

Abstract
The early inflammatory response to traumatic brain injury (TBI) may result in secondary damage. The purpose of this study was to evaluate the effects of a transient treatment employing a blocking monoclonal antibody (mAb) to the CD11d/CD18 integrin on histopathological outcome and macrophage infiltration following TBI. A parasagittal fluid percussion (FP) brain injury (1.8 – 2.1 atmosphere) was induced in male Sprague-Dawley rats. Rats were randomized into two trauma groups, treated (N=7) and nontreated (N=8) animals. In the treated group, a mAb to the CD11d subunit of the CD11d/CD18 integrin was administered 30 minutes, 24 and 48 hours after brain injury. Control animals received an isotype-matched irrelevant mAb using the same dose and treatment regimen. At three days after TBI, animals were perfusion-fixed for histopathological and immunocytochemical analysis. The anti-CD11d mAb treatment reduced contusion areas as well as overall contusion volume compared to vehicle-treated animals. For example, overall contusion volume was reduced from 2.7 ± 0.5 mm3 (mean ± SEM) to 1.4 ± 0.4 with treatment (p<0.05). Immunocytochemical studies identifying CD68 immunoreactive macrophages showed that treatment caused significant attenuation of leukocyte infiltration into the contused cortical areas. These data emphasize the beneficial effects of blocking inflammatory cell recruitment into the injured brain on histopathological outcome following traumatic brain injury.
Keywords: Traumatic brain injury, microglia/macrophages, CD11d/CD18 integrin, adhesion molecules, inflammation, histopathology
1. Introduction
Traumatic brain injury leads to both primary and secondary injury mechanisms (McIntosh et al., 1998). Secondary injury mechanisms include inflammatory events that are believed to contribute to outcome (Shohami et al., 1994,1996; Stahel et al., 2000). Acute inflammatory responses including alterations in the blood-brain barrier (BBB), infiltration and accumulation of polymorphonuclear leukocytes (PMNLs) and hematogenous macrophages with the subsequent production of proinflammatory cytokines and reactive free radicals can contribute to the pathologic process (Morganti-Kossmann et al., 2001; Raghupathi, 2004; for review see Scholz et al., 2007). Cytokines synthesized by the injured brain as well as invading circulating leukocytes are capable of altering the vascular permeability and exacerbate subsequent brain edema formation (Shohami et al., 1994). These acute inflammatory events can lead to accumulation of other cell types and the production of harmful reactive oxygen species, nitric oxide and other detrimental molecules leading to cell necrosis and apoptosis (Nguyen et al., 2007).
Various strategies have been utilized to target the acute inflammatory response to CNS injury (Barone et al., 1999; Bethea and Dietrich, 2002; Bye et al., 2007; Tehranian et al., 2002; Toulmond et al., 1995). It is known that the infiltration and accumulation of activated white blood cells into extravascular tissues depends on leukocyte-endothelial interactions between cell surface integrins and adhesion molecules such as endothelial selectins, leading to rolling, adhesion and ultimately transmigration of the circulating cell (Del Zoppo et al., 2000; Loddick et al., 2002). For example, increased expression of intercellular adhesion molecule (ICAM-1) and platelet endothelial adhesion molecule 1 (PCAM-1) after brain injury accounts partly for neutrophil adhesion to endothelial cells and transendothelial migration (Smith, 1993). Thus, treatments that block the upregulation of these adhesion molecules and reduce inflammatory processes have been shown in some studies to improve outcome in models of brain trauma (Chatizipantelli et al., 2002; Otto et al., 2002; Shohami et al., 1996).
After brain injury, leukocyte and monocyte infiltration requires tethering on the surface of endothelial cells (Bevilacqua, 1993; Whalen et al., 1999) followed by the interaction of endothelial cell-adhesion molecules with integrins on the PMNL or monocyte surface (Clark et al., 1996; Mabon et al., 2000; Neish et al., 1995; Shanley et al., 1998) facilitating leukocyte extravasation through the BBB. To prevent this interaction, previous studies in spinal cord injury (SCI) have utilized a monoclonal antibody (mAb) to the CD11d subunit of the CD11d/CD18 integrin (Grayson et al., 1999; Van der Vieren et al., 1999) to improve outcome. This integrin is expressed on both PMNLs and monocytes (macrophages). The anti-CD11d mAb treatment has been reported to decrease the number of PMNLs and macrophages in a spinal cord lesion two to three days after injury (Mabon et al., 2000; Saville et al., 2004). More recently, Gris and colleagues (2004) reported that treatment with anti-CD11d mAb reduced secondary damage and improved motor and sensory outcomes in a clip-compression SCI model. To determine whether this specific treatment would improve outcome in a model of TBI, we tested the efficacy of anti-CD11d mAb treatment starting 30 minutes after fluid percussion brain injury. Histopathological and immunocytochemical studies were conducted to determine whether treatment affected overall histopathological damage and leukocyte infiltration in the injured brain.
2. Results
2.1 Animal Physiology
Physiological measurements of pCO2, pO2, mean arterial blood pressure (MAP), pH and brain and rectal temperature are given in Table I. Physiological variables were taken immediately prior to TBI and 15 minutes and one hour after injury, only one hour post-injury is shown. pCO2 was significantly different between groups (p<0.02). The TBI-Vehicle treated animals had slightly lower pCO2 values compared to anti-CD11d treated animals. Although lower pCO2 can indicate hypocapnia (Muizelaar et al., 1991), our values were within normal range. pH was significantly different across time (p<0.008) as well with the post-trauma time point having lower pH values in both groups. Again, the pH numbers were within normal range and do not indicate metabolic alkalosis. All other physiological variables were not significantly different as assessed by two-way ANOVA. Controlled mechanical ventilation as utilized in this study is the best procedure for maintaining blood gases within normal ranges before and after TBI (Zausinger et al., 2002).
2.2 Histopathology
Representative cortical contusion areas of vehicle and anti-CD11d mAb treated animals are shown in Figure 1. Micrographs are taken from hematoxylin and eosin-stained sections of brain regions at the epicenter of the injury. Figure 1A shows a large, well-defined hemorrhagic contusion in the isotype-matched irrelevant mAb treated animal 3 days post-trauma. Following anti-CD11d treatment, the focal area of injury at the gray-white matter interface is much smaller (Figure 1B) and there appears to be less hemorrhagic than in the isotype control rats.
Figure 1.

Hematoxylin and eosin (H&E) stained paraffin sections of cortical contusions at 3 days post-TBI in vehicle and anti-CD11d treated animals. (A) Vehicle treated animal shows a large hemorrhagic contusion at the gray-white matter interface. (B) anti-CD11d treated animal has a much more focal area of injury after TBI. The upper boundaries of the contusion area are denoted with arrowheads (10X).
Micrographs of CD68 positive cells in the cerebral cortex are shown in Figure 2. Low magnification (10x) of the overlying cerebral cortex of injured animals treated with the isotype control (A,B) or anti-CD11d mAb (C,D) are shown. There is an observable increase in CD68 immunoreactive cells after TBI treated with the isotype control antibody compared to the anti-CD11d treated animals showing a reduction in the infiltration/activation of inflammatory cells. Higher magnification (40x) of the cortical area clearly denotes this change in cellular frequency as indicated by the arrow heads.
Figure 2.

CD68 stained immunopositive cells in the medial cortex (A) TBI vehicle animal with a large number of infiltrating/activated cells (10x) (B) Higher magnification of the medial cortex of a vehicle treated TBI animal. Arrowheads point to representative CD68 immunopositive cells (40X). (C) TBI anti-CD11d mAb treated animal. There are a smaller number of CD68 immunopositive cells within the medial cortex of antibody treated animals (10X). (D) Higher magnification of anti-CD11d treated animal showing reduced number of microglia/macrophges in this vulnerable cortical area (40x).
2.3 Contusion areas and volume
Contusion area and volumes are shown in Figure 3. Repeated measures ANOVA for contusion areas was significant for group (p<0.02), bregma level (p<0.001) and group X bregma level (p<0.03). Tukey’s post hoc comparisons were significantly (p<0.05) different between isotype control and anti-CD11d mAb-treated animals at bregma level 3.3, 4.3, 5.8 and 6.8. T-test for contusion volume assessment was significantly different (p<0.05) between groups. Groups treated with anti-CD11d mAb post-TBI showed a 50% reduction in the degree of contusional injury compared to isotype control TBI animals. This pattern of damage was present throughout the majority of areas assessed. It appears that blocking the CD11d portion of the integrin provides protection after brain trauma in reducing contusion injury.
Figure 3.
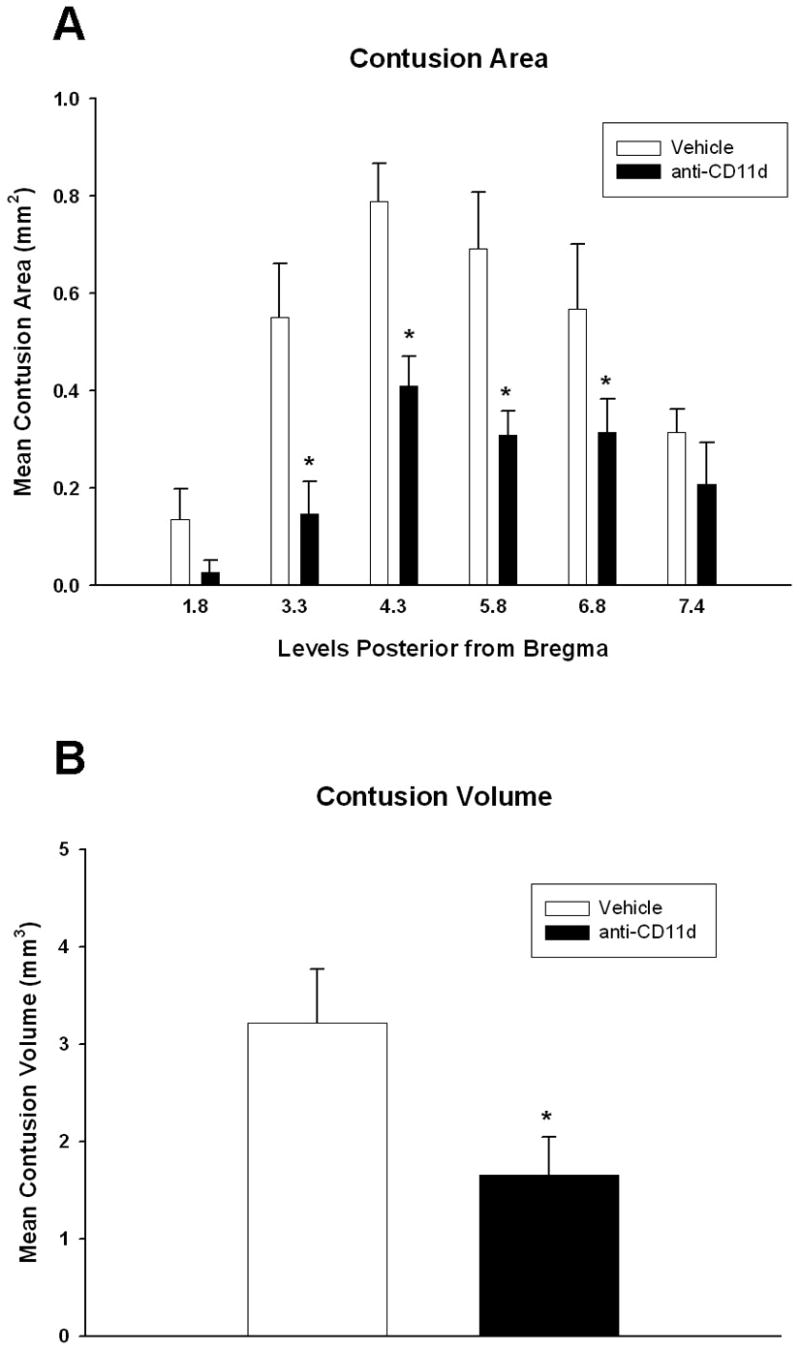
Bar graph of mean contusion areas and volume +SEM. Repeated measures ANOVA was significant (p<0.05) for group, bregma level and group x level. (A) Anti-CD11d treatment significantly reduced contusion area at bregma level −3.3, −4.3, −5.8 and −6.8 compared to vehicle treated animals. (*p<0.05). (B) Animals treated with the anti-CD11d antibody had smaller contusion volumes compared to vehicle treated animals. (*p<0.05)
2.4 CD68 cell counts
CD68 positive cells were quantitatively counted using a non-biased stereological approach in the medial and lateral cortex (Figure 4) and hippocampus (Figure 5) at posterior bregma levels 3.3, 4.3, and 5.8. Total cell counts were statistically analyzed for each structure using t-tests. Animals treated with the anti-CD11d mAb were significantly (p<0.009) different from isotype control mAb-treated animals for both the medial and lateral cerebral cortex for the number of CD68 positive cells present. Anti-CD11d mAB treated animals had a 40% reduction in the number of CD68 positive cells compared to isotype-control animals. In addition, the number of CD68 positive cells in the hippocampus was significantly reduced by 45% (p<0.02) in the anti-CD11d mAb group compared to the isotype control treated group. The numbers of infiltrating and activated inflammatory cells (microglia/macrophages) within vulnerable brain regions following FP injury are reduced by the anti-CD11d blocking mAb treatment.
Figure 4.
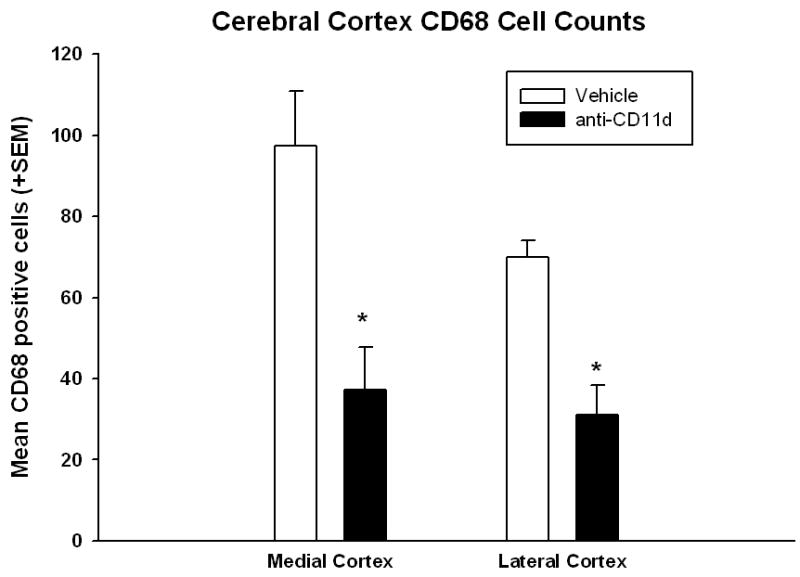
Bar graph of mean +SEM of total number of CD68 immunopositive cells in the medial and lateral cerebral cortex. Anti-CD11d treatment significantly (*p<0.05) reduced the number of activated microglia/macrophages within each cortical area ipsilateral to the injury site.
Figure 5.
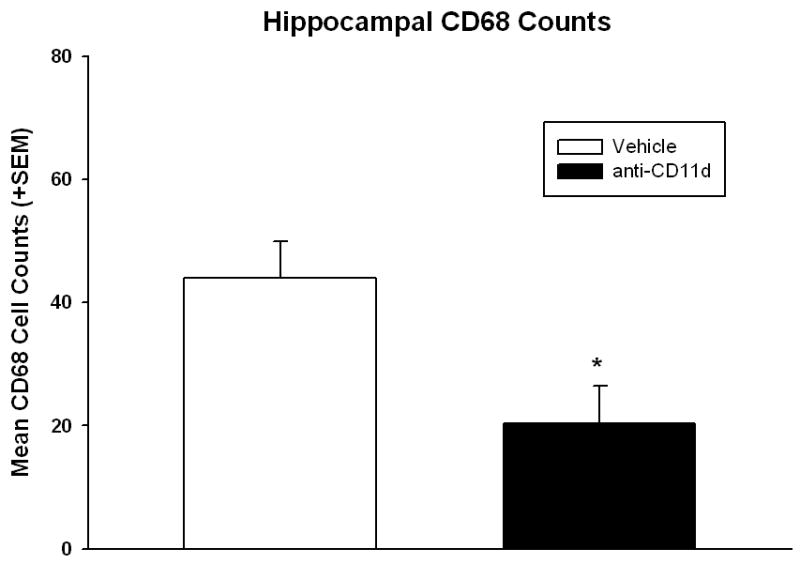
Bar graph of mean +SEM of total number of CD68 immunopositive cells in the ipsilateral hippocampus. Anti-CD11d treated animals had significantly (p<0.05) lower numbers of microglia/macrophages compared to vehicle treated animals.
A correlation analysis of infarct areas to number of CD68 positive cells was also conducted (Table 2). A significant correlation between isotype control mAb-treated animals and the number of CD68 positive cells was found for the lateral cerebral cortex (r=0.78, p<0.04). In the anti-CD11d group, a significant correlation was observed compared to the medial cerebral cortex (r=0.875, p<0.03). There appears to be a modest but significant relationship between the amount of damage present within the cerebral cortex and the number of infiltrating CD68 positive cells. Overall, these data indicate that this treatment strategy may reduce the amount of damage after TBI because of a reduction in the number of invading inflammatory cells.
Table 2.
Pearson Correlation Analysis Between Contusion Volume and CD68 Counts
| CD68 Counts | ||
|---|---|---|
| Contusion Volume | Middle Cerebral Cortex | Lateral Cerebral Cortex |
| Anti-CD11d mAb Group | r = 0.88* | r = 0.69 |
| Isotype-matched mAb Group | r = −0.35 | r = 0.78* |
p<0.05
3. Discussion
In the present study, treatment with a monoclonal antibody against the CD11d subunit of the CD11d/CD18 integrin reduced overall contusion size and limited the accumulation of CD68 immunoreactive inflammatory cells. Quantitative assessment of contusion volume showed a 50% reduction in this indicator of structural damage after traumatic injury. These observations support the hypothesis that early transient treatment with a blocking mAb specific for adhesion molecules can limit secondary inflammatory processes and improve histopathological outcome in this moderate TBI model.
Recent studies have emphasized the complexity of the inflammatory response following TBI and a rationale to therapeutics targeting this acute response for neuroprotection (Rancan et al., 2004; Scherbel et al., 1999; Sinz et al., 1999). The infiltration and accumulation of activated PMNLs and monocytes into extravascular tissues depend on endothelial interactions between cell surface PMNL/monocyte integrins and adhesion molecules such as endothelial selectins (E- and P- selectins), intercellular adhesion molecule (Carlos et al., 1997; Carlos and Harlan 1994; Hamada et al., 1996; Noti et al., 2000; Van der Vieren et al., 1995) and vascular adhesion molecule-1 (Mabon et al., 2000). The increased expression of intercellular adhesion molecule (ICAM-1) and platelet endothelial adhesion molecule-1 (PECAM-1) after CNS injury can account partly for neurotrophil adhesion to endothelial cells and trans-endothelial migration (Hamada et al., 1996; Smith 1993). Over a period ranging from minutes to hours following TBI, the self-destructive process begins and contributes to secondary or delayed processes that in turn contribute to irreversible tissue damage (Rodriguez-Paez et al., 2005). Although precise cascades remain to be elucidated, it has been recognized, both clinically and experimentally, that secondary insults serve to exacerbate the initial injury, often occurring in a delayed fashion (Csuka et al., 1999). It has previously been shown that treatment with a mAb to CD11d decreases macrophage and neutrophil infiltration into the damaged spinal cord (Mabon et al., 2000; Saville et al., 2004). It has also been demonstrated that endothelial ICAM-1 is expressed on the endothelial surface when exposed to various cytokines and mediates PMNL and endothelial adhesion (Whalen et al., 1999). In addition, the adhesion molecule, E-selectin, is also rapidly expressed on endothelial cells and neutrophils (Carlos et al., 1997). In a study by Whalen and colleagues (1998), soluble P-selectins in the cerebrospinal fluid (CSF) was shown to be increased five times that of control subjects 12 hours after TBI. Therefore, the infiltration of PMNLs and monocytes into damaged tissue is clearly a complex pathway.
In models of cerebral ischemia, factors that directly antagonize adhesion molecules have been shown to reduce reperfusion injury (Del Zoppo et al., 2000). In one study, an anti-ICAM-1 mAb was reported to reduce PMNL infiltration and brain damage after reperfusion (Zhang et al., 1995). Nevertheless, conflicting roles for ICAM-1 in the pathogenesis of ischemic and traumatic injury have also been reported, and continued investigations are required to clarify the role of these adhesion molecules and inflammatory cascades in secondary injury. In a study that supported the role of PMNL inflammatory response after head injury as a contributor of morbidity, Grady and colleagues (1999), blocked leukocyte-endothelial cell adhesion molecules by utilizing a mAb directed against a functional or nonfunctional epitope of endothelial P-selectin. In that study utilizing FP brain injury, treatment with P-selectin blockade reduced PMNL migration in the brain, preserved cholinergic immunolabeling in the medial septal nucleus, and alleviated some of the behavioral consequences of this insult.
Several laboratories have also used transgenic mice that are genetically engineered to overexpress or knockout various genes of interest in the inflammatory cascade (Tehranian et al., 2002; Whalen et al., 1999). For example, Whalen and colleagues (1999) tested mutant mice deficient in ICAM-1 to assess the importance of neutrophil accumulation on the pathogenesis of TBI. In that study, no difference between the wild-type and the ICAM-1 knockouts was demonstrated, in terms of neutrophil accumulation, behavioral scores, opr lesion volume. These investigations therefore did not support the role of ICAM-1 in the pathogenesis of TBI. However, more recently, investigations using double knock-out mice lacking both ICAM-1 and P-selectin have assessed inflammatory reactions following CNS trauma. In one study, Farooque and colleagues (1999) used double knock-out mice for ICAM-1 and P-selectin to investigate the importance of these adhesion molecules on functional outcome following spinal cord injury. Their studies demonstrated that ICAM-1/P-selectin knock-outs had better functional recovery during a 14 day observation period compared with wild-type animals. The caveat that a double target of ICAM-1 and P-selectin were necessary to improve recovery after injury is important as we consider the translation of these findings to other injury states. In terms of clinical stroke, this factor may have contributed to the failure of the Enlimomab (anti-human ICAM-1 antibody) clinical stroke trial (Enlimomab Acute Stroke Trial Investigators, 2001; Furuya et al., 2001) because of only targeting ICAM-1. An additional reason for the negative finding is that stroke patients were administered an antibody that was a murine monoclonal that may have produced a host anti-mouse response resulting in fever and infection (Becker, 2002; Furuya et al., 2001). A phase III trial using an antibody against the CD18 subunit of CD11/CD18 integrins (LeukArrest) was also conducted in acute myocardial infarction treated with direct angioplasty (Faxon et al., 2002). Although no adverse effects were observed, LeukArrest failed to reduct infarct size. This antibody would also have widespread effects on all members of the β2 integrin family, perhaps contributing to its lack of efficacy. In contrast, the present study employs a blocking mAb directed against the alpha chain, CD11d, of the integrin heterodimer CD11d/CD18, and may be more potent and specific in preventing the infiltration of invading inflammatory cells. Further investigation of the use of this type of treatment strategy in other injury conditions is warranted based on the robust infiltration of blood borne macrophages that does occur after CNS injury.
Although we did not assess the affects of treatment on hemorrhage, there appeared to be a reduction in the amount of hemorrhage within the contusion following the administration of the anti-CD11d mAb. The presence of intracerebral hemorrhage has been associated with an increase in PMNLs, microglia and ICAM-1 (Gong et al., 2000). Therefore, the ability of the anti-CD11d mAb treatment to stabilize the BBB and prevent neutrophil infiltration could be speculated in the present study. Specific blockade of anti-CD11d-mediated neutrophil infiltration also is likely. Further studies are necessary to determine if this treatment strategy has a specific effect on neutrophil influx and on BBB permeability. Also, because we initiated treatment early after TBI (30 min) it will be important in future studies to delay treatment and determine the therapeutic window for targeting this inflammatory process. Finally, the determination of whether this reduction in contusion volume translates into a functional improvement is also warranted. Clinically relevant outcome measures are important as we consider translating this treatment paradigm into patients with acute traumatic brain injury.
4. Experimental Procedure
4.1 Animals
Male Sprague-Dawley rats (N=7 per group) weighing 280 to 340 grams obtained from Charles River breeders were used for all experiments. Animal care was in accordance with the guidelines set by the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the University of Miami Animal Care and Use Committee, and all animal protocols were approved by the committee prior to study initiation. All animals were kept at a constant temperature (23–25° C) in an air-conditioned room for at least seven days before the study, and exposed to a 12 hr light-dark cycle. Rats were allowed free access to water, but food was withheld overnight before brain injury.
4.2 Traumatic brain injury (TBI)
Anesthesia was induced using 3.0 % halothane in a gas mixture of 70% nitrous oxide and a balance of oxygen to achieve deep sedation for surgical preparation for parasaggital fluid percussion brain injury as previously described (Dietrich et al., 1994). Briefly, a craniotomy (4.8 mm) was performed at 3.8 mm posterior to bregma, 2.5 mm lateral to the midline (Paxinos and Watson, 1982). A plastic injury tube was placed over the exposed dura and bonded by adhesive. Dental acrylic was used to affix the injury tube to the skull. The incision was closed and the animals were allowed to recover before returning to their home cages.
After fasting overnight, animals were anesthetized on a mixture of 70% nitrous oxide, 0.5 to 1.5% halothane with a balance of oxygen and endotracheally intubated and mechanically ventilated with a Harvard Rodent Ventilator. Pancuronium bromide (0.5ml) was administrated intravenously every hour during the surgical procedure to facilitate mechanical ventilation. The femoral artery and vein were cannulated with PE-50 tubing for drug administration, blood sampling for arterial blood gas determination and continuous blood pressure monitoring. Ane sthetic concentrations and ventilatory settings were adjusted to maintain normal values of all physiological variables (MAP =(mean arterial pressure), pH, PO2, and PCO2). Rectal and temporalis muscle thermistor probes maintained normothermic temperature using self-adjusting feedback external warming lamps. Arterial blood gases were measured 15 minutes before and 15 minutes and one hour after TBI. A FP device was used to induce moderate parasagittal FP injury (1.8 to 2.2atm) as previously described (Dietrich et al., 1994; Dixon et al., 1987; Suzuki et al., 2004).
4.3 Experimental Groups
The rats were randomized and divided into two groups. Group 1 control rats received an isotype-matched irrelevant monoclonal antibody (mAb) (1B7, 1 mg/kg, gift of the ICOS Corporation Bothell, WA/Eli Lilly Indianapolis, IN), and a second group received the anti-CD11d mAb (1.0 mg/kg; gift of the ICOS Corporation/Eli Lilly). We utilized a dose of CD11d mAb that has previously been reported to improve outcome in a SCI model (Gris et al., 2004; Saville et al., 2004). Intravenous treatment was initiated 30 minutes after TBI. Additional treatments were given at 24 and 48 hours after injury.
4.4 Histopathology/Immunocytochemistry
At three days post-TBI, rats were anesthetized and perfusion-fixed transcardially with isotonic saline at a pressure of 100–120 mmHg for 15 seconds and followed by fixative (4% paraformaldehyde in a 0.1 M phosphate buffer, pH 7.4) for 25 minutes. Following perfusion, brains were removed and immersed in fixative at 4° C for three days. Brains were then blocked and embedded in paraffin. Tissue sections 10 um thick were taken at 300 um intervals.
Sections were stained with hematoxylin and eosin (H&E) for histopathological assessment of contusion areas and volume. Coronal sections at various levels (0.8, 1.8, 3.3, 4.3, 5.8, 6.8, 7.3 mm posterior to bregma) were used for contusion area and volume measurements (Zilles, 1985). Volumes were determined by tracing the area of cortical contusion at a power of 2.5x using the Neurolucida software program (Microbrightfield Corp.) by an investigator (AU) blinded to the experimental groups. The cortical contusion boundaries were well demarcated in H&E stained sections. The contused area consisted of pyknotic neurons, reactive astrocytes and an area of shearing at the gray/white matter interface between the cortex and external capsule. Tissue appeared hemorrhagic in some areas and the extracellular space was edematous relative to regions outside the contused region. Areas were calculated and contusion volumes were computed by using numerical integration of successive areas.
For immunocytochemical studies alternate sections representing levels anterior and posterior from the epicenter (3.3, 4.3, 5.8 mm posterior to bregma) were immunostained for CD68, a marker of microglia and macrophages. Briefly, sections (10 um thick) were deparaffinized, rehydrated and incubated overnight at 4° C with CD68 (Serotec). After washing in 0.1 M PBS pH.7.4 with 0.4% Triton, secondary anti-mouse IgG antibody was applied for 90 minutes at room temperature. After further washing, ABC Elite was applied for 90 minutes. Slides were rinsed with PBS followed by Acetate-Imidasole buffer, (pH 7.2) and then reacted with NiDAB (2.5% Nickel Ammonium Sulfate Acetate-Imidisole Buffer, 0.05% DAB, 0.001% H2O2). Slides were then dehydrated, cleared and cover slipped for analysis.
The number of CD68 positive cells were quantified by an observer (LD) blinded to the experimental groups as previously described (Suzuki et al., 2004). The analysis was performed at levels 3.3, 4.3 and 5.8mm posterior to bregma. At each level, three regions were demarcated for analysis: medial cortex, lateral cortex and hippocampus. The medial cortex consisted of the cortical region beginning immediately above the contusion to the cortical region that parallels the ends of the dentate gyrus of the hippocampus. The mean areas of the medial cortex contours were 2.22±0.44, 1.87±0.17 and 1.50±0.18 mm2 at levels 3.3, 4.3 and 5.8 mm posterior from bregma, respectively. The lateral cortex consisted of the cortical region parallel to the contusion extending to the rhinal fissure. The mean areas of the lateral cortex contours were 5.96±0.93, 3.78±0.40 and 3.15±0.37 mm2 at levels 3.3, 4.3 and 5.8 mm posterior from bregma, respectively. The hippocampus consisted of the entire hippocampus, at level 5.8, the lower portion of the hippocampus was not included. The mean areas for the contours were 3.03±0.73, 3.04±0.53 and 3.98±0.45 mm2 at levels 3.3, 4.3 and 5.8 mm posterior from bregma, respectively. The contours were obtained at x5 magnification of the captured image and cell counts were performed at x68 magnification using oil immersion. CD68 positive cells were identified by their darkened brown color and activated appearance. The number of CD68 positive cells presented, are the averaged counts of two sections per each level. Stereological counts were performed using StereoInvestigator software (MicroBrightField, Inc., Williston, VT). For each contour region, 15 randomly positioned counting sites with a 75um x 75um counting frame were generated by the software.
4.5 Statistical analysis
Histopathological/immunocytochemical data were expressed as mean ± SEM. Group differences in contusion area and volume measurements were assessed by using repeated measures analysis of variance and t-test, respectively. Total CD68 cell counts were analyzed with t-tests. Pearson product moment correlations were performed between contusion volume data and cortical CD68 counts. Physiological data were compared using repeated measures ANOVA. Tukey’s pairwise comparison was used for post-hoc analysis where appropriate. Differences were considered significant at p<0.05.
Table 1.
Physiological variables
| TBI-isotype control
(n=8) |
TBI-anti-CD11d mAb
(n=7) |
|
|---|---|---|
| Pre-trauma | ||
| pCO2 | 39.34 ± 0.68 | 41.32 ± 0.80 |
| pO2 | 138.60 ± 8.93 | 137.55 ± 7.35 |
| pH | 7.48 ±0.01 | 7.48 ± 0.01 |
| MAP | 119.59 ± 3.85 | 125.9 ± 3.94 |
| Body Temp | 37.0 ± 0.06 | 37.0 ± 0.07 |
| Head Temp | 36.7 ± 0.03 | 36.8 ± 0.05 |
| Post-trauma | ||
| pCO2 | 39.47 ± 0.71 | 40.87 ± 0.89 |
| pO2 | 126.11 ± 3.50 | 141.65 ± 4.32 |
| pH | 7.46 ± 0.01 | 7.45 ± 0.01 |
| MAP | 117.65 ±2.81 | 124.13 ± 3.59 |
| Body Temp | 36.9 ± 0.06 | 36.9 ± 0.06 |
| Head Temp | 36.8 ± 0.05 | 36.7 ± 0.05 |
Values are expressed as mean ±SEM. MAP = mean arterial pressure.
Acknowledgments
This study was supported in part by grants NS030291, NS042133, NS056072 from NIH. We wish to thank Jeremy Lytle for editorial assistance and manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–34. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Becker KJ. Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin. 2002;18(Suppl 2):s18–s22. doi: 10.1185/030079902125000688. [DOI] [PubMed] [Google Scholar]
- Bethea JR, Dietrich WD. Targeting the host inflammatory response in traumatic spinal cord injury. Curr Opin Neurol. 2002;15:355–360. doi: 10.1097/00019052-200206000-00021. [DOI] [PubMed] [Google Scholar]
- Bevilacqua MP. Endothelial-leukocyte adhesion molecules. Annu Rev Immunol. 1993;11:767–804. doi: 10.1146/annurev.iy.11.040193.004003. [DOI] [PubMed] [Google Scholar]
- Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A, Kossmann T, Morganti-Kossmann MC. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol. 2007;204:220–233. doi: 10.1016/j.expneurol.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol. 1997;61:279–285. doi: 10.1002/jlb.61.3.279. [DOI] [PubMed] [Google Scholar]
- Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- Clark RS, Carlos TM, Schiding JK, Bree M, Fireman LA, Dekosky ST, Kochanek PM. Antibodies against Mac-1 attenuate neutrophil accumulation after traumatic brain injury in rats. J Neurotrauma. 1996;13:333–341. doi: 10.1089/neu.1996.13.333. [DOI] [PubMed] [Google Scholar]
- Csuka E, Morganti-Kossman MC, Lenzlinger PM, Joller H, Trentz O, Kossmann T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-alpha, TGF-beta1 and blood-brain barrier function. J Neuroimmunol. 1999;101:211–221. doi: 10.1016/s0165-5728(99)00148-4. [DOI] [PubMed] [Google Scholar]
- Del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: Putative role for cytokines, adhesion molecules and iNOS in brain responses to ischemia. Brain Path. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WD, Alonso O, Busto R, Globus MY-T, Ginsberg MD. Post-traumatic brain hypothermia reduces histopathological damage following concussive brain injury in the rat. Acta Neuropathol. 1994;87:250–258. doi: 10.1007/BF00296740. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:101–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Enlimomab, Acute Stroke Trial Investigators. Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. 2001;57:1428–1434. doi: 10.1212/wnl.57.8.1428. [DOI] [PubMed] [Google Scholar]
- Farooque M, Isaksson J, Olsson Y. Improved recovery after spinal cord trauma in ICAM-1 and P-selectin knockout mice. Neuroreport. 1999;10:131–134. doi: 10.1097/00001756-199901180-00024. [DOI] [PubMed] [Google Scholar]
- Faxon DP, Gibbons RJ, Chronos NAF, Gurbel PA, Sheehan F for the HALT-MI Investigators. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: The results of the HALT-MI Study. J Amer Coll Card. 2002;40:1199–1204. doi: 10.1016/s0735-1097(02)02136-8. [DOI] [PubMed] [Google Scholar]
- Furuya K, Takeda H, Azhar S, McCarron RM, Chen Y, Ruetzler CA, Wolcott KM, DeGraba TJ, Rothlein R, Hugli TE, del Zoppo GJ, Hallenbeck JM. Examination of several potential mechanisms for the negative outcome in a clinical stroke trial of Enlimomab, a murine anti-human intercellular adhesion molecule-1 antibody. Stroke. 2001;32:2665–2674. doi: 10.1161/hs3211.098535. [DOI] [PubMed] [Google Scholar]
- Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000;871:57–65. doi: 10.1016/s0006-8993(00)02427-6. [DOI] [PubMed] [Google Scholar]
- Grady MS, Cody RF, Jr, Maris DO, McCall TD, Seckin H, Sharar SR, Winn HR. P-selectin blockade following fluid-percussion injury: behavioral immunochemical sequelae. J Neurotrauma. 1999;16:13–25. doi: 10.1089/neu.1999.16.13. [DOI] [PubMed] [Google Scholar]
- Grayson MH, Van der Vieren M, Sterbinsky SA, Gallantin WM, Hoffman P, Staunton D, Bochner BS. Alphadbeta2 integrin is a ligand for vascular cell adhesion molecule-1. Int Arch Allergy Immunol. 1999;118:263–264. doi: 10.1159/000024094. [DOI] [PubMed] [Google Scholar]
- Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC. Transient blockage of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neuroscience. 2004;24:4043–4051. doi: 10.1523/JNEUROSCI.5343-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada Y, Ikata T, Katoh S, Nakauchi K, Niwa M, Kawai Y, Fukuzawa K. Involvement of an intercellular adhesion molecule 1-dependent pathway in the pathogenesis of secondary changes after spinal cord injury in rats. J Neurochem. 1996;66:1525–1531. doi: 10.1046/j.1471-4159.1996.66041525.x. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Rothwell NJ. Cytokines and neurodegeneration. In: Rothwell NJ, Loddick SA, editors. Immune and Inflammatory Responses in the Nervous System. Oxford University Press; Oxford: 2002. pp. 90–105. [Google Scholar]
- Mabon PJ, Weaver LC, Dekaban GA. Inhibition of monocyte/macrophage migration to a spinal cord injury site by an antibody to the Integrin alphaD: a potential new anti-inflammatory treatment. Exp Neurol. 2000;166:52–64. doi: 10.1006/exnr.2000.7488. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Saatman KE, Raghupathi R, Graham DI, Smith DH, Lee VM, Trojanowski JQ. The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenic mechanisms. Neuropathol Appl Neurobiol. 1998;24:251–267. doi: 10.1046/j.1365-2990.1998.00121.x. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165–177. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- Muizelaar JP, Marmarou A, Ward JD, Kontos HA, Choi SC, Becker DP, Gruemer H, Young HF. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731–739. doi: 10.3171/jns.1991.75.5.0731. [DOI] [PubMed] [Google Scholar]
- Neish AS, Read MA, Thanos D, Pine R, Maniatis T, Collins T. Endothelial interferon regulatory factor 1 cooperates with NF-kappa B as a transcriptional activator of vascular cell adhesion molecule 1. Mol Cell Biol. 1995;15:2558–2569. doi: 10.1128/mcb.15.5.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HX, O’Barr TJ, Anderson AJ. Polymorphonuclear leukocytes promote neurotoxicity through release of matrix-metalloproteinases, reactive oxygen species, and TNF-alpha. J Neurochem. 2007;102:900–912. doi: 10.1111/j.1471-4159.2007.04643.x. [DOI] [PubMed] [Google Scholar]
- Noti Jd, Johnson AK, Dillon JD. Structural and functional characterization of the leukocyte integrin gene CD11d. J Biol Chem. 2000;275:8959–8969. doi: 10.1074/jbc.275.12.8959. [DOI] [PubMed] [Google Scholar]
- Otto VI, Stahel PF, Rancan M, Rariya K, Shohami E, Yatsiv I, Eugster HP, Kossmann T, Trentz O, Morganti-Kossmann MC. Regulation of chemokines and chemokines receptors after experimental closed head injury. Neuroreport. 2001;12:2059–2064. doi: 10.1097/00001756-200107030-00053. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York: 1982. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancan M, Bye N, Otto VI, Trentz O, Kossmann T, Frentzel S, Morganti-Kossmann MC. The chemokines fractalkine in patients with severe traumatic brain injury and a mouse model of closed head injury. J Cereb Blood Flow Metab. 2004;24:1110–1118. doi: 10.1097/01.WCB.0000133470.91843.72. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Paez AC, Brunschwig JP, Bramlett HM. Light and electron microscopic assessment of progressive atrophy following moderate traumatic brain injury in the rat. Acta Neuropathol. 2005;109:603–616. doi: 10.1007/s00401-005-1010-z. [DOI] [PubMed] [Google Scholar]
- Saville LR, Pospisil CH, Mawhinney LA, Bao F, Simedrea FC, Peters AA, O’Connell PJ, Weaver LC, Dekaban GA. A monoclonal antibody CD11d reduces the inflammatory infiltrate into the injured spinal cord: A potential neuroprotective treatment. J NeuroImmunology. 2004;156:42–57. doi: 10.1016/j.jneuroim.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. PNAS. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz M, Cinatl J, Schädel-Höpfner M, Windolf J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med Res Rev. 2007;27:401–416. doi: 10.1002/med.20064. [DOI] [PubMed] [Google Scholar]
- Shanley TP, Warner RL, Crouch LD, Dietsch GN, Clark DL, O’Brien MM, Gallatin WM, Ward PA. Requirements for αd in IgG immune complex-induced rat lung injury. J Immunol. 1998;160:1014–1020. [PubMed] [Google Scholar]
- Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–619. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- Shohami E, Bass R, Wallach D, Yamin A, Gallily R. Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16:378–384. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- Sinz EH, Kochanek PM, Dixon CE, Clark RSB, Carcillo JA, Schiding JK, Chen M, Wisniewski SR, Carlos TM, Williams D, DeKosky ST, Watkins SC, Marion DW, Biliar TR. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J Clin Invest. 1999;104:647–656. doi: 10.1172/JCI6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CW. Leukocyte-endothelial cell interactions. Semin Hematol. 1993;30:45–53. [PubMed] [Google Scholar]
- Stahel PF, Shohami E, Younis FM, Kariya K, Otto VI, Lenzlinger PM, Grosjean MB, Eugster HP, Trentz O, Kossmann T, Morganti-Kossmann MC. Experimental closed head injury: analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for proinflammatory cytokines. J Cereb Blood Flow Metab. 2000;20:369–380. doi: 10.1097/00004647-200002000-00019. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Bramlett HM, Ruenes G, Dietrich WD. The effects of early post-traumatic hyperthermia in female and ovariectomized rats. J Neurotrauma. 2004;21:842–853. doi: 10.1089/0897715041526186. [DOI] [PubMed] [Google Scholar]
- Tehranian R, Andell-Jonsson S, Beni SM, Yatsiv I, Shohami E, Bartfai T, Lundkvist J, Iverfeldt K. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J Neurotrauma. 2002;19:939–951. doi: 10.1089/089771502320317096. [DOI] [PubMed] [Google Scholar]
- Toulmond S, Rothwell NJ. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995;671:261–266. doi: 10.1016/0006-8993(94)01343-g. [DOI] [PubMed] [Google Scholar]
- Van der Vieren M, Crowe DT, Hoekstra D, Vazeux R, Hoffman PA, Grayson MH, Bochner BS, Gallatin WM, Staunton DE. The leukocyte Integrin αDβ2 binds VCAM-1: evidence for a binding interface between I domain and VCAM-1. J Immunol. 1999;163:1984–1990. [PubMed] [Google Scholar]
- Van der Vieren M, Le Trong H, Wood CL, Moore PF, St John T, Staunton DE, Gallatin WM. A novel leukointegrin, alpha d beta 2, binds preferentially to ICAM-3. Immunity. 1995;3:683–690. doi: 10.1016/1074-7613(95)90058-6. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Clark RS, Marion DW, DeKosky MS, Heineman S, Schiding JK, Memarzadeh F, Dixon CE, Kochanek PM. The relationship between brain temperature and neutrophil accumulation after traumatic brain injury in rats. Acta Neurochir Suppl. 1997;70:260–261. doi: 10.1007/978-3-7091-6837-0_80. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Dixon CE, Schiding JK, Clark RSB, Baum E, Yan HQ, Marion DW, Kochanek PM. Effect of traumatic brain injury in mice deficient in intercellular adhesion molecule-1: Assessment of histopathologic and functional outcome. J Neurotrauma. 1999;16:299–309. doi: 10.1089/neu.1999.16.299. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Kochanek PM, Wisniewski SR, Bell MJ, Carcillo JA, Clark RS, Dekosky ST, Adelson PD. Soluble adhesion molecules in CSF are increased in children with severe head injury. J Neurotrauma. 15:777–787. doi: 10.1089/neu.1998.15.777. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Jiang N, Tang WX, Prostak J, Manning AM, Anderson DC. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke. 1995;26:1438–1442. doi: 10.1161/01.str.26.8.1438. [DOI] [PubMed] [Google Scholar]
- Zilles L. The cortex of the rat A stereotaxic Atlas. Springer; New York: 1985. [Google Scholar]
- Zausinger S, Baethmann A, Schmid-Elsaesser R. Anesthetic methods in rats determine outcome after experimental focal cerebral ischemia: mechanical ventilation is required to obtain controlled experimental conditions. Brain Res Protoc. 2002;9:112–121. doi: 10.1016/s1385-299x(02)00138-1. [DOI] [PubMed] [Google Scholar]