

Abstract
Here we report the facile synthesis of a designed multi-pharmacophore ligand derived from the linkage of a delta selective peptide antagonist (Dmt-Tic) and a mu / kappa morphinan agonist butorphan (MCL 101) through a two methylene spacer. The new compound MCL 450 maintains the same characteristics as the two reference compounds. MCL 450 represents a useful starting point for the synthesis of other multiple opioid ligands endowed with analgesic properties with low tolerance and dependence.
Introduction
For many years, clinicians have treated unresponsive patients by combining therapeutic mechanisms with a cocktail of drugs. The design of ligands that act on specific multiple targets (multiple ligands) is a more recent trend, as indicated by the substantial increase over the past few years in the number of publications describing such approaches. Numerous terms are currently used to describe ligands that have multiple activities: the words dual, binary, bivalent, dimeric, mixed, triple or balanced are used in conjunction with numerous suffixes, for example, ligand, inhibitor, agonist, antagonist, conjugate or blocker. To improve communication and awareness of this emerging field within the drug discovery community, some authors propose using the term “designed multiple” (DM) ligands as a generic phrase to describe compounds that are rationally designed to modulate multiple targets of relevance to a disease, with the overall goal of enhancing efficacy and / or improving safety.1, 2-4 Compared with drug combinations, there are some advantages associated with multiple ligands, such as the more predictable pharmacokinetic and pharmacodynamic relationship that is a consequence of the administration of a single drug, as well improved patient compliance. The molecular starting point for a multiple-ligand project is generated using one of two distinct approaches - either rational design by a combination of pharmacophores or the screening of compounds libraries of known drugs. The methodical combination of pharmacophores from selective ligands is currently the predominant technique used for the generation of multiple ligands. The pharmacophores are joined together by a cleavable or non-cleavable linker (termed “conjugates”) or, more commonly, they are overlapped by taking advantage of structural commonalities (“overlapping pharmacophore”). The majority of reported examples of cleavable conjugates contain an ester linker that is cleaved by plasma esterases to release two individual drugs that then act independently. Although the pharmacokinetic-pharmacodynamic relationship could become complex after cleavage of the linker, at the same time of administration cleavable conjugates are a single molecule, which is one potential advantage that this approach has over drug cocktails.3, 4 The prevalence in the literature of “designing in” new activities, that is the synthesis of designed multiple (DM) ligands from selective ligands, indicates that this approach is certainly more popular, and probably more feasible than “designing out” activities from non-selective ligands. As an example, a mixed μ agonist / δ antagonist pseudopeptide was obtained linking tail to tail a selective δ antagonist (H-Tyr-TicΨ[CH2-NH]Cha-Phe-OH) with a selective μ agonist (H-Dmt-D-Arg-Phe-Lys-NH2).5 Recently, Neumeyer et al. reported the synthesis and pharmacological evaluation of “bivalent ligands” containing homo- and heterodimeric pharmacophores related to morphinans.6 Ligands having two pharmacophores connected by a spacer have the potential for binding vicinal receptors.7-9 Such bridging should be manifested by a substantial increase in potency due to the high concentration of the pharmacophore in the vicinity of the recognition site when the ligand is bound in a monovalent mode. Portoghese et al. reported a range of “homo-“ and “heterodimeric ligands” with varying linker lengths designed to investigate pharmacodynamic and organizational features of opioid receptors.10 For example, recently reported heterodimeric ligands containing δ antagonist (naltrindole) and κ1 agonist (ICI-199,441) pharmacophores joined by variable length oligoglycyl-based linkers were demonstrated to possess significantly greater potency and selectivity compared to their monomer congeners, providing further evidence for the opioid receptor hetero-oligomerization phenomena.11 Considering earlier data in this field, we report the synthesis and pharmacological evaluation of the first opioid designed multiple ligand obtained through the linkage of the δ-selective peptidic Dmt-Tic pharmacophore and the κ agonist / μ partial agonist morphinan compound MCL 101 (butorphan).
Chemistry
The synthesis of compound MCL 450 is reported in Scheme 1. Boc-β-ala-OH was condensed with MCL 10112 via EDC/DMAP13 to yield crude MCL 438, which after solvent evaporation, was purified by column chromatography as reported in the experimental section. After N-terminal Boc deprotection with TFA / CH2Cl2,14 the product was condensed with Boc-Dmt-Tic-OH15 via EDC/HOBt. Boc deprotection with TFA / CH2Cl2 gave MCL 450. Condensation of Boc-Dmt-Tic-OH with HCl.H-β-ala-OMe via EDC/HOBt gave the corresponding protected tripeptide which was hydrolyzed at C-terminus methyl ester by NaOH and deprotected at Boc N-terminus with TFA to yield crude MCL 451. Final compounds were purified by reverse phase preparative HPLC.

Pharmacological Results and Discussion
Affinity and Selectivity of the Synthesized Ligands
MCL 450 and MCL 451 were evaluated for their affinity at and selectivity for μ, δ and κ opioid receptors with Chinese hamster ovary (CHO) cell membranes stably expressing the human opioid receptors. Data are summarized in Table 1. For comparison purposes, opioid binding affinity data for MCL 101and Dmt-Tic are included. The designed multiple ligand MCL 450 derives from the ester bond formation between MCL 451 and MCL 101. As expected, its selectivity derives from a combination of affinity data from MCL 101 (essentially a κ / μ ligand) and MCL 451 (δ ligand). In fact, μ and κ affinity drop 3 and 3.5 fold from MCL 101 to MCL 450, respectively; while δ affinity increases 3.9 fold. Considering MCL 451 in comparison with MCL 450, μ affinity increases about 1600 fold and unexpectedly δ affinity increases 2.8 fold. Moreover, MCL 451 confirms once again the importance of the Dmt-Tic pharmacophore in the induction of δ affinity and selectivity, especially if a free carboxylic function is present at C-terminus.16 Starting from a quite selective κ / μ ligand (MCL 101) and a selective δ ligand (MCL 451) a nonselective MCL 450 was obtained.
Table 1.
Ki values Inhibition of μ, δ, and κ Opioid Binding to CHO Membranes
| Ki (nM) ± SE | Selectivity | ||||||
|---|---|---|---|---|---|---|---|
| Compound | Structure | [3H]DAMGO (μ) | [3H]Naltrindole (δ) | [3H]U69,593 (κ) | Kiδ/Kiμ | Kiδ/Kiκ | Kiμ/Kiκ |
| MCL 101a |  |
0.23 | 5.9 | 0.079 | 26 | 75 | 2.9 |
| Dmt-Ticb | 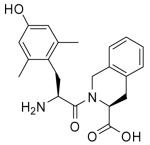 |
894 | 1.6 | 37500 | 559c | 23438d | 42e |
| MCL 450 |  |
0.69±0.04 | 1.5±0.03 | 0.28±0.03 | 2.2 | 5.4 | 2.5 |
| MCL 451 |  |
1100±33 | 4.2±0.58 | 62±0.82%* | 262c | - | - |
Maximum inhibition of [3H]U69,593 binding in the presence of 10 μM MCL 451.
Data from Peng, X. et al. J. Med. Chem. 2006, 49, 256-262.
Data from Pagé, D. et al. J. Med. Chem. 2001, 41, 2387-2390.
Selectivity Kiμ/Kiδ.
Selectivity Kiκ/Kiδ.
Selectivity Kik/Kiμ.
Functional Activity
Table 2 shows the agonist and antagonist properties of MCL 450 and MCL 451 in stimulating [35S]GTPγS binding mediated by the μ, δ and κ opioid receptors. The binding assays (Table 1), MCL 450 summarizes binding properties of its constituents MCL 101 (μ partial agonist / κ agonist) and MCL 451 (δ antagonist). The data show a decrease of EC50 for μ and κ receptors of 5.6 and 4.6 fold respectively, in comparison to MCL 101; at the same time the corresponding Emax (%) values increase of 1.58 and 1.63 fold (transforming it into an almost full μ agonist). On the other hand, MCL 450 is characterized by a δ antagonist activity furnished by MCL 451, moreover it is a δ antagonist more potent (2.6 fold) than the δ selective counterpart. As expected, MCL 451 results in a potent and selective δ antagonist, in line with all other δ antagonists containing the Dmt-Tic pharmacophore.15, 17-19
Table 2.
Functional Activity at the μ, δ and κ Receptorsa
| [35S]GTP-γ-S binding (Agonism) | ||||||
|---|---|---|---|---|---|---|
| μ | δ | κ | ||||
| EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | |
| MCL 101b | 1.6±0.2 | 50±2.5 | NT | NT | 1.3±0.44 | 80±6.8 |
| MCL 450 | 8.9±0.76 | 79±2.3 | 2.8±0.75 | 18±1.7 | 6.0±1.0 | 130±5.1 |
| MCL 451 | NT | NT | NT | NT | NA | 0.95±2.2 |
| [35S]GTP-γ-S binding (Agonism) | ||||||
|---|---|---|---|---|---|---|
| μ | δ | κ | ||||
| IC50 (nM) | Imax (%) | IC50 (nM) | Imax (%) | IC50 (nM) | Imax (%) | |
| MCL 101b | 20±3 | 50±3 | NT | NT | NT | NT |
| MCL 450 | 97±21 | 47±1.7 | 3.9±0.34 | 84±5.2 | No Inhibition | No Inhibition |
| MCL 451 | NT | NT | 10±1.5 | 93±1.0 | NT | NT |
Membranes from CHO that stably expressed only the μ, δ, or κ receptor were incubated with various concentrations of the compounds. The stimulation of [35S]GTPγS binding was measured as described in the Experimental Section. The Emax value is the maximal percent stimulation obtained with the compound. The EC50 value is the concentration of compound needed to produce 50% of the Emax value. When the Emax value was 30% or lower, it was not possible to calculate an EC50 value. To determine the antagonistic properties of a compound, membranes were incubated with 100 nM of the κ agonist U50,488 or 200 nM of the μ agonist DAMGO or 100 nM of the δ agonist SNC-80 in the presence of varying concentration of the compound. The Imax value is the maximal percent inhibition obtained with the compound. The IC50 value is the concentration of compound needed to produce half-maximal inhibition. NT, Not Tested; NA, Not Active.
Data from Peng, X. et al. J. Med. Chem. 2006, 49g , 256-262.
Conclusion
We have demonstrate the possibility of obtaining a designed multiple ligand in the opioid peptide field with the Dmt-Tic pharmacophore. Contrary to other researcher reports, in this case the distance between the two pharmacophores constituting the designed multiple ligand, seems not to be important. In fact, we selected a two methylene spacer simply because in a similar compound, synthesized by Schiller et al., he reported the same spacer between a μ agonist and a δ antagonist (H-Dmt→D-Arg→Phe→Lys-NH-CH2-CH2-NH-Phe←Cha[NH-CH2]ΨTic←Tyr-H), was reported the same spacer.5 We synthesized a very similar compound incorporating a δ antagonist and a μ agonist (H-Dmt→Tic-NH-CH2-CH2-NH-Phe←Phe←Pro←Tyr-H) and obtained very similar pharmacological results (personal data). On the other and, when the δ antagonist H-Dmt-Tic-Glu is linked to a fluorophore through a five methylene spacer, it maintains selectivity and δ antagonist properties similar to the reference tripeptide.20, 21 MCL 450 could represent a useful starting point in the synthesis of other designed multiple ligands from peptidic and morphinan pharmacophores; as an example some interesting compound could be obtained by esterification of δ antagonists (from the Dmt-Tic pharmacophore) and morphine. Such a compound, as for example diacetylmorphine (heroin), should be able to cross the blood brain barrier and then hydrolyzed by esterases to give morphine and a δ antagonist potentially useful to decreases or eliminate morphine induced side effects such as tolerance and dependence.22 Vice versa, MCL 101 or morphine could be esterified by the carboxylic function of arginine derivatives (NO synthase inhibitors) to prevent tolerance induced by morphinan μ- and κ-agonists.23 It is interesting to note that cleavable conjugates, such as these reported here, do not need spacers because their individual activities can be affected just after hydrolysis.
Experimental Section
(-)-3-Hydroxy-N-cyclobutylmethylmorphinan (MCL 101)
(-)-3-Hydroxy-N-cyclobutylmethylmorphinan free base was made from commercially available levorphanol tartrate (Mallinckrodt Inc.) by a procedure previously reported.12
1-((-)N-Cyclobutylmethylmorphinan-3-yl)-N-Boc-β-Alanine. (Boc-β-ala-O-MCL 101). (MCL 438)
MCL-101 (0.16 g, 0.5 mmol) and Boc-β-ala-OH (0.11 g, 0.6mmol) were dissolved in anhydrous dichloromethane (10 mL) under nitrogen. A catalytic amount of 4-dimethylaminopyridine (0.006 g, 0.05 mmol) was added, followed by EDC (0.12 g, 0.6 mmol). The mixture was stirred at room temperature overnight, after solvent evaporation, the crude product was purified by column chromatography on silica gel (EtOAc/Et3N, 100:1, v/v) to afford a colorless oil: yield 0.12 g (50%); Rf(B) 0.77; HPLC K’ 8.68; oil; [α]20D -22.6; MH+ 484; 1H-NMR (CDCl3) δ 1.05-2.83 (m, 35H), 3.02 (d, J=18.6Hz, 1H), 3.47-3.53 (m, 2H), 6.85(dd, J=8.1Hz, 2.4Hz, 1H), 6.94 (d, J=2.4Hz, 1H), 7.11(d, J=8.1Hz, 1H).
2TFA.H-β-ala-O-MCL 101
Boc-β-ala-O-MCL 101 (0.12 g, 0.25 mmol) was treated with 50% TFA in CH2Cl2 (2 mL) for 2 h at room temperature. After solvent evaporation, Et2O/Pe (1:5, v/v) was added to the solution until the product precipitated: yield 0.14 g (90%); Rf(A) 0.68; HPLC K’ 7.26; mp 121-123 °C; [α]20D -15.1; MH+ 384.
Boc-Dmt-Tic-β-ala-O-MCL 101
To a solution of Boc-Dmt-Tic-OH (0.05 g, 0.11 mmol) and 2TFA.H-β-ala-O-MCL 101 (0.07 g, 0.11 mmol) in DMF (10 mL) at 0 °C, NMM (0.02 mL, 0.22 mmol), HOBt (0.02 g, 0.12 mmol), and EDC (0.02 g, 0.12 mmol) were added. The reaction mixture was stirred for 3 h at 0 °C and 24 h at room temperature. After DMF was evaporated, the residue was dissolved in EtOAc and washed with NaHCO3 (5% in H2O), and brine. The organic phase was dried (Na2SO4) and evaporated to dryness. The residue was precipitated from Et2O/Pe (1:9, v/v): yield 0.07 g (83%); Rf(B) 0.75; HPLC K’ 9.52; mp 112-114 °C; [α]20D -15.6; MH+ 834; 1H-NMR (DMSO-d6) δ 1.05-3.17 (m, 46H), 3.47-4.92 (m, 6H), 6.29-7.02 (m, 9H).
TFA.H-Dmt-Tic-β-ala-O-MCL 101 (MCL 450)
Boc-Dmt-Tic-β-ala-O-MCL 101 (0.07 g, 0.08 mmol) was treated with 50% TFA in CH2Cl2 (2 mL) for 2 h at room temperature. After solvent evaporation, Et2O/Pe (1:1, v/v) was added to the solution until the product precipitated: yield 0.07 g (92%); Rf(A) 0.65; HPLC K’ 7.21; mp 128-130 °C; [α]20D -17.9; MH+ 734; 1H-NMR (DMSO-d6) δ 1.05-3.17 (m, 37H), 3.47-4.92 (m, 6H), 6.29-7.02 (m, 9H).
Boc-Dmt-Tic-β-ala-OMe
To a solution of Boc-Dmt-Tic-OH (0.14 g, 0.31 mmol) and HCl.H-β-ala-OMe (0.04 g, 0.31 mmol) in DMF (10 mL) at 0 °C, NMM (0.03 mL, 0.31 mmol), HOBt (0.05 g, 0.34 mmol), and EDC (0.06 g, 0.34 mmol) were added. The reaction mixture was stirred for 3 h at 0 °C and 24 h at room temperature. After DMF was evaporated, the residue was dissolved in EtOAc and washed with citric acid (10% in H2O), NaHCO3 (5% in H2O), and brine. The organic phase was dried (Na2SO4) and evaporated to dryness. The residue was precipitated from Et2O/Pe (1:9, v/v): yield 0.15 g (88%); Rf(B) 0.62; HPLC K’ 8.21; mp 124-126 °C; [α]20D +7.2; MH+ 555; 1H-NMR (DMSO-d6) δ 1.40-3.17 (m, 21H), 3.59-4.92 (m, 9H), 6.29 (s, 2H), 6.96-7.02 (m, 4H).
Boc-Dmt-Tic-β-ala-OH
To a solution of Boc-Dmt-Tic-β-ala-OMe (0.15 g, 0.27 mmol) in MeOH (10 mL) was added 1N NaOH (0.32 mL). The reaction mixture was stirred for 24 h at room temperature. After solvent evaporation, the residue was dissolved in EtOAc and washed with citric acid (10% in H2O) and brine. The organic phase was dried (Na2SO4) and evaporated to dryness. The residue was precipitated from Et2O/Pe (1:9, v/v): yield 0.12 g (82%); Rf(B) 0.58; HPLC K’ 7.92; mp 138-140 °C; [α]20D +9.1; MH+ 541.
TFA.H-Dmt-Tic-β-ala-OH (MCL 451)
Boc-Dmt-Tic-β-ala-OH (0.12 g, 0.22 mmol) was treated with TFA (1 mL) for 30 min. at room temperature. After solvent evaporation, Et2O/Pe (1:1, v/v) was added to the solution until the product precipitated: yield 0.11 g (92%); Rf(A) 0.46; HPLC K’ 6.12; mp 145-147 °C; [α]20D +12.3; MH+ 441; 1H-NMR (DMSO-d6) δ 2.35 (s, 6H), 2.51-3.59 (m, 8H), 3.95-4.92 (m, 4H), 6.29 (s, 2H), 6.96-7.02 (m, 4H).
Acknowledgment
This work was supported in part by NIH Grants RO1-DA14251 (JLN), K05-DA 00360 (JMB), University of Cagliari (PRIN 2004), University of Ferrara, and the Intramural Research Program of the NIH, and NIEHS. The authors appreciate the professional services of the library staff of the NIEHS. Levorphanol tartrate was generously donated by Mallinckrodt Inc.
Supporting Information Available. Chemistry general methods, biological general methods and elemental analysis. This material is available free of charge via the Internet at: http://pubs.acs.org.
References
- (1).J. Biol. Chem. 1985;260:14–42. Abbreviations. In addition to the IUPAC-IUB Commission on Biochemical Nomenclature. this paper uses the following additional symbols and abbreviations: Boc, t-butyloxycarbonyl; CHO, Chinese hamster ovary; DMAP, 4-dimethylaminopyridine; DAMGO, [D-Ala2,N-Me-Phe4,Gly-ol5]-enkephalin; DMF, N,N-dimethylformamide; DMSO-d6, hexadeuteriodimethyl sulfoxide; Dmt, 2′,6′-dimethyl-L-tyrosine; EDC, 1-ethyl-3-[3′-dimethyl)aminopropyl]-carbodiimide hydrochloride; EtOAc, ethyl acetate; Et3N, triethyl amine; Et2O, diethyl ether; HOBt, 1-hydroxybenzotriazole; HPLC, high performance liquid chromatography; MALDI-TOF, matrix assisted laser desorption ionization time-of-flight; MeOH, methanol; NMM, 4-methylmorpholine; Pe, petroleum ether; TFA, trifluoroacetic acid; Tic, 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid; TLC, thin-layer chromatography; U69,593, (5alpha,7alpha,8beta)-(+)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide. [Google Scholar]
- (2).Hopkins AL, Mason JS, Overington JP. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006;16:127–136. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- (3).Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005;48:6523–65433. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- (4).Morphy R, Kay C, Rankovic Z. From magic bullets to designed multiple ligands. Drug Discov Today. 2004;9:641–651. doi: 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]
- (5).Weltrowska G, Lemieux C, Chung NN, Schiller PW. A chimeric opioid peptide with mixed μ agonist / δ antagonist properties. J. Peptide Res. 2004;63:63–68. doi: 10.1111/j.1399-3011.2003.00108.x. [DOI] [PubMed] [Google Scholar]
- (6).Peng X, Knapp BI, Bidlack JM, Neumeyer JL. Synthesis and preliminary in vitro investigation of bivalent ligands containing homo- and heterodimeric pharmacophores at μ, δ, and κ opioid receptors. J. Med. Chem. 2006;49:256–262. doi: 10.1021/jm050577x. [DOI] [PubMed] [Google Scholar]
- (7).Jordan BA, Cvejic S, Devi LA. Opioid and their complicated receptor complexes. Neuropsycopharmacology. 2000;23:S5–S18. doi: 10.1016/S0893-133X(00)00143-3. [DOI] [PubMed] [Google Scholar]
- (8).George SR, Fan T, Xie Z, Tse R, Tam V, Vargase G, O’Dowd F. Oligomerization of μ- and δ-opioid receptors. Generation of novel functional properties. J. Biol. Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- (9).Portoghese PS, Nagase H, Takemori AE. Only one pharmacophore is required for the κ opioid antagonist selectivity of norbinaltorphimine. J. Med. Chem. 1988;31:1344–1347. doi: 10.1021/jm00402a015. [DOI] [PubMed] [Google Scholar]
- (10).Portoghese PS. From models to molecules: Opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J. Med. Chem. 2001;44:2259–2269. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- (11).Daniels DJ, Kulkarni A, Xie Z, Bhushan RG, Portoghese PS. A bivalent ligand (KDAN-18) containing δ-antagonist and κ-agonist pharmacophores bridges δ2 and κ1 opioid receptor phenotypes. J. Med. Chem. 2005;48:1713–1716. doi: 10.1021/jm034234f. [DOI] [PubMed] [Google Scholar]
- (12).Neumeyer JL, Bidlack JM, Zong R, Bakthavachalam V, Gao P, Cohen DJ, Negus SS, Mello NK. Synthesis and opioid receptor affinity of morphinan and benzomorphan derivatives: mixed κ agonists and μ agonists / antagonists as potential pharmatherapeutic for cocaine dependence. J. Med. Chem. 2000;43:114–122. doi: 10.1021/jm9903343. [DOI] [PubMed] [Google Scholar]
- (13).Ogawa T, Araki M, Miyamae T, Okayama T, Hagiwara M, Sakurada S, Morikawa T. Synthesis and antinociceptive activity of orally active opioid peptides: improvement of oral bioavailability by esterification. Chem. Pharm. Bull. 2003;51:759–771. doi: 10.1248/cpb.51.759. [DOI] [PubMed] [Google Scholar]
- (14).Baxter AJG, Robinson DH, Brown RC. Pharmaceutically active compounds. 1990. EP0384636.
- (15).Salvadori S, Guerrini R, Balboni G, Bianchi C, Bryant SD, Cooper PS, Lazarus LH. Further studies on the Dmt-Tic pharmacophore: hydrophobic substituents at the C-terminus endow δ antagonists to manifest μ agonism or μ antagonism. J. Med. Chem. 1999;42:5010–5019. doi: 10.1021/jm990165m. [DOI] [PubMed] [Google Scholar]
- (16).Balboni G, Salvadori S, Guerrini R, Negri L, Giannini E, Bryant SD, Jinsmaa Y, Lazarus LH. Direct influence of C-terminally substituted amino acids in the Dmt-Tic pharmacophore on δ-opioid receptor selectivity and antagonism. J. Med. Chem. 2004;47:4066–4071. doi: 10.1021/jm040033f. [DOI] [PubMed] [Google Scholar]
- (17).Salvadori S, Attila M, Balboni G, Bianchi C, Bryant SD, Crescenzi O, Guerrini R, Picone D, Tancredi T, Temussi PA, Lazarus LH. δ Opioidmimetic antagonists: prototypes for designing a new generation of ultraselective opioid peptides. Mol. Med. 1995;1:678–689. [PMC free article] [PubMed] [Google Scholar]
- (18).Salvadori S, Balboni G, Guerrini R, Tomatis R, Bianchi C, Bryant SD, Cooper PS, Lazarus LH. Evolution of the Dmt-Tic pharmacophore: N-terminal methylated derivatives with extraordinary δ opioid antagonist activity. J. Med. Chem. 1997;40:3100–3108. doi: 10.1021/jm9607663. [DOI] [PubMed] [Google Scholar]
- (19).Balboni G, Guerrini R, Salvadori S, Negri L, Giannini E, Bryant SD, Jinsmaa Y, Lazarus LH. Conversion of the potent δ-opioid agonist H-Dmt-Tic-NH-CH2-Bid into δ-opioid antagonists by N1-benzimidazole alkylation. J. Med. Chem. 2005;48:8112–8114. doi: 10.1021/jm058259l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Balboni G, Salvadori S, Dal Piaz A, Bortolotti F, Argazzi R, Negri L, Lattanzi R, Bryant SD, Jinsmaa Y, Lazarus LH. Highly selective fluorescent analogue of the potent δ-opioid receptor antagonist Dmt-Tic. J. Med. Chem. 2004;47:6541–6546. doi: 10.1021/jm040128h. [DOI] [PubMed] [Google Scholar]
- (21).Vázquez ME, Blanco JB, Salvadori S, Trapella C, Argazzi R, Bryant SD, Jinsmaa Y, Lazarus LH, Negri L, Giannini E, Lattanzi R, Colucci M, Balboni G. 6-N,N-Dimethylamino-2,3-naphthalimide a new environment-sensitive fluorescent probe in δ-selective and μ-selective opioid peptides. J. Med. Chem. doi: 10.1021/jm060343t. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991;258:299–303. [PubMed] [Google Scholar]
- (23).Bargava HN, Cao Y-J, Zhao G-M. Effect of 7-nitroindazole on tolerance to morphine, U-50,488H and [D-Pen2, D-Pen5]enkephalin in mice. Peptides. 1997;18:797–800. doi: 10.1016/s0196-9781(97)00021-1. [DOI] [PubMed] [Google Scholar]