

Abstract
The proinflammatory cytokine interleukin-1β (IL-1β) is induced rapidly after traumatic brain injury (TBI) and contributes to the inflammatory events that lead to neuronal loss. Although an important source of IL-1β is from the injured brain itself, in patients with multiple organ trauma (polytrauma) IL-1β is also released into the bloodstream which may potentially influence brain vulnerability. The purpose of this study was to determine the effects of systemic inflammation induced by peripheral administration of IL-1β on histopathological and behavioral outcome after moderate fluid percussion (FP) brain injury in rats. At 30 min or 24 hr after TBI, saline, 20μg/kg or 40μg/kg of IL-1β was injected (n=4-9/group) intraperitoneally (IP). Sham operated animals (n=9) received either saline or IL-1β (20 or 40 μg/kg) injections. The somatosensory tactile placing test was administered at 1, 2 and 3 days posttrauma. IL-1β treated animals showed significant placing deficits compared to vehicle-treated TBI animals. Three days after injection, contusion areas and volumes were significantly increased (p<0.05) with both IL-1β doses and at both treatment times compared to vehicle treated animals. IL-1β treated rats showed more contusion injury and hippocampal neuronal damage as well as enhanced perivascular neutrophil accumulation. Cortical IL-1r1 mRNA increased as early as 1 hr following TBI, peaking at 24 hr and remained elevated 3 days posttrauma. These data show that the posttraumatic administration of IL-1β significantly aggravates behavioral outcome and increases overall contusion volume after TBI. Increased systemic inflammatory processes, including extravasation of activated leukocytes and proinflammatory cytokines could participate in this detrimental outcome. Because peripherally circulating cytokines and other neurotoxic factors may be increased following multi-organ trauma, these findings may be important in targeting therapeutic interventions in this patient population.
Keywords: brain trauma, cytokine, leukocytes, IL-1β, inflammation, polytrauma
Introduction
Secondary injury mechanisms including posttraumatic inflammation are believed to participate in the pathophysiology of traumatic brain injury (TBI) (Stahel et al., 2000). Published data demonstrate that TBI initiates a complex pattern of acute and more chronic inflammatory events that may either aggravate outcome or promote reparative processes (Lucas et al., 2006, Werner and Engelhard, 2007). Treatment strategies targeting the more acute inflammatory events have demonstrated that a reduction in leukocyte infiltration into the post-injured brain improves both histopathological and functional outcome measures (Wong et al., 2007). Thus, secondary injury mechanisms including free radical production, lipid peroxidation and altered blood-brain barrier (BBB) function can be attenuated by treatments that reduce or inhibit acute post-injury inflammatory processes (Feuerstein, et al., 1997, Stahel et al., 2000).
Various endogenous inflammatory signaling pathways are activated in the brain after TBI. TBI increases levels of pro-inflammatory cytokines and chemokines and activates a variety of endogenous inflammatory cells including microglia and astrocytes (Smith, 1994, Shahbazian et al., 1999, Stamatovic et al., 2006, Whalen et al., 2000). These parenchymal cells contribute to an increase in endogenous proinflammatory modulators that can promote cellular responses to trauma and contribute to neuronal death. In particular, the proinflammatory cytokine interleukin-1β (IL-1β) is induced rapidly after TBI (Fassbender et al., 2000, Kinoshita et al., 2002, Taupin et al., 1993), and released by both activated astrocytes and microglia. Previously, strategies that target IL-1β, including the administration of anti-cytokine agents including the naturally-occurring interleukin-1 receptor antagonist (IL-1RA) have been reported to reduce damage caused by TBI (Sanderson et al., 1999, Taupin et al., 1993, Tehranian et al., 2002, Toulmond and Rothwell, 1995).
Posttraumatic inflammatory processes may also lead to the upregulation of endothelial and leukocyte adhesion molecules and promote leukocyte recruitment into the injured brain parenchyma (Ando et al., 2007, Gavins et al., 2007, Wong, et al., 2007). Vascular and perivascular events including the activation of matrix metalloproteinases lead to altered vascular permeability and the recruitment and extravasation of blood-born cells and circulating molecules (Truettner et al., 2005). IL-1β is also involved in regulating the expression and activation of MMPs at sites of inflammation (Schonbeck et al., 1998). Invading leukocytes and circulating factors may influence neuronal vulnerability and functional outcome of the posttraumatic brain (Schmidt et al., 2005; Whalen et al., 2000). Thus, in addition to primary parenchymal changes in cytokine levels and expression, TBI can also produce systemic alterations that may influence brain vulnerability and outcome. For example, experimental and clinical studies have measured circulating cytokines and chemokines in blood and cerebrospinal fluid (CSF) samples after trauma (Morganti-Kossman et al., 1997). In a clinical study of pediatric brain trauma, a positive correlation between plasma and CSF levels of IL1-β and injury severity as measured by the Glasgow Coma Scale and Outcome Score was reported (Chiaretti et al., 2005).
In some trauma patients, multi-organ damage can lead to elevated circulating levels of inflammatory cells and mediators. Total plasma levels of IL-1β from peripheral blood mononuclear cells in patients who suffered TBI plus polytrauma were found to be elevated by 650%, 48-60 hours after injury (Shahbazian et al., 1999). Importantly, if these cells and modulators gain access to the damaged brain through a leaky BBB, they may significantly contribute to the pathogenesis of TBI. Brain trauma leads to acute BBB permeability that may persist for days after injury (Dietrich et al., 1994, Habgood et al., 2007, Cortez et al., 1989).
The importance of systemic inflammation and circulating cytokines in the blood after TBI has received limited experimental investigation in terms of its potential effect on traumatic outcome. In one study by Whalen and colleagues (2000), administering granulocyte colony stimulating factor prior to controlled cortical impact injury (CCI) in rats increased absolute neutrophil counts along with post-TBI blood brain barrier permeability. The present study was therefore conducted to determine the consequences of artificially increasing systemic levels of the proinflammatory cytokine IL-1β on histopathological and behavioral outcome after FP injury to mimic one aspect of polytrauma events in a controlled setting. In addition, the upregulation of mRNA for the IL-1 receptor 1 was determined to insure that there was an injury induced upregulation of this receptor in our model of TBI. IL-1β was chosen to investigate because previous clinical and experimental studies have documented increased levels of this proinflammatory cytokine after experimental and clinical brain trauma.
Materials and methods
Animal Groups
Male Sprague-Dawley rats weighing 280-340 gm obtained from Charles River Breeders were used for all experiments. Animal care was in accordance with the guidelines set by the University of Miami Animal Care and Use Committee. All animals were kept at a constant temperature (23-25°C) for at least seven days before the study and exposed to a 12 hour light-dark cycle. Rats were allowed free access to water, but food was withheld overnight before brain injury.
Eight different animal groups were utilized for the peripheral IL-1β administration portion of this study. Sham (n=8-9) operated control animals received either saline or IL-1β and underwent all surgical procedures but did not receive the FP injury. TBI animals received saline infusions at 30 min after TBI (n=9) or either 20 μg/kg or 40 μg/kg of IL-1β (R&D Systems) injected interperitoneally at 30 min (n=8-9/group) or 24 hr (n=6/group) after FP injury. For histopathological evaluation, all animals were perfusion-fixed 3 days after injury for quantitative histopathological analysis. Additional TBI groups of animals (n=4-8 per group) were produced to document the temporal profile of IL-1 receptor 1 (IL-1r1) expression in the cerebral cortex after TBI. For this study, IL-1r1 expression was determined by quantitative RT-PCR at 1 hr, 3 hr, 24 hr, and 3 days after TBI.
Surgical preparation and FP injury
The basic surgical preparation for the FP brain injury was performed according to methods previously described (Dietrich et al., 1994, Suzuki et al., 2004). Rats were initially anesthetized with 3.0% halothane and a mixture of 70% nitrous oxide and 30% oxygen. The animals were placed in a stereotaxic frame and a 4.8 mm craniotomy was made overlying the right parietal cortex (3.8 mm posterior to bregma and 2.5 mm lateral to the midline) (Zilles, 1985). A plastic injury tube was then placed over the exposed dura, was bound by adhesive as well as general acrylic and hardened, and the scalp was closed. Animals were returned to their home cage and fasted overnight. The next day, the animals were initially anesthetized with 3.0% halothane and a mixture of 70/30% nitrous oxide/oxygen. The femoral artery and vein were cannulated with PE-50 tubing. Rats were then immobilized with pancuronium bromide (0.5 mg/kg IV). An endotracheal tube was inserted orally, and rats were mechanically ventilated and maintained on a mixture of 70% nitrous oxide, 0.12 – 1.5% halothane, with a balance of oxygen. In TBI animals, a FP device was used to induce moderate parasaggital FP injury (1.8 – 2.2 atmospheres) (Dixon et al., 1987). Physiological variables were maintained in normal ranges and core and temporalis temperature was controlled at 37°C with a feedback heat lamp system.
Quantitative RT-PCR
To determine if an upregulation of IL-1 receptor 1 (IL1r1) was present after TBI, changes in mRNA levels of expression of this receptor were quantified by Real Time RT-PCR in a separate group of animals. This will allow a determination of whether gene expression for the IL-1 receptor which is necessary for signaling in this inflammatory cascade is increased. Animals (n= 4-8 per group) underwent FP injury and were sacrificed after 1 hour, 3 hours, 24 hours, and 3 days. Sham animals underwent all surgical procedures except the FP injury. The injured cerebral cortex was removed and total RNA was extracted using the RNA NOW™ kit from Biogentex. cDNA was reverse transcribed from 1 μg total RNA per sample using random primers and MultiScribe™ Reverse Transcriptase (Applied Biosystems). Real-time PCR was performed on these cDNA samples using TaqMan® Gene Expression Assays from Applied Biosystems and the 7300 Real-Time PCR System. Primer pairs and MBG probes for IL1r1 and GAPDH were purchased from Applied Biosystems. GAPDH was used as an endogenous control. Relative quantification was analyzed by the ΔΔCt method yielding results as “fold increase or decrease versus sham”.
Sensorimotor testing
The ability of animals to place their forelimbs reflexively using tactile cues was assessed using procedures similar to those previously described (De Ryck et al., 1989). Tactile placing was conducted by gently placing the dorsal surface of the animal's paw (forelimb) against the table edge. This procedure was repeated on the lateral surface of each paw. Normal animals reflexively placed their paws on the table after contact with the table edge. During tests of tactile placing, the animal's head was held upward to prevent the use of visual cues. Animals were pretested for tactile placing to determine normal placing in the animal prior to injury. Animals were tested on days 1, 2 and 3 post-injury. Each animal's forepaw was tested twice (dorsal and lateral) and scored as follows: a score of 0 was given if the animal exhibited complete immediate placing, a score of 1 was given if there was incomplete and/or delayed placing, and a score of 2 was given if there was an absence of placing. An animal's total score for tactile placing was computed by adding up both test scores for both forepaws (score for maximum impairment = 8). Although the number of vehicle treated TBI animals with ipsilateral forelimb deficits was minimal on testing day 1(3 out of 10 animals) with no deficits in this forelimb on days 2 and 3, a total score is presented for both limbs as the IL-1β administration slightly increased ipsilateral forelimb deficits. However, the majority of forelimb deficits with this injury paradigm are on the contralateral forelimb as expected. Mean and SEM of each treatment group were calculated (n=6-9 per group).
Histopathology
Three days after TBI, animals were deeply anesthetized and transcardially perfused with isotonic saline for 2 min (75 ml) followed by 30 min (350 ml) of 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Following perfusion, brains were removed and immersed in paraformaldehyde at 4° C for 3 days. Brains were then blocked and embedded in paraffin. Tissue sections 10 μm thick were taken at 150 μm intervals.
Sections were stained with hematoxylin and eosin (H&E) for stereological quantification of cortical contusions by an investigator blinded to the experimental groups. Coronal sections at specific bregma levels (-0.8, -1.8, -3.3, -4.3, -5.8, -6.8, -7.3 mm posterior to bregma) were used for contusion area and volume measurements (Zilles, 1985). Contusion volumes were determined by tracing the area of the cortical contusion boundaries that were well demarcated in H&E-stained sections using the Neurolucida 7.50.1 software program (MicroBrightField Inc., Williston, VT, USA). The contused areas consisted of pyknotic neurons, edema, reactive astrocytes and areas of tissue shearing at the gray/white matter interface of the parietal cerebral cortex and hippocampus. Contusion volumes were then computed from the contusion area measurements using numerical integration of successive areas (Suzuki et al., 2004).
Statistical analysis
Histopathological, behavioral and RT-PCR data were expressed as mean + SEM. Differences in contusion area and volume measurements were assessed by using two-way repeated measure analysis of variance and one-way analysis of variance (ANOVA, respectively). Physiological data were compared using two-way repeated measures ANOVA. The tactile placing task was assessed by two-way repeated measures ANOVA. RT-PCR data were analyzed using one-way ANOVA. Holm-Sidak was used for post-hoc analysis where appropriate. Differences were considered significant if p< 0.05.
Results
Physiological Data
Physiological measurements of pCO2, pO2, mean arterial blood pressure (MAP), pH, and brain and rectal temperature are given in Table 1. Physiological variables were taken 15 min prior to TBI and 15 min after. All physiological variables were within normal ranges. There was no significant difference between the IL-1β-treated and saline treated animals after TBI. Although IL-1β has been implicated in the febrile response that can follow infection, seizure, and neurotrauma (Alheim and Bartfai, 1998, Conti,et al., 2004, Dinarello, 1984), in this study, normothermia was maintained and the administration of IL-1β did not induce a fever response during the limited observation period.
Table 1. Physiological Variables.
| 15′ pre | saline | 30min(20ug/kg) | 30 min(40 ug/kg) | 24 hr(20ug/kg) | 24hr(40ug/kg) |
|---|---|---|---|---|---|
| weight | 324.4±11.1 | 327.4±9.7 | 325.1±3.7 | 324±10.4 | 320±6.4 |
| Temp body | 36.8±0.0 | 36.7±0.0 | 36.8±0.0 | 37.1±0.0 | 37.1±0.0 |
| Temp brain | 36.7±0.0 | 36.7±0.0 | 36.7±0.0 | 36.8±0.0 | 36.7±0.0 |
| pH | 7.5±0.0 | 7.5±0.0 | 7.5±0.0 | 7.5±0.0 | 7.5±0.0 |
| pCO2 | 40.3±0.8 | 39.6±0.8 | 39.1±0.6 | 40.7±0.8 | 39.5±1.1 |
| pO2 | 150.8±5.8 | 160.4±11.8 | 154.7±8.3 | 133.2±8.1 | 154.7±7.3 |
| MAP | 129.4±2.8 | 125.2±2.2 | 129.3±2.9 | 122.4±3.3 | 122.4±2.5 |
| 15′ post | saline | 30min(20ug/kg) | 30 min(40 ug/kg) | 24 hr(20ug/kg) | 24hr(40ug/kg) |
|
| |||||
| weight (24hr) | 319.1±10.4 | 320.8±9.8 | 310.1±5.0 | 317±8.9 | 313±4.1 |
| Temp body (1hr) | 36.8±0.0 | 36.7±0.0 | 36.9±0.1 | 36.9±0.1 | 37.0±0.1 |
| Temp brain (1hr) | 36.7±0.0 | 36.7±0.0 | 36.7±0.0 | 36.7±0.1 | 36.9±0.1 |
| pH | 7.5±0.0 | 7.5±0.0 | 7.5±0.0 | 7.5±0.0 | 7.5±0.0 |
| pCO2 | 39.0±0.6 | 39.1±0.7 | 38.8±0.8 | 40.0±1.1 | 40.0±0.8 |
| pO2 | 164.8±7.5 | 140.4±6.6 | 141.4±4.1 | 121.1±4.2 | 140.7±2.8 |
| MAP | 123.9±3.0 | 123.2±2.3 | 126.1±2.3 | 118.6±3.3 | 123.9±3.2 |
IL-1β receptor changes
The level of expression of the IL1r1 increased rapidly following TBI in the ipsilateral cerebral cortex (Figure 1). The values are representative of fold increase versus the sham control animals. One-way ANOVA was significant for group (p<0.001). Posthoc multiple comparisons versus the Sham group (Holm-Sidak methods) were also significant at multiple time periods. Within 1 hr, mRNA levels almost doubled over sham values (1.9 fold). mRNA levels continued to increase at 3 hr posttrauma to 3.7 fold (p<0.05). By 24 hr, the expression peaked at 5 fold higher than sham (p<0.05). IL-1r1 expression remained elevated over sham at 3 days after TBI, but had begun to decline (2.8 fold over sham, p<0.05).
Figure 1.
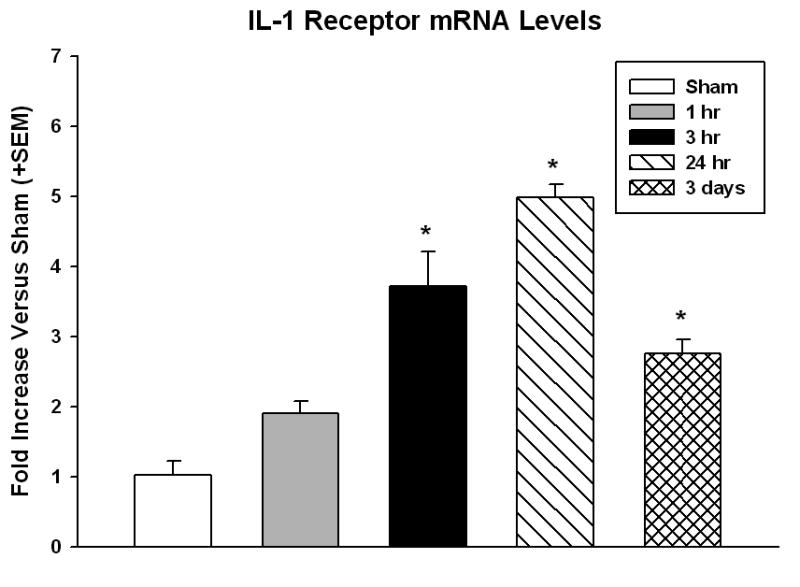
Real-Time RT-PCR of IL-1r1 (Mean +SEM). The timecourse of the major IL-1 receptor mRNA levels was determined by quantitative PCR. Data represent fold increase over sham. By 1 hr after TBI, IL-1r1 mRNA levels doubled over sham and levels continued to increase until 24 hr (5-fold increase). mRNA levels decreased by 3 days, but were still higher than sham. *p<0.05 versus Sham.
Behavior
To assess the effects of peripheral administration of IL-1β on sensorimotor behavior, forelimb placing reactions to tactile stimuli were measured. Two-way repeated measures ANOVA was significant for group (p<0.01), testing days (p<0.001) and group x testing days (p<0.001). Holm-Sidak posthoc analysis demonstrated significant differences between groups on days 1, 2, and 3. Specifically, IL-1β administered 30 min after injury resulted in a significantly impaired (p<0.05, 20 μg/kg, and p<0.001, 40 μg/kg) forelimb placing deficit on Day 1 compared to IL-1β (40 μg/kg) administered at 24 hrs (Figure 2). There was a trend for the IL-1β administered 30 minutes groups to perform worse on this task compared to TBI vehicle treated group. IL-1β (40 μg/kg) treated at 30 minutes was also significantly different (p<0.05) from TBI-Vehicle animals with less successful placements at 2 and 3 days after TBI. In contrast, when IL1-β was given 24 hr after injury, the effect on sensorimotor behavior between IL-1β treated and saline treated rats was less evident. Only the 40μg/kg dose had a significant detrimental effect (p<0.05) at 3 days after TBI.
Figure 2.
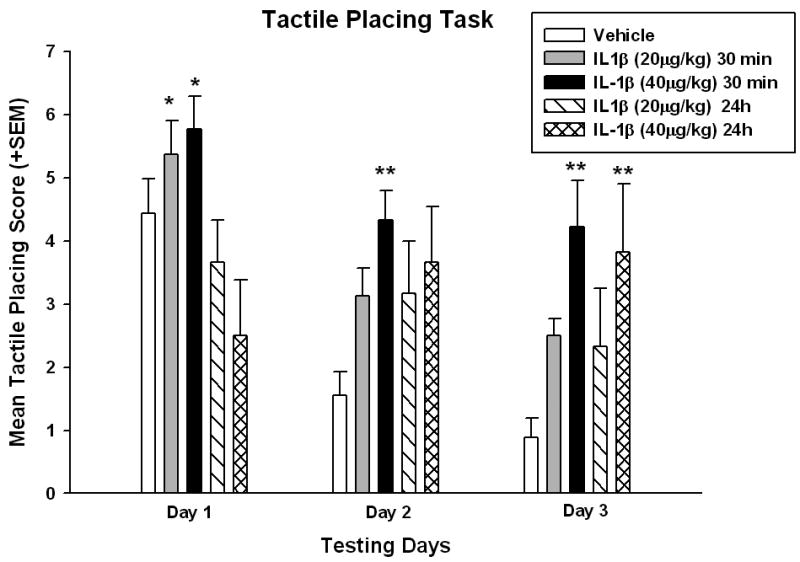
Tactile placing deficits are exacerbated by IL-1β treatment. Mean tactile placing scores +SEM represent both ipsilateral and contralateral forelimb scores. Forelimb tactile placing was measured for both dorsal and lateral stimulation. IL-1β (20 μg/kg) administration at 30 min after TBI increased deficits on day 1 as compared to IL-1β (40 μg/kg) treatment at 24 hrs (p<0.05). IL-1β (40 μg/kg) administered at 30 min after TBI significantly impaired forelimb placing on day 1 compared to the 24 hr administration group along with day 2 and 3 compared to TBI-vehicle. Animals administered the IL-1β 40 μg/kg at 24 hrs were also significantly different from vehicle treated animals (p<0.05). These data demonstrate that IL-1β administration exacerbates tactile placing deficits after TBI. *p<0.05 versus IL-1β (40 μg/kg) at 24 hrs; **p<0.05 versus TBI-Vehicle.
Histopathology
In the sham surgery animals that received 40 μg/kg IL-1β (Fig 3A), no obvious pathology was observed. In contrast, cortical contusion measurements showed an increase in contusion areas and volumes in the IL-1β treated animals as compared to saline treatment (Fig 3). Animals that underwent TBI but were treated with vehicle (saline) had small contusions visible along the gray/white matter interface (outlined) with visible hemorrhage (Fig 3B). When TBI animals received 20 μg/kg IL-1β at 30 min after injury, the size of the cortical contusion was markedly larger than saline alone (Fig 3C) and with administration of 40μg/kg IL-1β, this was even more pronounced (Fig 3D).
Figure 3.
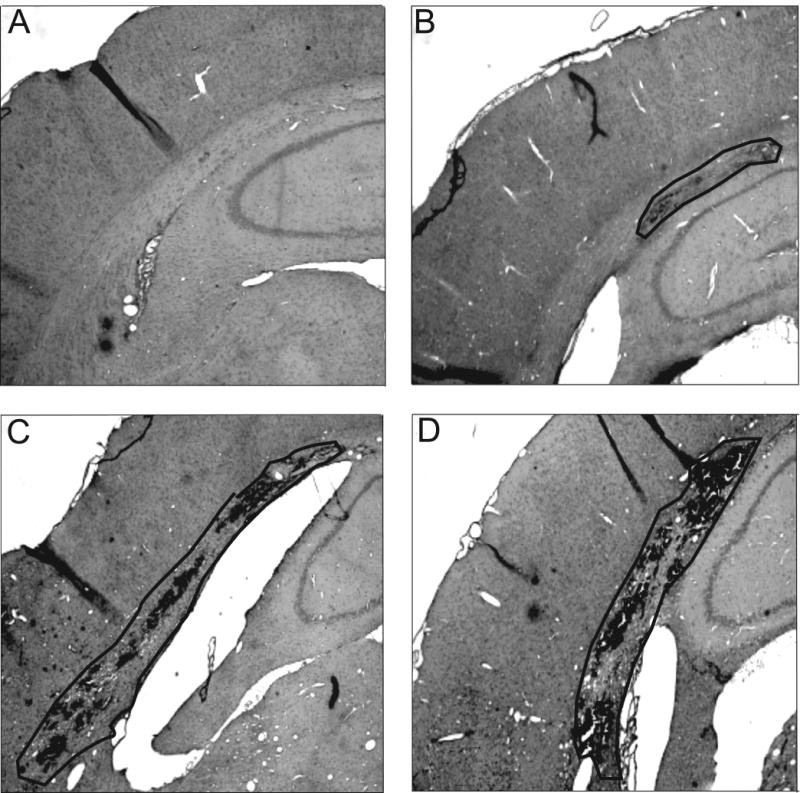
Cortical contusions are worsened by systemic IL-1β. Representative H&E stained coronal sections of sham surgery animals treated with 40 μg/kg IL-1β (A). The administration of this compound did not result in brain damage in the uninjured animal. TBI animals treated with saline (B), TBI animals treated with 20 μg/kg IL-1β (C), and TBI treated with 40 μg/kg IL-1β (D) administered 30 min after TBI. The contusion area is outline showing the extent of damaged tissue. Magnification - 4X.
To assess the extent of contusion size at various bregma levels, contusion areas at 7 different bregma levels were measured. Two-way repeated measures ANOVA was significant for group (p<0.01), bregma level (p<0.001) and group x bregma level (p<0.009). IL-1β (20μg/kg) given 30 min after injury resulted in a significant increase in contusion area from bregma levels -4.3mm to -5.8mm relative to vehicle treated animals (Fig 4A) (p<0.05). At the higher dosage (40 μg/kg), the affected area was more extended with significant increases over TBI-vehicle animals extending from bregma -3.3 to -6.8 (p< 0.05). When given 24 hours after injury, IL-1β increased the contusion areas as well. Both the 20μg/kg and 40 μg/kg doses increased the contusion areas from bregma levels -3.3mm to -4.3mm (p<0.05) and -4.3mm to -5.8mm (p<0.05), respectively, as compared to saline treated TBI animals.
Figure 4.
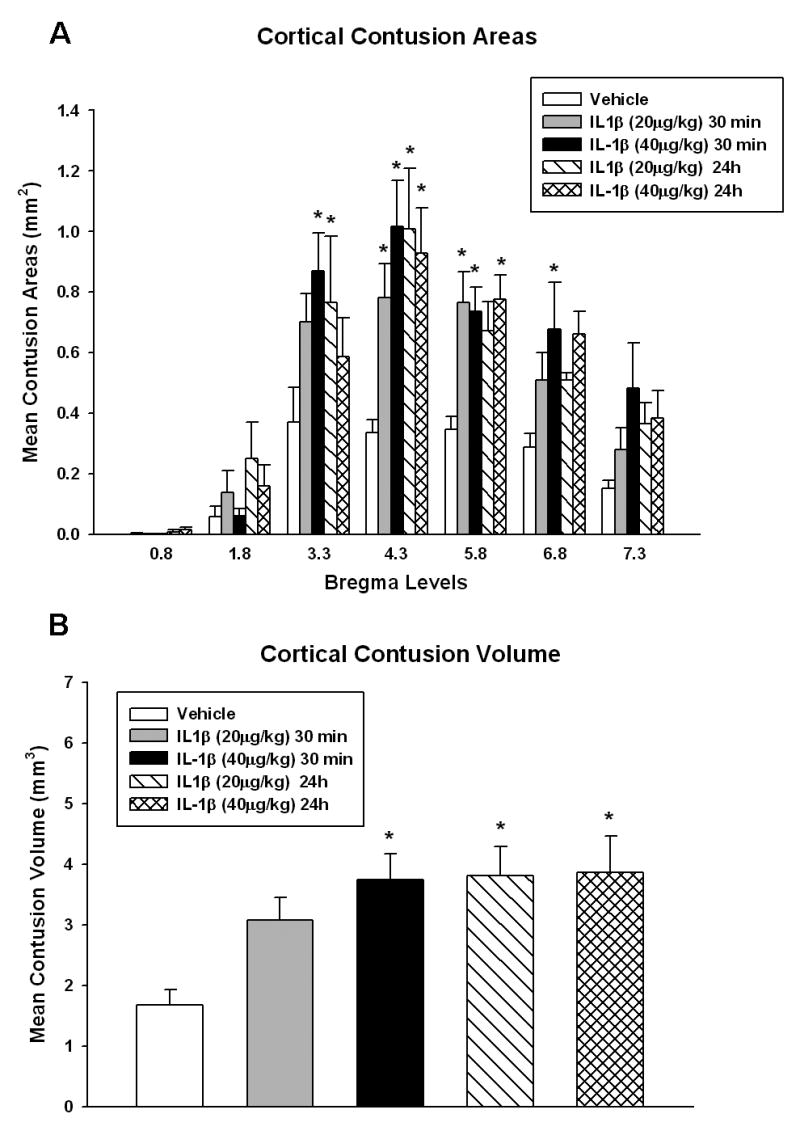
Bar graph of contusion areas and volume. Mean +SEM cortical contusion areas measured at 7 bregma levels after TBI showed widespread increases over several bregma levels for both IL-1β doses (p<0.05) compared to sham (Figure 4A). Mean +SEM contusion volumes were computed using serial integration procedures (Figure 4B). IL-1β (20 μg/kg) administered at 24 hrs post TBI was significantly different than TBI-vehicle animals (p<0.05). Although this concentration was not significantly different from vehicle animals when given at 30 min post-injury, there was a trend for a larger contusion volume in these animals. IL-1β (40 μg/kg) treatment at both 30 min and 24 hour after TBI increased contusion volume over vehicle treated TBI animals. *p<0.05 versus TBI-vehicle.
Administration of peripheral IL-1β increased the contusion volume in the FP injured rats (Figure 4B). One-way ANOVA was significant between groups (p<0.01). When administered 30 min post injury, IL-1β increased contusion volume significantly versus saline alone at the 40 μg/kg dose with a trend for the 20 μg/kg dose to worsen contusion volume (contusion volume: vehicle 1.67±0.25 mm3, n=9; 20μg/kg IL-1β 3.08±0.37 mm3, n=8; 40 μg/kg IL-1β 3.74±0.43 mm3, n=9, p<0.05). There was no difference between the two doses relative to each other. When given 24 hr after injury, IL-1β more than doubled the contusion volume at both the 20 and 40 μg/kg doses (p<0.005) compared to saline treated alone (contusion volume: vehicle 1.67±0.25 mm3, n=9; 20 μg/kg IL-1β 3.82±0.47 mm3, n=6; 40μg/kg IL-1β 3.86±0.60 mm3, n=6). Consistent with the 30 min treatment group data, there was no dosage difference in terms of contusion volumes.
Peripheral administration of IL-1β resulted in increased neuronal loss in the CA3 region of the hippocampus. In H&E stained sections of the brain, no neuronal loss was observed in the sham operated (Fig 6A), and saline treated TBI animals demonstrated modest CA3 damage (Fig 6B). However, the administration of 40μg/kg of IL-1β resulted in apparent loss of CA3 neurons 3 days after injection (Fig 5C). Evidence of extravasation of blood was also evident on the ipsilateral hemisphere along the contusion site and also at the gray/white matter interface (Fig 3 and 5D). The presence of hemorrhage in the gray/white matter interface is typical for fluid percussion injury. Blood vessels in the overlying cerebral cortex of damaged areas showed a perivascular accumulation of neutrophils (Fig 5E), indicative of a local breakdown of the blood-brain barrier (BBB). BBB breakdown has been previously reported in this model and occurs throughout the injured cortex (Dietrich et al., 1994). Finally, peripheral administration of IL-1β caused an accumulation of leukocytes in cortical vessels (Fig 5F) in areas of the cerebral cortex that had a marked loss of neurons.
Figure 5.

Hematoxylin and eosin stained sections after TBI. CA3 regions of the hippocampus of sham (A), saline (B), and 40 μg/kg IL-1β (C) show an apparent loss of CA3 neurons with IL-1β administration (arrow). Breakdown of the BBB and extravasation of blood into surrounding tissue is seen in the contusion in a vehicle treated TBI animal (D). Perivascular accumulation of neutrophils was observed in TBI vehicle treated animal (E). Leukocyte accumulation in blood vessels following TBI and IL-1β administration at 30 minutes (F) was noted.
Discussion
The present study demonstrates that artificially elevated levels of systemic IL-1β can markedly influence the extent of histopathological damage and behavioral outcome in a clinically relevant model of TBI. Increased levels of systemic IL-1β at two different doses worsened traumatic outcome when administered at either 30 min or 24 hr after TBI. A potential mechanism for this increased vulnerability to circulating levels of IL-1β was shown to be a trauma-induced increase in IL-1r1 expression in the vulnerable cerebral cortex, a critical receptor underlying the biological consequences of IL-1β on cell signaling. These data emphasize the complexity of systemic factors that may influence traumatic outcome under a controlled experimental condition.
Proinflammatory cytokine levels and expression patterns are significantly increased in a variety of TBI models. Previous studies have shown that both moderate and severe levels of TBI can lead to increased levels of mRNA for IL-1β in both cortical and subcortical brain areas as early as 1 hr after TBI (Kinoshita et al., 2002, Taupin et al., 1993). Protein levels of IL-1β are also increased and remain elevated for up to several days after injury (Kinoshita et al., 2002). Although, IL-1 receptor protein levels were not measured in the present study, IL-1r1 mRNA levels were still elevated at day 3 post-TBI indicating the potential for continuous upregulation of IL-1 receptor for activation. As previously discussed, therapies including the administration of the endogenous IL-1r1 antagonist have been reported to protect against neuronal injury after FP brain injury (Sanderson et al., 1999, Toulmond and Rothwell, 1995), controlled cortical impact injury (Tehranian et al., 2002), and cerebral ischemia (McColl et al., 2007, Relton and Rothwell, 1992). These treatment strategies have primarily targeted endogenous sources of IL-1β that have been proposed to increase neuronal vulnerability and worsen behavioral outcome. Other treatments that have targeted IL-1β mediated damage include minocycline (Bye et al., 2007) and cell cycle inhibition (Tian et al., 2007). However, another important source of IL-1β is from circulating leukocytes that are known to synthesize and release a number of cytokines under a variety of pathological conditions (Smith, 1994). This may result in a vicious cycle of the inflammatory response. McColl and colleagues (2007) have recently demonstrated that the mobilization of PMNLs may be through the activation of IL-1 induced CXC chemokines. This potentiates neutrophil activation resulting in further IL-1β release from these circulating cells. Immunocytochemical and biochemical analyses of these activated cells have shown them to be a rich source for proinflammatory cytokines. Thus, when circulating leukocytes gain access to the injured parenchyma via a breakdown of the BBB which occurs in this model (Dietrich et al., 1994), adverse effects may occur as a consequence of this systemic response (Scholz et al., 2007). We showed in the present study the presence of leukocytes in the traumatized cortex overlying the contusion site along with an increased amount of extravasated blood. These observations are therefore indicative of a breakdown of the BBB and a pathway for entry of peripherally-derived cytokines such as IL-1β which can exacerbate outcome especially in the early posttraumatic period.
Trauma-induced primary vascular damage leading to the upregulation of vascular endothelial adhesion molecules may allow circulating cells to accumulate within injured sites and enter the brain parenchyma. Thus, treatments that protect against abnormal vascular permeability (Whalen et al., 1997, Jiang et al., 1992, Smith and Hall, 1996, Dietrich et al., 1990) and leukocyte accumulation have been shown to improve traumatic outcome. For example, in a spinal cord injury study, the transient blockage of the CD11d/CD18 integrin on circulating leukocytes improved neurological recovery and reduced early inflammation (Gris et al., 2004). In addition to pharmacological treatments, therapeutic hypothermia, which also improves histopathological and functional traumatic outcome, has also been shown to protect against BBB disruption and inflammatory responses in a number of experimental models (Dietrich et al., 1990, Jiang et al., 1992). In the present study, the IL-1r1 message was shown to increase as early as 30 min after trauma and remained elevated up to 3 days in the vulnerable cerebral cortex. Because this receptor is primarily responsible for the biological actions of IL-1β, the sustained elevation provides a mechanism by which circulating IL-1β levels could influence traumatic outcome. Although, increased message does not necessarily imply there there are similar increases in IL-1r1 protein expression, these data emphasize the complex nature of TBI and potential mechanisms underlying neuronal vulnerability. In terms of therapeutic strategies, treatments that target circulating sources of proinflammatory cytokines or their receptors may improve outcome by removing or blocking a systemic source of potentially neurotoxic substances.
These studies are important because treatment strategies targeting systemic factors may be necessary to promote maximum benefit and outcome in a subset of TBI patients coping with polytrauma. Increasingly, it is being recognized that a specific population of TBI patients suffer from additional injuries or polytrauma. The Veterans Health Administration, in its handbook, defines polytrauma as consisting of 2 or more injuries to physical regions or organ systems. In addition, 85.5% of VA polytrauma patients between May 2003 and Oct 2006 suffered brain injuries (Department of Veterans Affairs, 2005). While brain injury is the primary injury that drives treatment of patients with polytrauma, often mild TBI is overlooked in the face of externally evident life-threatening bodily injuries that require immediate treatment (Lew, 2005). This opens a window of time following injury where secondary injury mechanisms become more important in TBI outcome. In a review by Schmidt and colleagues (2005), secondary brain injury was seen at autopsy in 70-90% of all fatal head injury patients. In a 1990 study of 328 patients, those patients with multiple injuries aside from TBI had significantly less favorable outcomes than TBI alone (Groswasser et al., 1990). In a separate study, patients with extra-cranial injuries in addition to head trauma had a mortality level almost twice of those with TBI alone (TBI 11.1%, polytrauma 21.8%) (Wilson and Tyburski, 2001). Thus, the pathophysiology of polytrauma as it relates to outcome after TBI is a topic of current clinical interest (Keel and Trentz, 2005).
In summary, the present data support and extend findings from other models of CNS injury that emphasize a critical role of systemic inflammation in the vulnerability of the post-injured brain and spinal cord. Inflammatory cytokines contribute to the pathogenesis of CNS injury in both the early and late time frames with early consequences being a target for neuroprotection. The traumatized brain was shown to be highly vulnerable to elevated levels of peripherally derived IL-1β up to 24 hr post injury. This traumatic response is associated with the increased expression of the IL-1r1 message in vulnerable cortical areas. These findings regarding the detrimental consequences of early systemic inflammation on traumatic outcome do not address the later and possible beneficial response of inflammation after trauma (for review see Lucas et al., 2006). Nonetheless, the present study may have more important implications in the pathobiology and treatment of early TBI as a significant number of brain-injured patients sustain multi-organ trauma.
Acknowledgments
This work was supported by Grants NS30291 and NS42133 from NIH. We wish to thank Jeremy Lytle and Coleen Atkins for editorial assistance and manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alheim K, Bartfai T. The interleukin-1 system: receptors, ligands, and ICE in the brain and their involvement in the fever response. Ann N Y Acad Sci. 1998;840:51–58. doi: 10.1111/j.1749-6632.1998.tb09548.x. [DOI] [PubMed] [Google Scholar]
- Ando T, Langley RR, Wang Y, Jordan PA, Minagar A, Alexander JS, Jennings MH. Inflammatory cytokines induce MAdCAM-1 in murine hepatic endothelial cells and mediate alpha-4 beta-7 integrin dependent lymphocyte endothelial adhesion In Vitro. BMC Physiol. 2007;7:10. doi: 10.1186/1472-6793-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A, Kossmann T, Morganti-Kossmann MC. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuates microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol. 2007;204:220–233. doi: 10.1016/j.expneurol.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Chiaretti A, Genovese O, Aloe L, Antonelli A, Piastra M, Polidori G, Di Rocco C. Interleukin 1beta and interleukin 6 relationship with paediatric head trauma severity and outcome. Childs Nerv Syst. 2005;21:185–193. doi: 10.1007/s00381-004-1032-1. discussion 194. [DOI] [PubMed] [Google Scholar]
- Conti B, Tabarean I, Andrei C, Bartfai T. Cytokines and fever. Front Biosci. 2004;9:1433–1449. doi: 10.2741/1341. [DOI] [PubMed] [Google Scholar]
- Cortez SC, McIntosh TK, Noble LJ. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989;482:271–282. doi: 10.1016/0006-8993(89)91190-6. [DOI] [PubMed] [Google Scholar]
- Department of Veterans Affairs, Veterans Health Administration Directive. Polytrauma Rehabilitation Centers. Washington (DC): Veterans Health Administration; Jun 8, 2005. p. 2. [Google Scholar]
- De Ryck M, Van Reempts J, Borgers M, Wauquier A, Janssen PA. Photochemical Stroke Model: Flunarizine Prevents Sensorimotor Deficits After Neocorticl Infarcts in Rats. Stroke. 1989;20:1383–1390. doi: 10.1161/01.str.20.10.1383. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J Neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Busto R, Halley M, Valdes I. The importance of brain temperature in alterations of the blood-brain barrier following cerebral ischemia. J Neuropathol Exp Neurol. 1990;49:486–497. doi: 10.1097/00005072-199009000-00004. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin 1 as mediator of the acute-phase response. Surv Immunol Res. 1984;3:29–33. doi: 10.1007/BF02918594. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Fassbender K, Schneider S, Bertsch T, Schlueter D, Fatar M, Ragoschke A, Kuhl S, Kischka U, Hennerici M. Temporal profile of release of interleukin-1beta in neurotrauma. Neurosci Lett. 2000;284:135–138. doi: 10.1016/s0304-3940(00)00977-0. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ, Wang X, Barone FC. Inflammatory gene expression in cerebral ischemia and trauma. Potential new therapeutic targets. Ann N Y Acad Sci. 1997;825:179–193. doi: 10.1111/j.1749-6632.1997.tb48428.x. [DOI] [PubMed] [Google Scholar]
- Gavins F, Yilmaz G, Granger DN. The evolving paradigm for blood cell-endothelial cell interactions in the cerebral microcirculation. Microcirculation. 2007;14:667–681. doi: 10.1080/10739680701404903. [DOI] [PubMed] [Google Scholar]
- Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC. Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci. 2004;24:4043–4051. doi: 10.1523/JNEUROSCI.5343-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groswasser Z, Cohen M, Blankstein E. Polytrauma associated with traumatic brain injury: incidence, nature and impact on rehabilitation outcome. Brain Inj. 1990;4:161–166. doi: 10.3109/02699059009026161. [DOI] [PubMed] [Google Scholar]
- Habgood MD, Bye N, Dziegielewska KM, Ek CJ, Lane MA, Potter A, Morganti-Kossmann C, Saunders NR. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur J Neurosci. 2007;25:231–238. doi: 10.1111/j.1460-9568.2006.05275.x. [DOI] [PubMed] [Google Scholar]
- Jiang JY, Lyeth BG, Kapasi MZ, Jenkins LW, Povlishock JT. Moderate hypothermia reduces blood-brain barrier disruption following traumatic brain injury in the rat. Acta Neuropathol (Berl) 1992;84:495–500. doi: 10.1007/BF00304468. [DOI] [PubMed] [Google Scholar]
- Keel M, Trentz O. Pathophysiology of polytrauma. Injury. 2005;36:691–709. doi: 10.1016/j.injury.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Chatzipanteli K, Vitarbo E, Truettner JS, Alonso OF, Dietrich WD. Interleukin-1beta messenger ribonucleic acid and protein levels after fluid-percussion brain injury in rats: importance of injury severity and brain temperature. Neurosurgery. 2002;51:195–203. doi: 10.1097/00006123-200207000-00027. [DOI] [PubMed] [Google Scholar]
- Lew HL. Rehabilitation needs of an increasing population of patients: Traumatic brain injury, polytrauma, and blast-related injuries. J Rehabil Res Dev. 2005;42:xiii–xvi. [PubMed] [Google Scholar]
- Luca SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147 1:S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via Interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti-Kossman MC, Lenzlinger PM, Hans V, Stahel P, Csuka E, Ammann E, Stocker R, Trentz O, Kossmann T. Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol Psychiatry. 1997;2:133–136. doi: 10.1038/sj.mp.4000227. [DOI] [PubMed] [Google Scholar]
- Relton JK, Rothwell NJ. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res Bull. 1992;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- Sanderson KL, Raghupathi R, Saatman KE, Martin D, Miller G, McIntosh TK. Interleukin-1 receptor antagonist attenuates regional neuronal cell death and cognitive dysfunction after experimental brain Injury. J Cereb Blood Flow Metab. 1999;19:1118–1125. doi: 10.1097/00004647-199910000-00008. [DOI] [PubMed] [Google Scholar]
- Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury--an inflammatory disease? Brain Res Brain Res Rev. 2005;48:388–399. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Scholz M, cinatl J, Schädel-Höpfner M, Windolf J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med Res Rev. 2007;27:401–416. doi: 10.1002/med.20064. [DOI] [PubMed] [Google Scholar]
- Schonbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- Shahbazian LM, Jeevanandam M, Petersen SR. Release of proinflammatory cytokines by mitogen-stimulated peripheral blood mononuclear cells from critically ill multiple-trauma victims. Metabolism. 1999;48:1397–1401. doi: 10.1016/s0026-0495(99)90149-x. [DOI] [PubMed] [Google Scholar]
- Smith JA. Neutrophils, host defense, and inflammation: a double-edged sword. J Leukoc Biol. 1994;56:672–686. doi: 10.1002/jlb.56.6.672. [DOI] [PubMed] [Google Scholar]
- Smith SL, Hall ED. Mild pre- and posttraumatic hypothermia attenuates blood-brain barrier damage following controlled cortical impact injury in the rat. J Neurotrauma. 1996;13:1–9. doi: 10.1089/neu.1996.13.1. [DOI] [PubMed] [Google Scholar]
- Stahel PF, Shohami E, Younis FM, Kariya K, Otto VI, Lenzlinger PM, Grosjean MB, Eugster HP, Trentz O, Kossmann T, Morganti-Kossmann MC. Experimental closed head injury: analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J Cereb Blood Flow Metab. 2000;20:369–380. doi: 10.1097/00004647-200002000-00019. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV. Inflammation and brain edema: new insights into the role of chemokines and their receptors. Acta Neurochir Suppl. 2006;96:444–450. doi: 10.1007/3-211-30714-1_91. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Bramlett HM, Ruenes G, Dietrich WD. The effects of early post-traumatic hyperthermia in female and ovariectomized rats. J Neurotrauma. 2004;21:842–853. doi: 10.1089/0897715041526186. [DOI] [PubMed] [Google Scholar]
- Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol. 1993;42:177–185. doi: 10.1016/0165-5728(93)90008-m. [DOI] [PubMed] [Google Scholar]
- Tehranian R, Andell-Jonsson S, Beni SM, Yatsiv I, Shohami E, Bartfai T, Lundkvist J, Iverfeldt K. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J Neurotrauma. 2002;19:939–951. doi: 10.1089/089771502320317096. [DOI] [PubMed] [Google Scholar]
- Tian DS, Xie MJ, Yu ZY, Zhang Q, Wang YH, Chen B, Chen C, Wang W. Cell cycle inhibition attenuates microglia induced inflammatory response and alleviates neuronal cell death after spinal cord injury in rats. Brain Res. 2007;1135:177–185. doi: 10.1016/j.brainres.2006.11.085. [DOI] [PubMed] [Google Scholar]
- Toulmond S, Rothwell NJ. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995;671:261–266. doi: 10.1016/0006-8993(94)01343-g. [DOI] [PubMed] [Google Scholar]
- Truettner JS, Alonso OF, Dalton Dietrich W. Influence of therapeutic hypothermia on matrix metalloproteinase activity after traumatic brain injury in rats. J Cereb Blood Flow Metab. 2005;25:1505–1516. doi: 10.1038/sj.jcbfm.9600150. [DOI] [PubMed] [Google Scholar]
- Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Clark RS, Marion DW, DeKosky MS, Heineman S, Schiding JK, Memarzadeh F, Dixon CE, Kochanek PM. The relationship between brain temperature and neutrophil accumulation after traumatic brain injury in rats. Acta Neurochir Suppl. 1997;70:260–261. doi: 10.1007/978-3-7091-6837-0_80. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Wisniewski SR, Clark RS, Mellick JA, Marion DW, Kochanek PM. Effect of neutropenia and granulocyte colony stimulating factor-induced neutrophilia on blood-brain barrier permeability and brain edema after traumatic brain injury in rats. Crit Care med. 2000;28:3710–3717. doi: 10.1097/00003246-200011000-00029. [DOI] [PubMed] [Google Scholar]
- Wilson RF, Tyburski JG. Management of patients with head injuries and multiple other trauma. Neurol Res. 2001;23:117–120. doi: 10.1179/016164101101198415. [DOI] [PubMed] [Google Scholar]
- Wong D, Prameya R, Dorovini-Zis K. Adhesion and migration of polymorphonuclear leukocytes across human brain microvessel endothelial cells are differentially regulated by endothelial cell adhesion molecules and modulate monolayer permeability. J Neuroimmunol. 2007;184:136–148. doi: 10.1016/j.jneuroim.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Zilles L. A Stereotaxic Atlas. Springer; New York: 1985. The Cortex of the Rat. [Google Scholar]