

Abstract
Motivation
Annotation databases are widely used as public repositories of biological knowledge. However, most of these resources have been developed by independent groups which used different designs and different identifiers for the same biological entities. As we show in this paper, incoherent name spaces between various databases represent a serious impediment to using the existing annotations at their full potential. Navigating between various such name spaces by mapping IDs from one database to another is a very important issue which is not properly addressed at the moment.
Results
We have developed a web-based resource, Onto-Translate (OT), which effectively addresses this problem. OT is able to map onto each other different types of biological entities from the following annotation databases: Swiss-Prot, TrEMBL, NREF, PIR, Gene Ontology, KEGG, Entrez Gene, GenBank, GenPept, IMAGE, RefSeq, UniGene, OMIM, PDB, Eukaryotic Promoter Database, HUGO Gene Nomenclature Committee and NetAffx. Currently, OT is able to perform 462 types of mappings between 29 different types of IDs from 17 databases concerning 53 organisms. Among these, over 300 types of translations and 15 types of IDs are not currently supported by any other tool or resource. On average, OT is able to correctly map between 96% and 99% of the biological entities provided as input. In terms of speed, sets of approximatively 20,000 IDs can be translated in under 30 seconds, in most cases.
Availability
Onto-Translate is a part of Onto-Tools, which is freely available at http://vortex.cs.wayne.edu/Projects.html.
Contact
sorin@wayne.edu
1 INTRODUCTION
Gene annotations databases are widely used as public repositories of biological knowledge. Understanding the results of almost any molecular biology experiment involves consulting such annotation databases. Our current knowledge is spread out over a number of databases (DBs) such as: Entrez Gene [19], UniProt [3], Protein Data Bank [5], RefSeq [20], RGD, SGD, WormBase and Gene Ontology (GO) [2], to name just a few. Many such databases support multiple organisms but are specialized on a subset of specific biological entities. For instance, UniProt focuses on proteins, Entrez Gene focuses on genes, EPD focuses on eukaryotic promoters, etc. Other databases aim to provide a wider angle but focus on specific organisms. Examples could include RGD for rat, SGD for yeast, WormBase for C. Elegans, etc. Obtaining a complete understanding of an experiment, usually requires combining information from several such annotations databases. Unique key identifiers (IDs) in the internal structure of each such database represent biological entities such as genes, proteins, and mRNAs. Design and implementation restrictions specific to each database ensure that, within each database, the data are consistent, coherent and non-redundant. However, most of these annotation databases have been developed by independent groups which have used completely different designs and completely different sets of key identifiers for the same biological entities. Because of this, the ensemble of such annotation databases, which is the current repository of all our biological knowledge is inconsistent, incoherent and highly redundant.
At the same time, the old-fashion gene-centric approach of research in life sciences has been all but substituted by more high-throughput approaches involving entire sets of genes, sometimes entire genomes. In many current life science experiments, researchers obtain results identifying many genes that are interesting in a given condition. In order to fully interpret such results, researchers must combine annotations from several different databases which essentially requires mapping tens or hundreds of IDs across all databases involved. If performed manually, this mapping often leads to incomplete and incorrect results, and is time consuming and error prone even for short lists of genes. Even if performed automatically, querying various databases for the same data often yields different results. This represents a very important problem that has not been satisfactorily addressed yet.
2 NAME SPACE ISSUES IN ANNOTATION DATABASES
Identifiers used in different databases often represent different types of biological entities (e.g. genes, ESTs, mRNAs, proteins, etc.). Usually, there is a very clear and biologically meaningful mapping from one such entity to another. For instance, in the simplest case, a gene has a unique DNA sequence, which in turn can be mapped to an mRNA sequence, that is translated into a protein sequence, which perhaps has a known protein structure. However, the problem is further complicated by one-to-many mappings at various levels. For instance, several ESTs can represent the same gene, several alternatively spliced mRNAs can be constructed from the same gene DNA sequence, several structures corresponding to alternative folding patterns or different possible ligands can be associated with the same protein, etc. Specific annotations are available at each level (gene, mRNA, protein, structure, etc.). Given for instance a set of genes found to be differentially expressed in a specific condition of interest, one wishes to quickly find all known annotations about this set of genes, at all levels: the known GO categories associated with each of these genes, their proteins, the annotations associated with these proteins, etc. This information is currently spread out over many different databases, and each such database uses its own type of IDs. For instance, Table 1 shows 8 different IDs used to refer to the same XBP1 gene in 7 different databases, as well as 7 different probe IDs used on several Affymetrix arrays. Because the same biological entity is referred to by many different IDs, one needs to first map these IDs from one database to another and then query each database with its own specific IDs. This apparently trivial problem has become a challenge because various databases contain redundant information about the same biological entity. For instance, the GO categories known to be associated to a specific gene are stored in many databases such as UniProt [3]1, Entrez Gene [19], NetAffx[18] and GO itself [1, 2]. In spite of everybody's best efforts, because these databases are managed separately and they have different release and maintenance cycles, any data stored in more than one database creates very serious consistency and coherency problems.
Table 1.
Human gene XBP1 is represented by six additional distinct identifiers (IDs) in six different databases, as well as by one nucleotide sequence ID, one protein sequence ID and 7 different probe IDs on several different Affymetrix arrays.
| Database |
Identifier (ID) |
|---|---|
| UniGene | Hs.437638 |
| HGNC | 12801 |
| Entrez Gene | 7494 |
| Swiss-Prot | P17861 |
| ENSEMBL | ENSG00000100219 |
| PharmGKB | PA37400 |
| RefSeq | NM_005080, NP_005071 |
| NetAffx | RC_W90128_s_at (HU35ksubd), 200670_at (HGU 133), 71584_at (HGU95e), M31627_at (HG FL), 39756_g_at (HGU 95av2), 39755_at (HG U95a), g4827057_3p_s_at (U133 X3P) |
A brief example will hopefully illustrate the gravity of the issues involved. Let us consider for instance, the example of a microarray experiment involving Affymetrix's GeneChips. Let us assume that a specific probe ID, 39755_at, corresponding to the human gene XBP1, is found to be differentially expressed. The researcher may be interested in finding the corresponding UniGene [21] cluster ID for the selected probe ID, 39755_at. This can be achieved by querying NetAffx [18] with the given probe ID, 39755_at, which yields the Hs.437638 cluster ID. Alternatively, one can find the cluster ID by querying NCBI's UniGene database with the gene name, XBP1. In this example, there exist at least two paths which yield the required information and following both paths yields the same final result. However, let us now assume that one is interested in the GO annotations associated with this gene. Querying each of the resources above with the IDs representing the same gene, XBP1, yields very different results. UniProt queried with P17861 provides 2 unique GO terms: transcription factor activity and immune response; QuickGO queried with the same P17861 provides 8 unique GO terms: protein dimerization activity, sequence-specific DNA binding, immune response, DNA-dependent regulation of transcription, transcription, DNA binding, transcription factor activity and nucleus; NCBI's Entrez Gene entry XBP1 provides 7 unique GO terms: immune response, protein dimerization activity, sequence-specific DNA binding, DNA-dependent regulation of transcription, transcription, transcription factor activity and nucleus; PIR's iProClass [23] entry P17861 provides 5 unique GO terms: immune response, nucleus, DNA-dependent regulation of transcription, transcription factor activity, DNA binding, whereas GO (XBP1_HUMAN) provides only 2 unique GO terms: immune response and transcription factor activity. Essentially, querying 5 different resources can yield anything between 2 and 8 GO terms for the same gene. This situation is nothing short of disastrous. When one retrieves annotations for a set of genes from a particular source, one is always left to wonder whether the results obtained are really the entire picture or just a part of it, and whether one should continue to query other sources or just use the data retrieved so far.
Until the various resources currently available are organized into a real semantic web, free of coherency and consistency problems, arguably the best approach to retrieving annotations for a set of given biological entities is to query the authoritative source of such annotations for the given entity. In turn, in order to do this, one must map various types of IDs onto each other. This is also a tremendous challenge since various IDs can be mapped onto each other by traversing a number of alternative paths from one database to another. Since no unified map of the various databases exists, one is forced to rely on one's inherently limited personal understanding of the relationships between such databases in order to determine such a path on a case by case basis. Unfortunately, due to the lack of global consistency and coherency, the path used to travel from one resource to another often influences dramatically the results obtained.
Another important problem is related to the cross referencing between various annotation databases. Databases such as Entrez Gene and HGNC provide gene information and are supposed to cross-reference each other. For example, gene SMCR (Smith-Magenis syndrome chromosome region) has the identifier 11113 in HGNC. The same gene is identified by Entrez Gene as gene 6600. Entrez Gene cross-references HGNC i.e. the entry 6600 contains a field with the HGNC ID 11113. However, the reverse is not true. HGNCs entry for this gene does not contain the appropriate Entrez Gene ID. Here the data are mapped only one way, from Entrez Gene to HGNC number. If the user queries HGNC using its IDs, (s)he will not be able to link to NCBI and thus will not have access to all the annotations regarding this gene available in Entrez Gene.
This problem is more widespread than one would like to believe. For instance, both UniGene and Entrez Gene focus on non-redundant genes. However, only 69.53% of the genes in UniGene can be mapped on Entrez Gene entries. Furthermore, only 43.54% of the IDs in Entrez Gene can be mapped back to UniGene. An even more striking example is the mapping between GenBank dbEST and GenPept. GenPept is supposed to contain the protein translations of the sequences in GenBank dbEST, so going back and forth between these resources should be trivially simple. However, this is far from being the case. At the moment, 91.6% of the entries in dbEST can be mapped to GenPept entries but the reverse mapping is possible only for 1.82% of the entries. Clearly, translations and mappings that are theoretically both meaningful and useful, cannot always be performed just by querying the resources which are supposed to allow them. These examples strongly support the idea that ID mappings cannot be done casually, by ad-hoc, need-driven queries, or quick-and-dirty Perl scripts, as most researchers currently do. These quick solutions might satisfy an immediate need for a translation but offer no guarantees that the translation performed is the best possible mapping, nor that the results are correct or complete. At this time, the issues of incoherent name spaces between various databases represent a serious impediment to using the existing annotations at their full potential. Navigating between various such name spaces by mapping IDs from one database to another is a very important issue that must be addressed in a thorough and systematic way.
3 METHODS
In order to address the above problems, we undertook a thorough study of the following 17 annotations databases and their respective types of IDs: Swiss-Prot (IDs, accession numbers), TrEMBL (accession numbers, TrEMBL IDs), NREF (protein IDs), PIR (accession IDs), GO (GO IDs), KEGG (pathway IDs), Entrez Gene (Gene ID, gene symbol), GenBank (GI ID, accession and sequence numbers), GenPept (accession numbers), IMAGE (clone ID), RefSeq (protein, genome, mRNA accession number), UniGene (cluster ID), OMIM (OMIM number), PDB (PDB ID), Eukaryotic Promoter Database (accession number), HUGO Gene Nomenclature Committee (HGNC ID) and NetAffx (Affymetrix probe IDs). Based on the structure of these databases, we developed a relational database that allows meaningful mappings of various types of IDs onto each other. This meta-database was implemented in Oracle and all relevant data from the above databases was downloaded and used to populate the local database. Fig. 1 shows a simplified schema of the part of the Onto-Tools database that is used by Onto-Translate (the complete schema includes over 70 tables). As shown in the figure, Entrez Gene, RefSeq and iProclass databases are used as central hubs that link all other source databases.
Fig. 1.
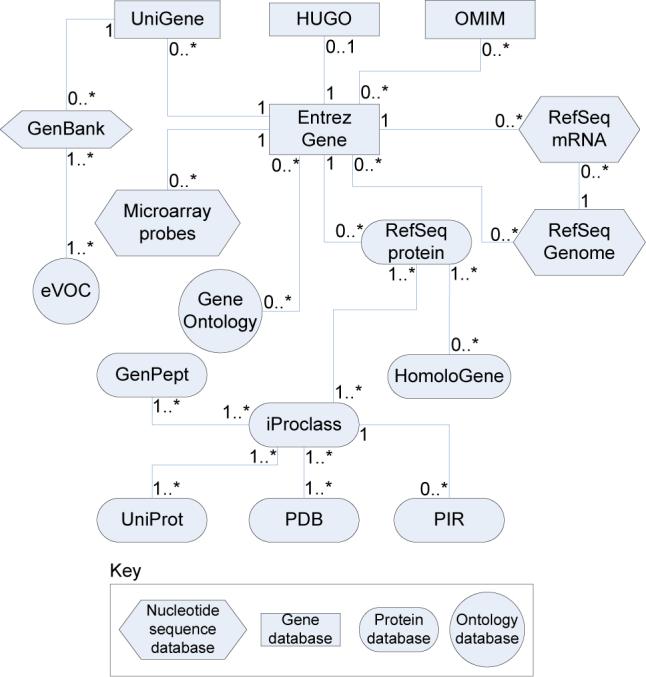
Onto-Translate relational database schema. This schema contains an entity for each of the source databases used by OT. The shapes represent the type of the given biological entity. A relationship between two databases is represented by a line connecting the two entities. The type of relationship between two entities is indicated by labels on the corresponding line. For instance, the relationship between Entrez Gene and Gene Ontology is many-to-many. In other words, a gene may be annotated using zero or more GO terms and a GO term may be used to annotate zero or more genes.
Using this database as a back-end resource, we developed a tool, Onto-Translate, that can perform arbitrary translations in an optimal manner. Given two types of IDs, a translation source ID and a translation destination ID, as well as a list of specific source IDs, the algorithm calculates an optimal route between the source type and the translation type and performs the translation. The optimality of the translation is not intended in the sense of finding the translation that involves the shortest path (i.e., the lowest number of intermediate translations) but rather by the trustworthiness of the data contained in various databases. A path is defined as trustworthy if for every ID type used in any of the necessary intermediate translations, the path passes through the tables corresponding to the database that is considered as the authoritative source for that particular type of ID. For instance, Entrez Gene is considered as the authoritative source for gene data, KEGG is considered the authoritative source for pathway data, PDB the authoritative source for protein structures, etc. Thus, even if the entries of many databases across the world contain protein structure IDs, for instance, a translation involving this type of ID must use data from PDB in order to be valid. Table 2 shows some examples of some ID types and their authoritative sources.
Table 2.
Authoritative database sources in Onto-Translate for different types of biological entities. A path is considered biologically valid if it either starts or ends in one of the authoritative sources. For instance, when converting from or to a gene ID, the path must start or end in Entrez Gene, respectively.
| Biological entity |
Authoritative source |
|---|---|
| Gene | Entrez Gene |
| Protein | UniProt |
| Protein structure | PDB |
| Nucleotide sequence | GenBank |
| Nucleotide sequence cluster | UniGene |
| Pathway | KEGG |
| Disease | OMIM |
The tables in the OT database and their relationships are represented as nodes and edges, respectively, in a graph structure. The basic relationships between IDs remain as given in the source databases. In a first step, the algorithm traverses the graph to find all possible paths between the source type and the destination type. This is done based on the semantic relationships between various source databases which are captured by the constraints of the Oracle database dictionary. After obtaining all possible paths between the source type and the destination type, OT removes the paths that are not trustworthy according to the criterion defined above. If the algorithm cannot find a trustworthy path between the source and the destination type, an error message is generated. If several trustworthy paths are found between the source and the destination ID, several criteria are used in order to rank them: i) a manually curated database will always be preferred to a database containing unverified data); ii) a database containing more entries will be preferred to a database with fewer entries and iii) everything else being the same, a shorter path (involving fewer intermediate translation) will be preferred to a longer one. These criteria are also centered around biological motivations. A manually curated database reflects our preference towards accuracy rather than coverage: fewer but accurate translations are deemed preferable to a larger number of translated IDs but potentially including some incorrect translations. The second criterion above is motivated by the fact that the a priori probability of finding a mapping for a given ID is directly proportional to the number of entries in a database. Thus, intermediate translation through a large database is more likely to successfully find mappings for all IDs required, compared to a smaller database that might contain the same types of IDs but fewer entries. Finally, the third ranking criterion is based on the assumption that the probability of losing some IDs in each intermediate translation is non-zero and constant. In these circumstances, a shorter translation path will minimize the number of IDs lost in translation and will be better than a longer one.
Once all trustworthy paths are ranked according to these criteria, the top path between the source and the destination type is chosen as the optimal one for the required translation. At this point, OT dynamically creates a database query that follows this translation path. Besides providing an output list with the translation of the input IDs into the desired type of IDs, the algorithm also identifies the specific IDs which could not be translated, as well as the exact source database which broke the intermediate chain of translations required for each such specific ID. This gives the user the ability to verify that indeed the translation of that specific ID failed because the source database lacks the necessary information rather than because of a bug or missing information in our database.
Since the name-space issues that motivated the creation of OT in the first place are caused by the existence of several databases that maintain arbitrary cross-links and contain redundant information, one might ask whether the addition of yet another database would not exacerbate the problem by adding yet another level of redundancy and many more cross-references (in essence, we created cross-references from our Onto-Translate database to each of the 17 databases above). This is not the case. There are two major aspects that differ between our resource and any other major resource currently available. Firstly, most other databases are focused on either some type of biological entity (e.g. Entrez Gene for genes, UniProt for proteins, etc.) or to some specific organism (e.g MGD for mouse, RGD for rat, etc). In contrast, our focus is on maintaining the ID mappings themselves rather than any specific annotations. The second aspect follows from this. If a database stores annotations, the maintenance and release cycle are dictated by the evolution of the annotation activities in that area. Since the Onto-Translate database does not store annotations as such, we only need to maintain the synchronization between IDs which can be done much more frequently and much more rapidly. In practice, this must be done every time any of the 17 mapped databases has a new release. In the future, this can be upgraded to an automatic overnight push of any new IDs from these databases to ours.
Onto-Translate currently supports biological categories such as genes, proteins, promoters, pathways, RNAs, OMIM, ESTs, and functional annotations. It can map between 29 different types of IDs which includes: Swiss-Prot protein ID, Swiss-Prot accession number, TrEMBL accession number, TrEMBL ID, non-redundant reference (NREF) protein ID from PIR, PIR accession ID from PIR, Gene Ontology (GO) ID, KEGG [13] pathway ID, GenBank GI ID, dbEST nucleotide accession number, Entrez Gene ID, Gene symbol, GenBank [4]'s dbEST [6] nucleotide accession number, GenPept protein accession number, RefSeq's protein, genome, mRNA accession number, UniGene cluster ID, clone IDs from UniGene, OMIM number, allelic variant from OMIM, Protein Data Bank (PDB) ID, Eukaryotic Promoter Database (EPD) accession number, EPD ID, HGNC ID, and probe IDs from commercial microarrays such as Affymetrix arrays, Agilent Technologies arrays, Amersham's CodeLink arrays, SuperArray, etc. The Onto-Translate tool is implemented in Java as a web application, fully integrated with the Onto-Tools [10, 11, 12, 14, 15, 16, 17].
4 RESULTS AND DISCUSSION
Clearly, the need for a reliable way of mapping IDs from one database to another has been felt in the past. In response to this needs, several approaches have been proposed to deal with this issue although none of them addressed the problem to its full extent. The best known resources currently able to perform a non-trivial mapping of various biological entities are: SOURCE [9] from Stanford University, MatchMiner [7] from NCI, RESOURCERER [22] from TIGR, and GeneMerge [8] from Harvard.
We compared Onto-Translate with each of these existing resources in terms of scope, accuracy of translation, speed and scaling capabilities. We define scope as the number of different mappings between types of IDs supported by a given resource. The comparison in Table 3 shows that OT has vastly larger capabilities compared to any of the existing resources. Figure 2 shows the specific translations that can be performed by each of the resources considered. Again, the difference in scope is striking.
Table 3.
A comparison of the scopes of Onto-Translate, SOURCE, MatchMiner, GeneMerge and RESOURCERER: types of input IDs supported and number of possible translation types.
| Software name |
Types of input IDs |
Number of translations |
|---|---|---|
| Onto-Translate | 22 | 462 |
| MatchMiner | 11 | 137 |
| SOURCE | 6 | 96 |
| GeneMerge | 12 | 30 |
| RESOURCERER | 1 | 16 |
Fig. 2.

A comparison of the scopes of Onto-Translate, RESOURCERER, MatchMiner, SOURCE, and GeneMerge. in terms of possible mappings between various types of IDs.
Of course, the scope is irrelevant if the accuracy of the mappings performed is inadequate. In order to compare the accuracy of the existing resources, we performed a number of translations using OT, SOURCE, and MatchMiner (top 3 in terms of scope), and compared the number of input IDs correctly mapped by each resource for each data set. OT consistently mapped more correct input IDs than both SOURCE and MatchMiner. The sets of genes to be translated were taken from popular human and mouse Affymetrix arrays. The set of genes contained on the HG-U133 Plus 2.0 array was used to test the translations from gene symbols to UniGene IDs, gene symbols to Entrez Gene IDs, and Entrez Gene IDs to gene symbols. Finally, for the translations involving mouse genes, we used the set of genes contained on the MG-430A 2.0 arrays. These genes were translated from gene symbols to UniGene IDs, gene symbols to Entrez Gene IDs and Entrez Gene IDs to gene symbols. Fig. 3 shows a comparison of the accuracy of these translations. OT was the most accurate resource in all cases, with accuracies between 96% and 99%. For human data, SOURCE is second best with an accuracy hovering around 93%. MatchMiner is weaker with an accuracy of around 70%. For mouse data, MatchMiner is better than SOURCE: 94−98% for MM, compared to 81−94% for SOURCE.
Fig. 3.

A comparison of the accuracy of Onto-Translate, MatchMiner and SOURCE. The input file included 19,248 gene symbols (19,562 Entrez Gene IDs) for human, and 12,991 gene symbols (13,023 Entrez Gene IDs) for mouse, from the respective Affymetrix arrays. The graph shows the percentages of the input genes successfully translated in each case.
Fig. 4 shows a comparison of the time (in seconds) necessary to perform a sample translation from gene symbols to gene IDs with Onto-Translate, MatchMiner and SOURCE. The time necessary to translate fewer than 1,000 genes is approximately the same for the 3 resources. However, when longer lists are involved, OT is approximately 2 times faster than SOURCE and approximatively 10 times faster than MatchMiner, in all translations performed.
Fig. 4.

Scaling properties of Onto-Translate (OT), MatchMiner (MM) and SOURCE. The graph shows the time (in sec) necessary to translate various sets containing between 10 and 19,119 distinct genes from Affymetrix 133 Plus 2.0. At fewer than 1,000 genes, the 3 resources have very comparable query times of under 10 seconds. When larger sets are involved, there is a substantial performance difference.
5 CONCLUSIONS
This paper discusses various issues related to name space inconsistencies between existing annotation databases. The distribution of our knowledge over several databases forces researchers to navigate from one such database to another, in order to construct the correct interpretation of any given experiment. Currently, the lack of the ability to map correctly various IDs from one DB to another creates very substantial problems in annotation retrieval. We have developed a resource that addresses this stringent need. This resource includes a back-end database as well as a web tool, Onto-Translate, that provides a convenient user interface. Currently, OT is able to perform 462 types of mappings between 29 different types of IDs from 17 databases concerning 53 organisms. This is better than the other resources we have investigated in terms of: i) number of translations possible, ii) types of IDs supported, iii) accuracy and iv) speed. Onto-Translate is a part of Onto-Tools, which is freely available at http://vortex.cs.wayne.edu/Projects.html.
ACKNOWLEDGMENTS
This work has been supported by the following grants: NSF DBI-0234806, NIH 1R01HG003491, NSF CCF-0438970, MLSC MEDC-538, NIH 1R21 CA10074001, 1R21 EB00990-01 and 1R01 NS045207-01. Onto-Tools currently runs on equipment provided by Sun Micro-systems under the grant EDU 7824-02344-US.
Footnotes
Swiss-Prot, TrEMBL and PIR have been recently merged as a single database in UniProt.
REFERENCES
- 1.Ashburner M, et al. Gene ontology: Tool for the unification of biology. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashburner M, et al. Creating the gene ontology resource: Design and implementation. Genome Research. 2001;11:1425–1433. doi: 10.1101/gr.180801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann SFB, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh L-SL. The universal protein resource (uniprot). Nucleic Acids Research. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler D. Genbank. Nucleic Acids Research. 2005;33:D34–D38. doi: 10.1093/nar/gki063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman HM, Westbrook J, Feng Z, Gilliland1 G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Research. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boguski MS, Lowe T. m. J., Tolstoshev CM. dbest - database for expressed sequence tags. Nature Genetics. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- 7.Bussey KJ, Kane D, Sunshine M, Narasimhan S, Nishizuka S, Reinhold WC, Zeeberg B, Ajay, Weinstein JN. Matchminer: a tool for batch navigation among gene and gene product identifiers. Genome Biology. 2003;4(4):R27. doi: 10.1186/gb-2003-4-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castillo-Davis CI, Hartl DL. Genemerge - post-genomic analysis, data mining, and hypothesis testing. Bioinformatics. 2002;19(7):891–892. doi: 10.1093/bioinformatics/btg114. [DOI] [PubMed] [Google Scholar]
- 9.Diehn M, Sherlock G, Binkley G, Jin H, Matese JC, Hernandez-Boussard T, Rees CA, Cherry JM, Botstein D, Brown PO, Alizadeh AA. Source: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Research. 2003;31(1):219–223. doi: 10.1093/nar/gkg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drăghici S, Khatri P, Bhavsar P, Shah A, Krawetz SA, Tainsky MA. Onto-tools, the toolkit of the modern biologist: Onto-express, onto-compare, onto-design and onto-translate. Nucleic Acids Research. 2003;31(13):3775–81. doi: 10.1093/nar/gkg624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drăghici S, Khatri P, Martins RP, Ostermeier GC, Krawetz SA. Global functional profiling of gene expression. Genomics. 2003;81(2):98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- 12.Drăghici S, Khatri P, Shah A, Tainsky M. BioTechniques. Microarrays and Cancer: Research and Applications; 2003. Assessing the functional bias of commercial microarrays using the onto-compare database. pp. 55–61. [PubMed] [Google Scholar]
- 13.Kanehisa M, Goto S, Kawashima S, Nakaya A. The kegg databases at genomenet. Nucleic Acids Research. 2002;30(1):42–46. doi: 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khatri P, Bhavsar P, Bawa G, Drăghici S. Onto-tools: an ensemble of web-accessible, ontology-based tools for the functional design and interpretation of high-throughput gene expression experiments. Nucleic Acids Research. 2004;32:W449–56. doi: 10.1093/nar/gkh409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21(18):3587–95. doi: 10.1093/bioinformatics/bti565. 1367−4803 (Print) Journal Article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khatri P, Drăghici S, Ostermeier GC, Krawetz SA. Profiling gene expression using onto-express. Genomics. 2002;79(2):266–270. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- 17.Khatri P, Sellamuthu S, Malhotra P, Amin K, Done A, Drăghici S. Recent additions and improvements to the onto-tools. Nucleic Acids Research. 2005;33(Web server issue) doi: 10.1093/nar/gki472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu G, Loraine AE, Shigeta R, Cline M, Cheng J, Valmeekam V, Sun S, Kulp D, Siani-Rose MA. Netaffx: Affymetrix probesets and annotations. Nucleic Acids Research. 2003;31(1):82–86. doi: 10.1093/nar/gkg121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez gene: gene-oriented information at ncbi. Nucleic Acids Research. 2005 doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pruitt KD, Maglott DR. Refseq and locuslink: Ncbi gene-centered resources. Nucleic Acids Research. 2001;30(1):137–140. doi: 10.1093/nar/29.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuler GD. Pieces of puzzle: Expressed sequence tags and the catalog of human genes. Journal of Molecular Medicine. 1997;75(10):694–698. doi: 10.1007/s001090050155. [DOI] [PubMed] [Google Scholar]
- 22.Tsai J, Sultana R, Lee Y, Pertea G, Karamycheva S, Antonescu V, Cho J, Parvizi B, Cheung F, Quackenbush J. Resourcerer: a database for annotating and linking microarray resources within and across species. Genome Biology. 2001;2(11):software0002.1–0002.4. doi: 10.1186/gb-2001-2-11-software0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu CH, Yeh L-SL, Huang H, Arminski L, Castro-Alvear J, Chen Y, Hu Z, Kourtesis P, Ledley RS, Suzek BE, Vinayaka C, Zhang J, Barker, C W. The protein information resource. Nucleic Acids Research. 2003;31(1)::345–347. doi: 10.1093/nar/gkg040. [DOI] [PMC free article] [PubMed] [Google Scholar]