VA), rabbit-phosphotyrosine STAT3, pY-STAT3 (1:1000 dilution, Cell Signaling Technology), total STAT1 and total STAT2 clone C-20 (1:2000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) and total STAT3 (Cell Signaling, Boston MA). Beta-actin was obtained from Abcam (Cambridge, MA) and used at a 1:5000 dilution. Following washing, immunoblots were incubated with the respective anti-mouse or anti-rabbit HRP-conjugated IgG secondary antibody (1:10000, Zymed, San Francisco, CA). Detection was performed using Supersignal West Pico Chemiluminescent substrate kit (Pierce, Rockford, IL).
Quantitative RT-PCR
Total cellular RNA was isolated with Trizol (Invitrogen, Carlsbad, CA) from cells and 5 μg of total RNA was reverse transcribed to generate cDNA using Superscript II reverse transcriptase (Invitrogen) as described previously 11. qRT-PCR primers for ISGs were obtained from Applied Biosystems (Foster City, CA). cDNA was mixed with Taqman 2× PCR master mix (Applied Biosystems), using primers with FAM reporter dyes, and qPCR reactions were carried out using the 7300 Real Time PCR system (Applied Biosystems). mRNA quantification was normalized by multiplexing with 18S-VIC MGB primers. Samples were amplified using the following PCR variables: 1 cycle of 55°C for 2 minutes, 1 cycle of 95°C for 10 minutes, then 40 cycles of 95°C for 30 seconds, 60°C for 1 minute.
Pulse chase tyrosine dephosphorylation
Cells were stimulated with IFN-α or IFN-λ for 30 min and washed once with complete DMEM medium to remove cytokine. Cells were then treated for various times with 500 nM of staurosporine (Sigma-Aldrich, St. Louis MO) and resuspended in complete DMEM medium to stop further tyrosine phosphorylation. Whole cell extracts were prepared and analyzed by Western blotting.
RESULTS
IFN-λ favors the induction of apoptosis in HaCaT cells
Although type I (IFN-α) and type III IFN (IFN-λ) signal through distinct receptors, both cytokines induce antiproliferative responses. While receptors to type I IFNs are ubiquitously expressed on all cells, receptors to IFN-λ, however, are only found on most but not all tumor lines. In this study, to determine if the level of the antiproliferative response induced by IFN-λ and IFN-α differed, we chose the human keratinocyte HaCaT cell line as it expresses receptors to both types of IFNs 6. Stimulation of HaCaT cells with increasing and saturating doses of IFN-α for 72 h led to a 25% reduction in cell proliferation (Fig. 1A). In marked contrast, IFN-λ induced a greater growth inhibitory effect of 50%, which is twice the effect seen with IFN-α (Fig. 1B). We also examined if the antiproliferative effect could be augmented when both IFN types were used in combination. Adding IFN-α and IFN-λ together augmented their antiproliferative effect, suggesting that there is no competition for intracellular JAK kinases or STATs (Fig. 1C). Next, we assessed whether IFN-λ-induced antiproliferative effects could be attributed to the induction of program cell death (apoptosis). As measured by annexin-V and propidium iodide dual staining, IFN-λ stimulated HaCaT cells to undergo apoptosis. Pretreatment with the broad spectrum caspase inhibitor ZVAD-fmk protected cells from the apoptotic effects of IFN-λ (Fig. 1D). Furthermore, it restored proliferation of IFN-λ treated HaCaT cells. Induction of apoptosis was also measured by assaying for caspase 3/7 activity and similar results were obtained (data not shown). Of note, even when HaCaT cells were incubated with saturating doses of IFN-α (15 ng/mL), these cells failed to undergo apoptosis (data not shown). Therefore, our data indicate that HaCaT cells respond differently to each type of IFN.
Figure 1.
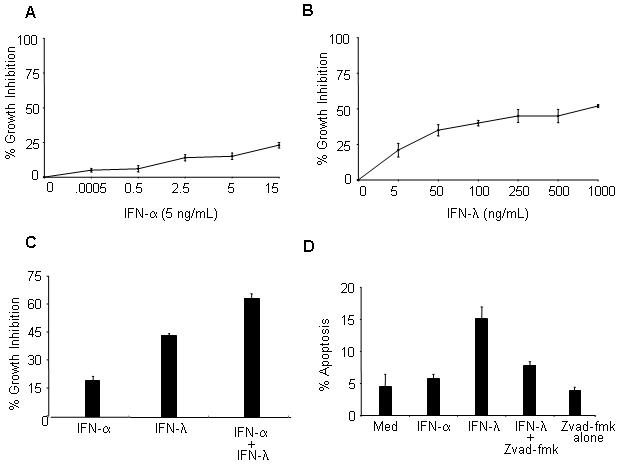
IFN-λ promotes an antiproliferative and apoptotic response in HaCaT cells. Cells were left untreated or treated with increasing doses of either (A) IFN-α or (B) IFN-λ for 72 h. Proliferation was measured by MTS assay. (C). Same as in B, except cells were treated for 72 h with either IFN-α (5 ng/mL) or IFN-λ (100 ng/mL) separately, or a combination of both at the stated concentrations. (D) Apoptosis was assessed dual Annexin V and PI staining and analyzed by flow cytometry. For caspase inactivation cells were pre-treated with 40 μM ZVAD-fmk for 2 h prior to stimulation. Data shown are the mean ± SD and are representative of at least 3 independent experiments.
IFN-λ extends STAT activation relative to IFN-α
The antiproliferative and apoptotic activities of IFNs are primarily driven via signaling through the JAK/STAT pathway. To begin characterizing the disparity of the antiproliferative response seen between IFN-λ and IFN-α, we first performed a dose-response curve of IFN-induced STAT activation. We chose this approach because of the unavailability of IFNλ-R1 antibodies with high affinity binding to measure surface expression of IFN-λ receptors. Immunoblot analysis showed that doses between 100 and 250 ng/mL of IFN-λ were optimal to activate approximately the same levels of tyrosine phosphorylated STAT1 and STAT2 achieved with 5 ng/mL IFN-α (Fig. 2A). Interestingly, treatment of HaCaT cells with higher doses of IFN-λ (500-1000 ng/mL) caused a reduction rather than an increase in STAT activation. Having chosen an optimal dose of IFN-λ (100 ng/mL), we observed that the kinetics of STAT tyrosine phosphorylation was different for each cytokine. Like with IFN-α, the level of IFN-λ activated STAT1 and STAT2 peaked between 30 min and 1 h (Fig. 2B) and decayed shortly after. However, treatment with IFN-λ led to sustained activation of STAT1 and STAT2, which was detectable over the course of 24 h. We also observed that at the later time points there was a larger pool of total STAT1 and STAT2 compared with that seen with IFN-α treatment (Fig. 2B). Of note, by using higher doses of each cytokine, this neither changed the level of duration and levels of STAT activation nor increased significantly the antiproliferative effects already seen with the chosen doses of IFN-α and IFN-λ. We also examined activation of STAT3 as this transcription factor plays a critical role in cell survival and tumorigenesis 12, 13. Interestingly, STAT3 activation by IFN-λ followed the same pattern and kinetics of activation as that of STAT1 and STAT2.
Figure 2.
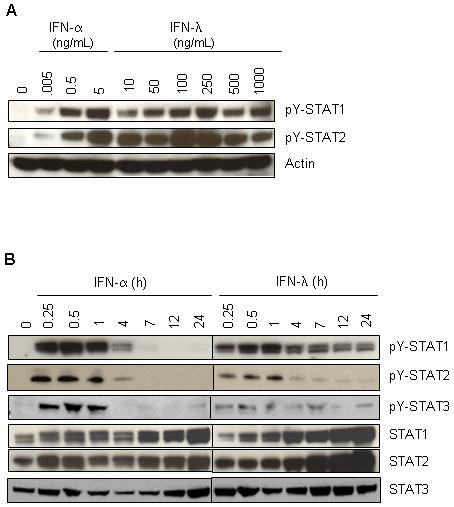
IFN-λ signals extended STAT activation in HaCaT relative to IFN-α. A. Cells were treated for 30 min with increasing concentrations of either IFN-α (0 - 5 ng/mL) or IFN-λ (0 - 1000 ng/mL). Following stimulation, cells extracts were prepared and immunoblot analysis was performed with antibodies against pY-STAT1, pY-STAT2, pY-STAT3, STAT1, STAT2 and STAT3. β-actin served as an internal control for equal loading. (B) Same as in (A) except cells were treated with either IFN-α (5 ng/mL) or IFN-λ (100 ng/mL) over a 24 h period. Blots are representative of at least 3 independent experiments.
IFN-λ stimulation leads to prolonged ISG expression
We next assessed whether as opposed to IFN-α, sustained IFN-λ induced STAT activation correlated with prolonged ISG expression. We chose to analyze by quantitative RT-PCR the expression of a panel of target ISGs involved in IFN-mediated biological processes including antiproliferative, antiviral, apoptosis, and negative regulation. IFN-λ treatment of HaCaT cells resulted, for the most part, in a delay, but soon after robust ISG expression (e.g. ISG15, MX-1, TRAIL, and SOCS1), relative to that observed with IFN-α (Fig. 3). Our results also show that while IFN-α-induced gene expression peaked at 6 h post stimulation, IFN-λ-induced gene expression continue to increase at 24 h (Fig. 3). Our data, therefore, demonstrate a distinct difference between the kinetics of types I and III IFN signaling.
Figure 3.

Compared to IFN-α, IFN-λ prolongs ISG expression in HaCaT cells. Cells were treated with either IFN-α (5 ng/mL) or IFN-λ (100 ng/mL) for the indicated times. Expression of the ISGs: ISG15, MX-1, TRAIL, SOCS1, and SOCS3 were determined by qRT-PCR. Results are presented as the relative fold induction and this is a representative experiment of 3 that were performed. Samples were normalized using the 18S ribosomal subunit.
Sustained IFN-λ-signaling is JAK/STAT dependent
Having shown that the kinetics of IFN-α and IFN-λ signaling was different in HaCaT cells, we next aimed to test whether components of the JAK/STAT pathway were essential in mediating the antiproliferative effects of IFN-λ. We proceeded to measure IFN-λ responsiveness in human fibrosarcoma 2fTGH cells. We used this cell line and its variants that have missing individual components of the JAK/STAT pathway as they have been used extensively in the study of type I IFN signaling. Treatment with IFN-α, but not IFN-λ, induced the activation of STAT1 or STAT2 (data not shown), thus indicating that these cells lack a functional IFN-λR1. To overcome this, 2fTGH cells were stably transfected with an HA-tagged IFN-λR1 expression vector. A number of individual clones were selected and designated thereafter as 2fTGH-IFN-λR1 for further studies. It is worth noting that 2fTGH cells expressing empty vector responded the same to IFN-α as parental cells. In addition, in response to IFN-α, 2fTGH-IFN-λR1 cells showed impaired STAT activation. Indeed, this phenomenon has been previously reported to occur with exogenous expression of cytokine receptors 14. As a result, for comparison analysis parental 2fTGH cells were used instead to measure IFN-α-induced signaling events.
As with HaCaT cells (Fig. 2A), an IFN-λ titration response curve was performed to select a dose of IFN-λ (5-10 ng/mL) that gave a level of STAT activation comparable to that induced with IFN-α (5 ng/mL) (data not shown). As expected, IFN-λ stimulation of 2fTGH-IFN-λR1 cells resulted in the prolonged tyrosine phosphorylation of STAT1 and STAT2 that could be detected at 24 h (Fig. 4A). In contrast, IFN-α-activated STAT tyrosine phosphorylation in 2fTGH cells decayed shortly after 6 h post-stimulation.
Figure 4.


The apoptotic effects of IFN-λ requires an intact JAK/STAT pathway (A) Parental 2fTGH cells and individual clones of 2fTGH IFN-λR1 cells were treated with either IFN-α (5 ng/mL) or IFN-λ (5 ng/mL), respectively, over a 24 h period. Detection of pY-STAT1, pY-STAT2, and β-actin expression levels were assessed by Western blot analysis. (B) parental 2fTGH and a panel of individual 2fTGH IFN-λR clones were treated for 72 h with either IFN-α or IFN-λ, respectively and proliferation was measured by MTS assay. (C). Apoptosis was measured in 2fTGH cells and 2fTGH IFN-λR1 clone 5 untreated or treated with IFN-α or IFN-λ for 24 or 48 h. In some experiments, 2fTGH IFN-λR1 cells were pretreated with 40 μM ZVAD-fmk prior to IFN-λ treatment. (D) 2fTGH cells and individual clones of U4A and U3A stably expressing IFN-λR1 were treated with IFN-α (5 ng/mL) or IFN-λ (500 ng/mL), respectively. Proliferation was measured by MTS assay. Data are plotted as the average ± SD of 3 independent experiments.
We also observed that IFN-λ was more effective than IFN-α in inhibiting cell proliferation. While IFN-λ stimulation led to growth inhibition levels ranging between 35%-80%, IFN-α showed a lesser antigrowth effect of 25% (Fig. 4B). Again, the antiproliferative response induced by IFN-λ and not IFN-α correlated with the induction of caspase-dependent apoptosis (Fig. 4C). Once we established that IFN-λ responsiveness in 2fTGH-λR1 cells was the same as in HaCaT cells, STAT1 deficient U3A and JAK1 deficient U4A were stably transfected with IFN-λR1. Our results showed that both cell lines had defective antiproliferative responses to IFN-α and IFN-λ (Fig. 4D). These data then confirmed that activation of the JAK/STAT signaling pathway remained essential for triggering the antiproliferative responses induced by type I and type III IFNs.
Sustained IFN-λ induced STAT2 activation requires de novo protein synthesis
One likely explanation for the prolonged duration of STAT1 and STAT2 activation induced by IFN-λ points to a defect in the rate of STAT tyrosine dephosphorylation. To test this, HaCaT cells were treated with IFN-α or IFN-λ for 30 min followed by a pulse chase with the kinase inhibitor staurosporine (STS) to block further increases in STAT tyrosine phosphorylation. STAT1 inactivation occurred at similar rates with either IFN-α or IFN-λ and was complete after 2 h of treatment with STS (Fig. 5A, compare lane 8 vs. lane 13). In contrast, STAT2 activation remained detectable; albeit weakly only in IFN-λ stimulated HaCaT cells at 4 h of treatment with STS (Fig. 5A, compare lane 9 vs. lane 14). These results demonstrated that sustained IFN-λ-induced ISGF3 signals could be attributed in part to a delay in STAT2 tyrosine dephosphorylation.
Figure 5.
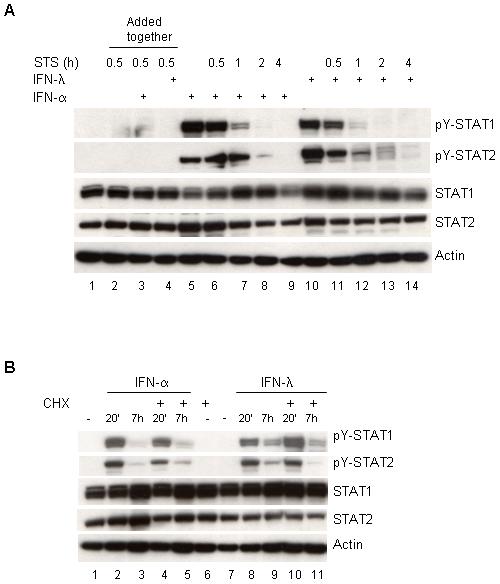
Sustained IFN-λ induced STAT activation requires de novo protein synthesis (A) HaCaT cells were stimulated with IFN-α (5 ng/mL) or IFN-λ (100 ng/mL) for 30 min followed by pulse chase with staurosporine (STS) for the indicated times. Cell extracts were prepared and immunoblot analysis was performed with antibodies against pY-STAT1, pY-STAT2, total STAT1, total STAT2. β-actin served as an internal control for equal loading (B) Same as in (A) except, cells were pretreated with cycloheximide (5μg/mL) followed with IFN-α or IFN-λ treatment.
We also chose to examine whether prolonged STAT activation induced by IFN-λ could be mediated by the actions of a newly synthesized protein(s). Therefore, HaCaT cells were pretreated with cycloheximide then followed by cytokine treatment of 20 min or 7 h. No obvious differences were noted in the activation of STAT1 with either cytokine. However, the level of tyrosine phosphorylated STAT2 was decreased only in IFN-λ but not in IFN-α stimulated cells (Figure 5B, second panel, compare lanes 3 and 5 vs. lanes 9 and 11). This finding pointed out that a specific IFN-λ induced protein is involved in the regulation of STAT2 activation and that IFN-α must activate a protein tyrosine phosphatase that IFN-λ does not.
DISCUSSION
Our study reports that growth inhibitory responses induced by type I and type III IFN are distinct and such effect correlated with differences in the duration of JAK/STAT pathway activation and prolonged ISG expression. Our findings also highlight that although type I and type III IFNs activate the same components of the JAK/STAT pathway and share many of the biological properties 1, 2, 6, 7, 15-19 they differ at least in the magnitude of the antiproliferative response.
IFNs induce the expression of many genes involved in the antiproliferative, antiviral, and apoptotic responses 1, 2. Unlike receptors for IFN-α/β, expression of IFN-λ receptors is restricted. The antiproliferative effects of IFN-λ have been demonstrated in various tumor cell lines that express endogenous or ectopic IFN-λ receptors 8, 16, 20. In our study, we found that IFN-λ was more efficient than IFN-α in inducing an antiproliferative effect that overlapped with the activation of apoptosis. The caspase-dependent apoptotic effects of IFN-λ was not restricted to HaCaT cells as this response was also detected in 2fTGH IFN-λR1 cells and in mouse melanoma B16 cells stably expressing IFN-λR1 (data not shown). Overall, the growth inhibitory effects of IFN-λ were more pronounced than that of IFN-α. We also observed that combination of IFN-α and IFN-λ had additive effects on the antiproliferative response. This indicated that there was no apparent intrinsic competition between the receptors for the JAKs or STATs, which may have been the case if there were differences in expression levels between the IFN-αR1 and the IFN-λR1. Furthermore, we confirmed that the expression of JAK1 and STAT1 was essential for the antigrowth effects of IFN-λ (Figure 5D).
In light of the different magnitude of the apoptotic and antiproliferative effects induced by IFN-α and IFN-λ, we examined the kinetics of STAT activation. Careful analysis of the STAT activation profiles showed that tyrosine phosphorylation of STAT1, STAT2 and STAT3 was sustained with IFN-λ as opposed to IFN-α. Therefore, we believe that the enhanced antiproliferative effects of IFN-λ cannot be simply explained by high surface IFN-λ receptor expression but rather differences in signal strength since higher doses of IFN-λ were required to achieve the same level of STAT activation with low doses of IFN-α. Of note, ectopic expression of the IFN-λR1 in 2fTGH cells did not account for the extended IFN-λ-mediated STAT activation, as IFN-λ treatment of HaCaT cells, which express endogenous and functional IFN-λR, yielded similar results. Moreover, the pattern of ISG expression induced by IFN-α and IFN-λ was also different (Figure 4). For instance, while IFN-α induced expression of Mx1 and ISG15 peaked at 6 h, expression of these same genes induced by IFN-λ continued to increase at 24 h. Recently, Marcello et al. 21 showed that in Huh7.5 cells compared to IFN-α stimulation, IFN-λ-induced activation of STAT1 and STAT2 peaked early and declined rapidly when examined within a 2 h time frame. This is in stark contrast to our results as we demonstrate that the pattern of activation of the JAK/STAT pathway induced by IFN-λ is sustained beyond 3 h post-stimulation. Perhaps this conflicting result could be best explained in that expression of HCV replicons in Huh7.5 tumor cells may alter responsiveness to each type of IFN. This same group performed microarray analysis of ISG expression demonstrating that IFN-λ-mediated gene activation was prolonged when compared to that observed with IFN-α. In our model, however, we postulate that persistent activation of STATs is what permits the continuous increase in ISG expression as observed at 24 h. The basis behind this sustained STAT activation is currently being studied in our laboratory.
Regulation of the JAK/STAT pathway occurs via a number of mechanisms. One of them involves the SOCS family of proteins that are upregulated by IFNs as they attenuate signaling by binding to tyrosine-phosphorylated JAKs and receptors, thereby facilitating their ubiquitin-dependent degradation 22. In our study, both IFN-α and IFN-λ induced the expression of SOCS1 at similar levels. We then explored a couple of possibilities that IFN-λ may extend STAT activation by affecting the rate of STAT tyrosine dephosphorylation and/or alternatively by inducing the synthesis of new proteins that maintain STATs activated. Our results showed that in response to both IFN-α and IFN-λ, and in the presence of STS, STAT2 and not STAT1 tyrosine dephosphorylation decayed slower when compared to IFN-α. We also observed that sustained STAT2 tyrosine phosphorylation required newly synthesized proteins specifically induced by IFN-λ and not by IFN-α. These findings help in part explain a distinct difference in the duration of expression of ISGF3 dependent genes induced by IFN-λ and highlight a role for STAT2 in prolonging IFN-λ signaling.
The fact that IFN-λ can prolong the duration of STAT activation and promote apoptosis is of clinical interest. Particularly since most patients receiving IFN-α must discontinue therapy due to severe side effects 10. This comes as no surprise since IFN-α/β receptors are virtually expressed on all cell types. In addition, IFN-α signals are short-lived and tumors have the capacity to become desensitized to this cytokine 23, 24. Notably, STAT1 and STAT2 are required for the growth inhibitory effects of type I IFNs 25, 26 and type III IFNs (our observations).
In conclusion, our findings strongly suggest that prolonged duration of IFN-λ induced STAT activation and gene expression may account for the enhanced antigrowth and apoptotic effects than were not achievable even with high dose IFN-α. Considering the adverse effects of IFN-α immunotherapy and that IFN-λR1 is not ubiquitously expressed as IFNAR1/IFNAR2, perhaps the combination of low dose IFN-α and high dose IFN-λ could be a more effective cancer therapy. Given that IFN-λ can induce a robust antiproliferative effect in tumor cells that respond less optimally to IFN-α, this highlights the potency of IFN-λ in suppressing the growth of tumors expressing both types of IFN receptors.
ACKNOWLEDGMENTS
We thank Dr. Howard Young for helpful discussions, Dr. Giorgio Trinchieri for critical reading of this manuscript and Dr. Veronica Hall for technical assistance with the quantitative RT-PCR system.
Footnotes
- IFN-λR
- IFN-lambda receptor
- IRF9
- interferon regulatory factor 9
- ISG
- interferon-stimulated gene
- STAT
- signal transducers and activators of transcription
- qRT-PCR
- quantitative reverse transcription-polymerase chain reaction
- STS
- staurosporine
- SOCS
- suppressors of cytokine signalling
- ZVAD-fmk
- N-benzyl-oxycarbonyl-valyl-alanyl-aspartic acid fluoromethyl ketone.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health under Contract No. N01-CO-12400.
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
REFERENCES
- 1.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 2.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 3.Pestka S. The interferon receptors. Semin Oncol. 1997;24:S9-18–S9-40. [PubMed] [Google Scholar]
- 4.Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 5.Ihle JN, Kerr IM. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995;11:69–74. doi: 10.1016/s0168-9525(00)89000-9. [DOI] [PubMed] [Google Scholar]
- 6.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 7.Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, Ostrander C, Dong D, Shin J, Presnell S, Fox B, Haldeman B, Cooper E, Taft D, Gilbert T, Grant FJ, Tackett M, Krivan W, McKnight G, Clegg C, Foster D, Klucher KM. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–8. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 8.Zitzmann K, Brand S, Baehs S, Goke B, Meinecke J, Spottl G, Meyer H, Auernhammer CJ. Novel interferon-lambdas induce antiproliferative effects in neuroendocrine tumor cells. Biochem Biophys Res Commun. 2006;344:1334–41. doi: 10.1016/j.bbrc.2006.04.043. [DOI] [PubMed] [Google Scholar]
- 9.Dumoutier L, Tounsi A, Michiels T, Sommereyns C, Kotenko SV, Renauld JC. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J Biol Chem. 2004;279:32269–74. doi: 10.1074/jbc.M404789200. [DOI] [PubMed] [Google Scholar]
- 10.Bleumer I, Oosterwijk E, De Mulder P, Mulders PF. Immunotherapy for renal cell carcinoma. Eur Urol. 2003;44:65–75. doi: 10.1016/s0302-2838(03)00191-x. [DOI] [PubMed] [Google Scholar]
- 11.Chi B, Dickensheets HL, Spann KM, Alston MA, Luongo C, Dumoutier L, Huang J, Renauld JC, Kotenko SV, Roederer M, Beeler JA, Donnelly RP, Collins PL, Rabin RL. Alpha and lambda interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J Virol. 2006;80:5032–40. doi: 10.1128/JVI.80.10.5032-5040.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sumita N, Bito T, Nakajima K, Nishigori C. Stat3 activation is required for cell proliferation and tumorigenesis but not for cell viability in cutaneous squamous cell carcinoma cell lines. Experimental Dermatology. 2006;15:291–9. doi: 10.1111/j.0906-6705.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 13.Pedranzini L, Leitch A, Bromberg J. Stat3 is required for the development of skin cancer. J Clin Invest. 2004;114:619–22. doi: 10.1172/JCI22800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dondi E, Pattyn E, Lutfalla G, Van Ostade X, Uze G, Pellegrini S, Tavernier J. Down-modulation of type 1 interferon responses by receptor cross-competition for a shared Jak kinase. J Biol Chem. 2001;276:47004–12. doi: 10.1074/jbc.M104316200. [DOI] [PubMed] [Google Scholar]
- 15.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006;80:4501–9. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, Diebold J, Diepolder H, Adler B, Auernhammer CJ, Goke B, Dambacher J. IL-28A and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am J Physiol Gastrointest Liver Physiol. 2005;289:G960–8. doi: 10.1152/ajpgi.00126.2005. [DOI] [PubMed] [Google Scholar]
- 17.Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, Cella M, Lande R, Uze G. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. 2004;34:796–805. doi: 10.1002/eji.200324610. [DOI] [PubMed] [Google Scholar]
- 18.Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits hepatitis B and C virus replication. J Virol. 2005;79:3851–4. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartlett NW, Buttigieg K, Kotenko SV, Smith GL. Murine interferon lambdas (type III interferons) exhibit potent antiviral activity in vivo in a poxvirus infection model. J Gen Virol. 2005;86:1589–96. doi: 10.1099/vir.0.80904-0. [DOI] [PubMed] [Google Scholar]
- 20.Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M. Biological activity of interleukins-28 and -29: Comparison with type I interferons. Cytokine. 2005 doi: 10.1016/j.cyto.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, Macdonald MR, Rice CM. Interferons [alpha] and [lambda] Inhibit Hepatitis C Virus Replication With Distinct Signal Transduction and Gene Regulation Kinetics. Gastroenterology. 2006;131:1887–98. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 22.Kile BT, Schulman BA, Alexander WS, Nicola NA, Martin HM, Hilton DJ. The SOCS box: a tale of destruction and degradation. Trends Biochem Sci. 2002;27:235–41. doi: 10.1016/s0968-0004(02)02085-6. [DOI] [PubMed] [Google Scholar]
- 23.Sakamoto S, Qin J, Navarro A, Gamero A, Potla R, Yi T, Zhu W, Baker DP, Feldman G, Larner AC. Cells previously desensitized to type 1 interferons display different mechanisms of activation of stat-dependent gene expression from naive cells. J Biol Chem. 2004;279:3245–53. doi: 10.1074/jbc.M309631200. [DOI] [PubMed] [Google Scholar]
- 24.Wormald S, Zhang JG, Krebs DL, Mielke LA, Silver J, Alexander WS, Speed TP, Nicola NA, Hilton DJ. The comparative roles of suppressor of cytokine signaling-1 and -3 in the inhibition and desensitization of cytokine signaling. J Biol Chem. 2006;281:11135–43. doi: 10.1074/jbc.M509595200. [DOI] [PubMed] [Google Scholar]
- 25.Battle TE, Lynch RA, Frank DA. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. 2006;66:3649–57. doi: 10.1158/0008-5472.CAN-05-3612. [DOI] [PubMed] [Google Scholar]
- 26.Qureshi SA, Leung S, Kerr IM, Stark GR, Darnell JE., Jr. Function of Stat2 protein in transcriptional activation by alpha interferon. Mol Cell Biol. 1996;16:288–93. doi: 10.1128/mcb.16.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]