

Abstract
With increasing age, the competence of the immune system to fight infections and tumors declines. Age-dependent changes have been mostly described for human CD8 T cells, raising the question of whether the response patterns for CD4 T cells are different. Gene expression arrays of memory CD4 T cells yielded a similar age-induced fingerprint as has been described for CD8 T cells. In cross-sectional studies, the phenotypic changes were not qualitatively different for CD4 and CD8 T cells, but occurred much more frequently in CD8 T cells. Homeostatic stability partially explained this lesser age sensitivity of CD4 T cells. With aging, naïve and central memory CD8 T cells were lost at the expense of phenotypically distinct CD8 effector T cells, while effector CD4 T cells did not accumulate. However, phenotypic shifts on central memory T cells were also more pronounced in CD8 T cells. This distinct stability in cell surface marker expression can be reproduced in vitro. The data show that CD8 T cells are age sensitive by at least two partially independent mechanisms: fragile homeostatic control and gene expression instability in a large set of regulatory cell surface molecules.
Keywords: immunosenescence, aging, T-cell subset, T-cell homeostasis, CD4, CD8, killer immunoglobulin-like receptors, CD85
Introduction
Failure of adaptive immunity with age is a major cause for morbidity and mortality in the elderly [1; 2]. As a highly dynamic organ, the immune system is in constant turnover, even in the absence of infections or obvious challenges with exogenous antigens [3]. Naïve T cells have a half-life of 6 to 12 months and memory T cells of 15 to 45 days [4; 5]. Thymic production of new T cells dwindles with age and does not meet the replenishment demand during adulthood [6; 7; 8]. After the ages of 40 to 50 years old, virtually the entire T-cell supply is generated from existing naïve and memory T cells [9]. In this setting, insufficient homeostatic mechanisms may lead to a progressive loss of naïve and memory T cells and contraction of T-cell receptor diversity [10; 11; 12; 13; 14]. In addition, the replicative stress associated with continuous turnover can induce cellular senescence and lead to phenotypic changes that impinge on the competence of the adaptive immune system [15; 16; 17; 18].
Both mechanisms contribute to failure to respond to new antigenic challenges, poor vaccine responses [19; 20; 21; 22] and increased morbidity with newly arising infections, such as is seen with antigenic shift or drift of the influenza virus [23; 24]. Moreover, memory T-cell responses to some persisting viruses wane—a prime example is the increased incidence of herpes zoster with age [25]. Epidemiological evidence suggests that signs of decreased immune competence first occur after the age of 50 and accelerate after the ages of 65 to 70 [20; 23; 25]. It is currently unclear which functional and phenotypic changes in the immune system occur at what ages and, in particular, whether different cell types are affected to distinct extents, possibly providing a model for the patterns of age-dependent susceptibilities for different viral infections.
Circumstantial evidence suggests that CD4 and CD8 T cells behave differently in response to aging. Oligoclonal expansions in the CD8 T-cell compartment can be readily detected with age and appear to be induced by chronic persisting viruses, in particular CMV, but also arise from uneven homeostatic proliferation [12; 15; 26; 27]. In contrast, oligoclonal expansions within the CD4 compartment are rare and preferentially found in patients with autoimmune diseases [28]; they do not reach the clonal size seen with CD8 T cells [18]. Even at the age of 65, decades after the involution of the thymus, both naïve and memory CD4 T cells are highly diverse and show no contraction in T-cell receptor diversity compared to young adults [10]. Also, the classical phenotypic change of CD28 loss, frequently encountered in CD8 T cells with age [17], is only inconsistently seen for CD4 T cells [11]. However, the molecular mechanisms regulating CD28 expression and loss appear to be identical in both T-cell subsets [29; 30], suggesting a fundamental difference in CD4 and CD8 T cells in response to aging, possibly in addition to distinct cell-specific transcriptional regulation. The objective of this study was to compare CD4 and CD8 T cells and to determine whether differences in their phenotypic response pattern to aging are explained by differences in T-cell subset homeostasis or whether CD4 and CD8 T cells are intrinsically different in controlling gene expression.
Materials and Methods
Study population
Peripheral blood was obtained from 140 individuals aged 20–90 years and immediately processed. The study cohort included 68 individuals age 20 to 39 years, 31 age 40 to 59, and 41 age 60 to 90 years. Exclusion criteria included the presence or a history of cancer, uncontrolled hypertension, diabetes mellitus, any chronic inflammatory or autoimmune disease, or any acute disease. Appropriate written informed consent was obtained, and the study was approved by the Emory Institutional Review Board.
Gene expression microarray studies
CD4 T cells were negatively enriched with human CD4+ T-cell enrichment cocktail (RosetteSep; StemCell Technologies, Vancouver, Canada) from PBMC of five 20- to 30- and five 70- to 75-year-old healthy volunteers. CD45RA− CD4+ memory T cells were isolated by depletion of CD45RA+ subsets with anti-CD45RA magnetic beads (Miltenyi Biotec, Auburn, CA). Purity was between 95–98%. Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Valencia, CA), amplified, and labeled using a modification of the technique described by Baugh et al [31]. A first round of in vitro transcription (IVT) was followed by a second round of reverse transcription, second strand synthesis, and IVT. aRNA was hybridized with Affymetrix U133A GeneChips by the Institute of System Biology (Seattle, WA). Gene expression signal was summarized by robust multi-array average RMA [32]. Genes that showed a 1.25-fold difference between young and old samples were identified.
In a second microarray series, CD4+ CD28+ and CD4+ CD28− T cells were purified from three donors by cell sorting using FACS vantage (BD Biosciences, San Jose, CA). Cells were expanded by stimulation with anti-CD3 (OKT3; Ortho Diagnostics, Rochester, NY) immobilized at 1µg/ml in IL-2-containing medium. RNA was extracted 2 weeks after stimulation. This time point was chosen because gene expression in T cells cultured under these conditions is stable and kinetic changes induced by the stimulation are no longer observed (data not shown). Labeled aRNA was hybridized with Affymetrix Hu-95Av2 GeneChips (Mayo Cancer Microarray Core Facility, Mayo Foundation; Rochester, MN). Data were analyzed with Microarray Analysis Software version 5.0 (Affymetrix). A gene was called differentially expressed if comparisons for all three individuals yielded a consistent change call, or if two comparisons yielded consistent change calls and the third yielded a "no change" call.
Immunophenotyping and flow cytometry analysis
To confirm age-dependent differential expression of cell surface markers identified in the gene arrays, PBMCs were stained with the following monoclonal antibodies in 5-color panels: FITC-conjugated anti-CD158b/j (GLI83; CH-L), PE-anti-CD85j (HP-F1), PerCP-Cy5.5-anti-CD28 (L293), APC-anti-CD8 (RPA-T8), APC-Cy7-anti-CD3 (SK7); FITC-anti-CD57 (HNK-1), PE-anti-CD26 (M-A261), PerCP-anti-CD4 (L200), APC-anti-CD3 (HIT3a); FITC-conjugated anti-HLA-DR (G46-6), PE-anti-CD3 (HIT3a), PerCP-anti-CD4 (L200), APC-anti-CD45R0 (4CHL1), PE-Cy7-anti-CD69 (FN 50). FITC- or APC-anti-CD45RA (RA5H9, HI100), PE- or APC-anti-CCR7 (150503), PerCP-anti-CD8 (SK1) were used to define the functional subset distribution.
To determine the frequency of cytomegalovirus (CMV)-specific CD8 T cells in different age groups, PBMCs from HLA-A2 individuals were stained with APC-conjugated HLA-A*0201/CMV pp65495–504 (NLVPMVATV) pentamer (Proimmune Inc, Springfield, VA) at room temperature for 10 min. Following washing, the cells were incubated with PE-anti-CD85j, PerCP-, or PE-Cy7-anti-CD8; FITC- or PerCP-Cy5.5-anti-CD28; and APC-Cy7-anti-CD3 antibodies for 20 min.
All samples were acquired with FACSort or LSRII (BD Biosciences), and data were analyzed by using CellQuest, FACS DIVA software (both BD Biosciences), or FlowJo (Tree Star, Inc., Ashland, OR). All antibodies were from BD Biosciences, except PE-, APC-anti-CCR7 (R&D Systems, Minneapolis, MN, USA), and anti-CD85j (Beckman Coulter, Fullerton, CA, USA).
In vitro cultures
CD28 positive cells were sorted from PBMC with biotin-labeled anti-CD28 antibody (BD Biosciences), and anti-biotin labeled microbeads (Miltenyi). Isolated cells were labeled with CFSE and stimulated with anti-CD3 (OKT3, 30ng/ml) in the presence of irradiated EBV transformed cells, rhIl-2 (50U/ml) and rhIl-15 (100ng/ml). Cultures were split every 4 days. Cells were analyzed at 6 to 8 day intervals for the expression of CD85j, CD28 and HLA-DR on CD4 and CD8 T cells that had undergone the same number of division as determined by CSFE dilution.
Statistical analysis
Results are expressed as medians and 25th/75th percentiles. Groups were compared using ANOVA or non-parametric Mann Whitney U test. p values <0.05 were considered statistically significant.
Results
Differential loss of CD28 in CD8 versus CD4 T cells
Loss of CD28 expression, the dominant phenotypic change of T cells with aging, is seen much more frequently in CD8 than in CD4 T cells. Figure 1shows a cross-sectional study of CD28 expression in different age groups. In the vast majority of individuals, lack of CD28 is a rare event in CD4 T cells, irrespective of age. There was a small increase in CD4+ CD28− T-cell frequencies (p<0.001) and increasing variance in healthy individuals older than 60 years; however, even in this age group only a median of 7.9 % of CD4 T cells lost CD28 expression, and individuals with more than 20% CD4+ CD28− T cells were the rare exception. In contrast, 21% of CD8 T cells were already negative for CD28 expression in individuals between the age of 20 and 39 years. The frequency increased to 25.5% in 40- to 59-year-old healthy individuals (p<0.001). After that age, CD28 loss accelerated to a median frequency of 53.9 % in the 60–90 year cohort (p<0.001). Again the variance increased with age, indicating higher heterogeneity in the older population.
Figure 1. Influence of age on CD28 loss within CD4+ and CD8+ T cells.
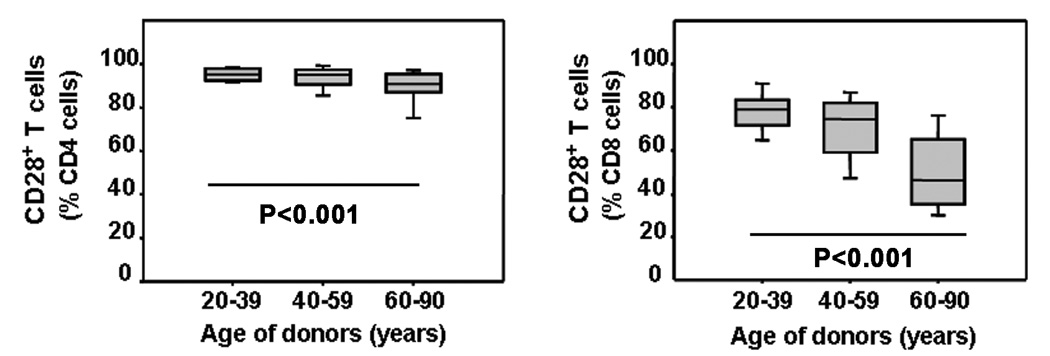
The frequency of CD28 loss in CD4 (left panel) and CD8 (right panel) T-cell subpopulations was determined by FACS in healthy individuals of different ages. Results are shown as box blots with medians, 25th and 75th percentiles as boxes and 10th and 90th percentile as whiskers for different age strata. Although decline was significant for both CD4 (p<0.001) and CD8 T cells (p<0.001), loss was much more dramatic and earlier in life in the CD8 population and was already significant for middle-aged individuals (p<0.001).
Age-dependent changes in the transcriptional profile of CD4 memory T cells
Regulation of CD28 may be cell specific, and CD8 T cells may lose CD28 more easily. Indeed, the DNA-binding proteins controlling the initiation of CD28 transcription differ in composition between CD4 and CD8 T cells [30]. However, the basic mechanism of CD28 repression is the same in both T-cell subsets, suggesting that factors not related to CD28 transcription play a role [29]. The CD4 memory T cells may be a more stable cell type, with lesser susceptibility to age-dependent phenotypic and functional changes. To determine whether CD4 T cells principally undergo the same changes in gene expression with age as CD8 T cells but do so at a substantially reduced or delayed rate, we performed gene expression arrays on (a) CD45RA−CD4+ cells purified from PBMC of five 20- to 30-year-old and five 70- to 75-year-old healthy donors.and (b) CD28+ CD4+ and CD28- CD4+ T cells from the same donor and compared them to published gene profiles for CD8 T cells [33]; The gene expression profiles in young and old memory CD4 T cells highly correlated indicating that age-dependent changes in CD4 T-cell transcription were small or only involved a minor subpopulation. Because even small changes in gene expression or emergence of a low-frequent subpopulation could have physiological consequences, we used a low cut-off of a 1.25-fold difference in expression levels between the two age groups to screen for genes that warrant further studies. 536 genes were identified, including the cell surface molecules listed in Table 1 Several of the genes over-expressed in the elderly mapped to the HLA region. CD28 was among the few genes that were less expressed in elderly memory CD4 cells. Hypothesizing that the differences were small because only a minor subset of CD4 T cells is affected by aging and these senescent cells are characterized by the loss of CD28, in analogy to CD8+ CD28− T cells, we compared the gene expression profiles of CD4+ CD28− to CD4+ CD28+ T cells. In general, identified differences in this subset were of larger magnitude than the age-dependent differences observed in the global memory CD4 T-cell pool. Cell surface molecules identified as differentially expressed partially overlapped with the genes identified in the global memory cell populations (Table 1). MHC class II genes again were among those that were overexpressed in the cells with the senescent CD4+CD28− phenotype. Also, the KLRC2 genes were underrepresented in both the elderly CD4+ memory cells and CD4+ CD28− T cells. As expected, CD27 transcripts that are known to correlate with CD28 loss in CD4 and CD8 T cells were reduced in CD4+ CD28− T cells. The most striking feature of these cells was the overexpression of several genes encoded in the leukocyte regulatory cluster, including several KIR genes.
Table 1.
Influence of age on gene expression in CD4 T cells
| Age of donor | 20–30 years | 70–75 years | 65–75 years | |
|---|---|---|---|---|
| Cell type | CD4+CD45RA− | CD4+CD45RA− | CD4+CD45RA−CD28+ | CD4+CD45RA−CD28− |
| Differentially | KLRB1 | HLA-DRA | TCRβ-chain | KIR2DL3 |
| overexpressed | CD28 | CCR4 | ICAM-2 | KIR2DL4 |
| cell surface | KLRC2 | CD26 | CD7 | KIR3DL2 |
| molecules | CCR6 | HLA-DPB1 | KLRC2 | CD70 |
| HLA-DPB5 | IL9R | HLA-DRA | ||
| HLA-DPB4 | IL7R | HLA-DQA | ||
| HLA-F | CD27 | CD74 | ||
| HLA-DPA1 | HLA-DRB1 | |||
| CCR8 | HLA-DPB1 | |||
| CD58 | HLA-DMA | |||
| IL17RB | HLA-DPA | |||
| LAIT1 | CCR5 | |||
| CD3E | HLA-DQB1 | |||
| CD2 | ||||
In comparing our results with those of Fann et al, it is evident that aging CD4 and CD8 T cells undergo similar transcriptional changes. In agreement with their findings for senescent CD8 T cells [33], we observed a significant decrease in CD28 expression in memory CD4 T cells from elderly donors. We found that expression of the costimulatory molecules CTLA4 and PD-1 did not differ with respect to age in CD4 memory cells, a finding which mirrored Fann et al’s results with respect to senescence phenotype in CD8 T cells. While CD40L is significantly downregulated in CD8+ CD28− T cells [33], we did not detect an age-dependent decline in memory CD4 cells; however, we have previously shown that CD28− CD4+ T cells lack the ability to induce CD40L [34]. Furthermore, there are many similarities in the expression of NK cell receptors on CD28− CD8 and CD4 T cells. For example, our results showed significant downregulation of KLRB1 (CD161) expression in old memory CD4 cells, as Fann et al demonstrated for CD8+ CD28− cells. Similarly, the expression of KIR2DL2, KIR2DL3, and KIR2DL4 increased in CD4+ CD28− cells, although these differences did not reach the significance found in CD8+ CD28− T cells. We did not detect any changes in expression of KLRD1, KLRG1, and KLRK1 between old and young memory CD4 cells, in contrast to the significant upregulation of these genes in CD8+ CD28− cells. Several chemokine receptors exhibited expression patterns in CD4 memory cells that are analogous to those found in CD8 cells. CCR2, CCR6, and CCR7, which are all significantly downregulated in CD8+ CD28− cells [33], decrease with age in memory CD4 T cells, with CCR6 reaching significance.
Preferential age-dependent expression of negative regulatory receptors on CD8 T cells
Candidate cell surface molecules implicated in the gene arrays were examined for their age-dependent expression in a cohort of 140 healthy individuals. One of the most striking findings of the gene array studies was the overexpression of several KIR genes in CD4+ CD28−T cells. KIR genes have allelic polymorphisms, and we selected the antibody to CD158b/j that recognizes the most frequent genetic variants KIR2DL2, KIR2DL3, and KIR2DS2 for further analysis [35]. In addition, we included CD85j, which is encoded in the leukocyte cluster near the KIR genes, in our analysis. Like most KIR genes, CD85j is a negative regulatory receptor that is irregularly expressed on T cells. Cross-sectional studies showed preferential expression of CD85j and CD158b/j in CD8 T cells, while they were infrequently found in CD4 T cells (Figure 2A). Expression of both molecules, but particularly CD85j, increased with age; the increase was already apparent in the age group of 40- to 59-year olds, but clearly accelerated after that age. Compared to the 20–39 year-old individuals, CD158b/j (p<0.001) and CD85j (p<0.001) expression was significantly more frequent in CD8 T cells from individuals older than 60 years In many of these individuals, the majority of CD8 T cells were positive for CD85j. The age-dependent increase was much less striking in CD4 T cells and did not reach significance for either molecule. However, in both T-cell subsets, the expression of these receptors was closely related to the loss of CD28. In both the CD4 and CD8 populations, the majority of CD28− T cells expressed CD85j. In contrast, CD4+CD28+ T cells expressed almost no CD85j. A small fraction of CD8+CD28+ T cells, however, was consistently found to express CD85j (Figure 2B). CD158b/j showed the same distribution pattern but was much less frequent and exceeded 10% expression in only a few individuals, even on CD28− T cells.
Figure 2. Age-dependent gain of CD57, CD85j, and CD158b/j expression in CD8+ T cells.
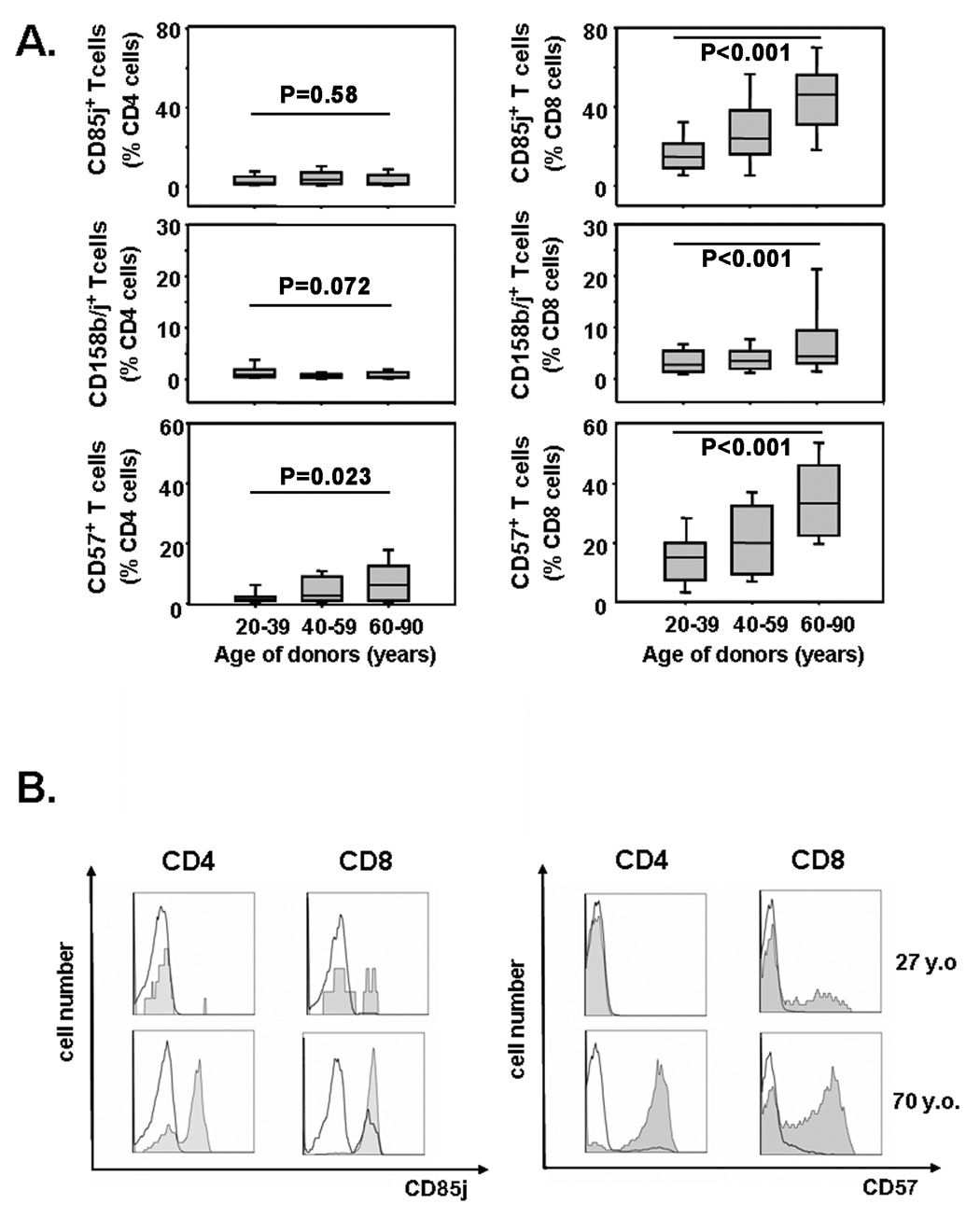
(A) Frequencies of CD85j, CD158b/j, and CD57 cells within CD4 (left panel) and CD8 (right panel) T-cells were determined by FACS. Results are shown as box plots with medians, 25th and 75th percentiles as boxes, and 10th and 90th percentiles as whiskers for healthy individuals representing different age strata. (B) In both CD4 and CD8 T cells, CD57 and CD85j were preferentially expressed on CD28− (shaded areas) and less on CD28+ (dark lines).
KIR and CD85j expression patterns resembled those of CD57, a generally accepted T-cell senescence marker [36]. Gain in CD57 expression showed a similar age dependence (Figure 2A), with a median of 5.1 % of CD4 T cells and 33.35 % of CD8 T cells being CD57-positive in 60 to 90 year olds. CD57 expression was mainly restricted to the CD28− cell population and occurred only infrequently in CD28+ cells (Figure 2B). Thus, in both CD4 and CD8 T cells, the gains in CD57, CD85j, and CD158b/j, and loss of CD28 with age were highly correlated, suggesting that the changes are qualitatively similar. However, they occurred much more frequently in CD8 T cells. Table 2 shows the slopes of the linear regression curves examining the relationship between age and frequency of cell surface expression. For each biomarker shown, the slope is significantly steeper for CD8 T cells, documenting a fundamental difference in CD4 and CD8 T cells in their responses to aging.
Table 2.
Linear regression analysis of the age dependency of cell surface molecule expression in CD4 and CD8 T cells
| CD4 T cells Δ frequency (%/year) | CD8 T cells Δ frequency (%/year) | F | P | |
|---|---|---|---|---|
| CD28 | −0.03±0.04 | −0.6±0.07 | F=36.798 | P<0.0001 |
| CD85j | 0.1±0.07 | 0.7±0.07 | F=25.75 | P<0.001 |
| CD57 | 0.2±0.07 | 0.4±0.07 | F=4.942 | P=0.03 |
| CD158b/j | −0.02±0.01 | 0.1±0.03 | F=12.483 | P=0.0005 |
| CD26 | 0.09±0.07 | −0.5±0.1 | F=12.46 | P=0.0006 |
| HLA-DR | 0.20±.03 | 0.6±0.8 | F=26.02 | P<0.0001 |
Defective T-cell homeostasis in CD8 T cells contributes to the preferential acquisition of negative regulatory receptors
Since CD28 loss is mostly seen on effector T cells, we addressed the question of whether the higher susceptibility of CD8 T cells to undergo age-dependent phenotypic changes reflected different stability in homeostatic control mechanisms. Quantification of naïve, central, and effector memory cells using the CD45RA and CCR7 markers in different age groups indeed showed considerable differences between CD4 and CD8 T cells. In both compartments, naïve T cells were reduced in elderly individuals (p<0.001); however, the loss was much more dramatic for CD8 (Figure 3B) than for CD4 T cells (Figure 3A). The functional subset distribution within the CD4 memory compartment was stable with age (Figure 3C). In contrast, the subset of central CD8 memory T cells was diminished (p=0.01), and the CD8 effector memory T cells increasingly acquired a CD45RA phenotype (p<0.001, Figure 3D) The CD4/CD8 ratio was not affected by age, contrary to the hypothesis that expansion of CD8 effector T cells outcompetes CD4 T cells.
Figure 3. Influence of age on functional T-cell subset distribution.
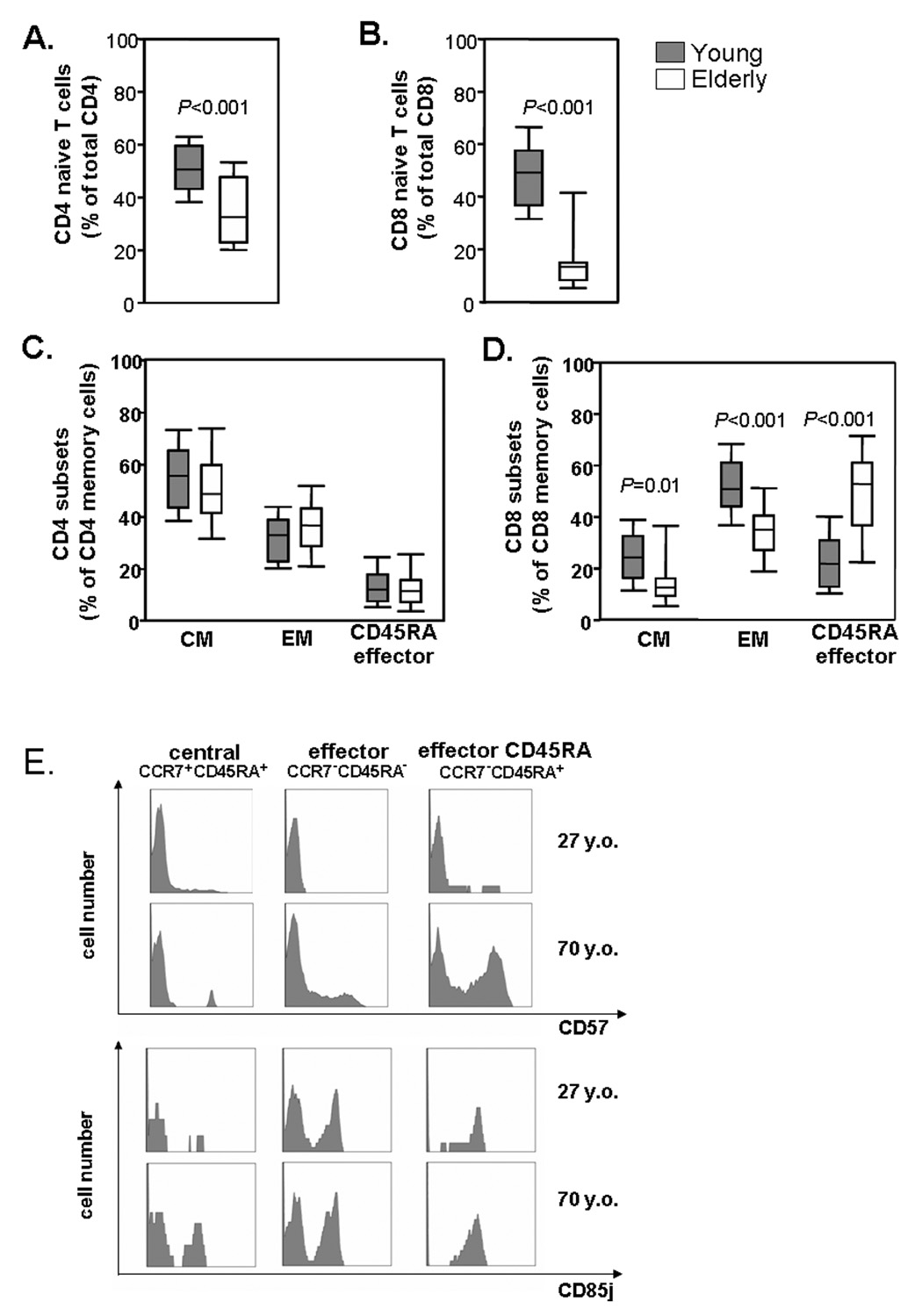
PBMCs were isolated from healthy young (n=31; 27.9± 5.5years) and elderly (n=26; 73.1± 4.2years) individuals and stained with anti-CD3, -CD4, -CD8, -CD45RA, and -CCR7 mAbs to determine frequencies of T-cell subsets. Results are shown for CD45RA+ CCR7+ naïve CD4 (A) and CD8 (B), CD45RA− CCR7+ central memory (CM), CD45RA− CCR7− effector memory (EM) and CD45RA+ CCR7-effector (CD45RA EM) CD4 (C) and CD8 cells (D). Expression of CD57, CD85j and CD158b/j in the elderly was highest in effector CD8 T cells, but CD57 and CD85j was also present in a central memory T cells. Representative histograms are shown (E).
The altered subset distribution in part accounted for the preferential phenotypic changes in CD8 T cells. CD158b/j was exclusively expressed on CD45RA terminal effector T cells, suggesting that the accumulation of this subset was responsible for the emergence of CD158b/j cells with age. CD57 and CD85j were also preferentially expressed on CD45RA effector T cells; however, expression was not subset specific, and all three memory subsets gained expression of these molecules with age (Figure 3E).
Expansion of CMV-specific T cells does not account for the preference of age-dependent changes for the CD8 compartment
Several recent studies have attributed disordered CD8 T-cell homeostasis to chronic persistent anti-CMV responses and the accumulation of oligoclonal CMV-specific effector T cells [14; 15; 27]. To examine the relationship between CMV responses and age-dependent phenotypic changes, we selected HLA-A2+ individuals and determined the frequency and phenotype of HLA-A2-restricted T cells specific for the CMV peptide NLVPMVATV (A2/NLV). While the majority of individuals in our population did not have any detectable (<0.1%) CMV pp65495–504 peptide-specific clones irrespective of age, clonal expansions of more than 1% were seen in about one half of the middle-aged and elderly populations (Figure 4A). In contrast to the global CD8 T-cell compartment, CMV-specific clones did not undergo phenotypic changes with age; approximately 60% had lost CD28 expression and 50% had gained CD85j expression in young and elderly individuals (Figure 4B). Individuals with CMV-specific clones had overall higher frequencies of CD8+CD28−CD85j+ T cells than individuals without detectable CMV T-cell responses; however, both subject populations exhibited the same age-dependent phenotypic shift in their CD8 compartment (Figure 4C). These data suggest that CMV-specific responses are not the sole and, in most individuals, not a major cause for the observed age-dependent phenotypic conversions and the disequilibrium in functional T-cell subset distribution that mostly affect CD8 T cells.
Figure 4. Expansion of CMV-specific T cells does not account for the preference of age-dependent phenotypic changes for the CD8 compartment.
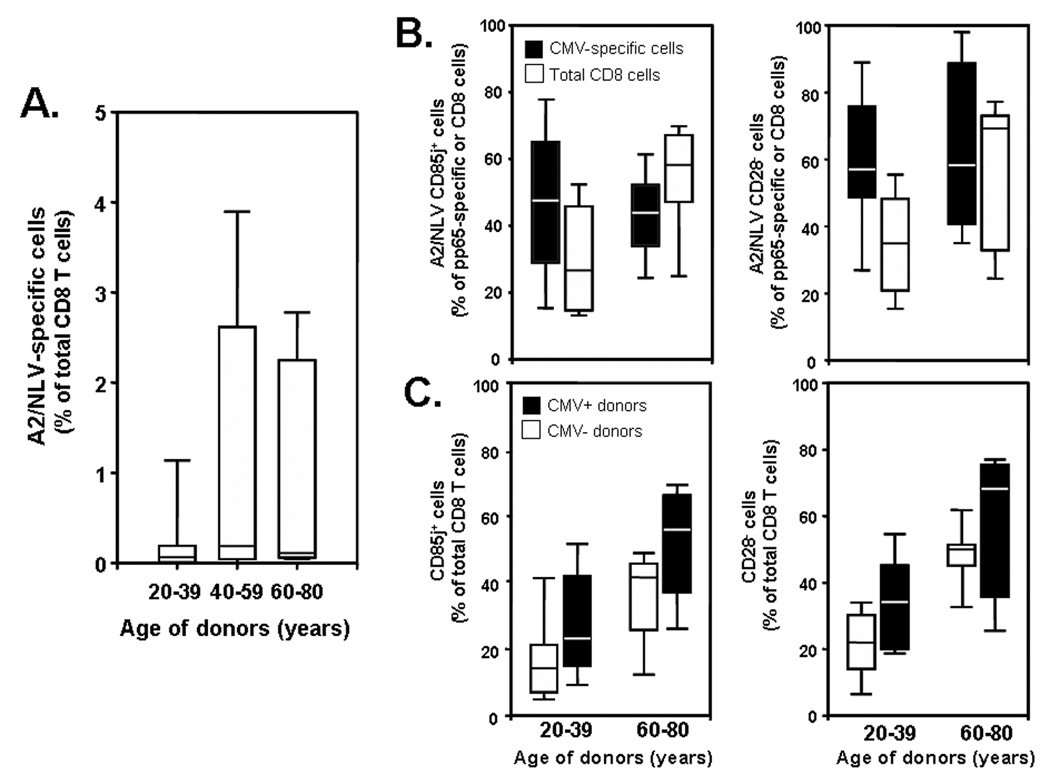
PBMCs were isolated from HLA-A2+ individuals and stained with HLA-A2-CMV peptide (NLVPMVATV) (A2/NLV) pentamer, anti-CD3, -CD8, -CD28, and -CD85j mAbs. (A) Frequencies of CMV peptide-specific CD8 T cells are shown in correlation to age. (B) CMV-peptide-specific (solid boxes) and total CD8 T cells (open boxes) from young and elderly individuals who had <0.1% of CMV-peptide specific cells, were compared. (C) Individuals who had (solid bars) or did not have expanded CMV-peptide specific T cells (open bars) were compared for the frequency of CD85j and CD28 expression on CD8 T cells.
Preference for phenotypic conversions in CD8 T cells independent of homeostatic imbalance
Several genes including CD26 and MHC class II molecules were found to be overexpressed in the gene array of elderly memory T cells independent of CD28 loss, raising the issue of whether their expression was independent of homeostatic disregulation. Flow cytometric studies were performed to compare the effect of age on CD4 and CD8 T cells for these markers. A complex CD26 expression pattern was found, particularly on CD8 T cells. Three separate populations of CD26-expressing CD8 T cells could be distinguished: high, medium, and low expression. Most CD8 cells that had lost CD28 expression were also negative for CD26 (Figure 5B, right panel). In addition, CD26high CD8 T cells virtually disappeared with age. Both phenomena contributed to an age-dependent decrease in CD26 expression in CD8 T cells (p<0.001, Figure 5A). CD4 T cells were mostly CD26intermediate (Figure 5B, left panel) and, contrary to the gene array, expression did not change with age (Figure 5A, left panel). Thus, again there were striking differences between CD4 and CD8 T cells in age-dependent phenotypic changes.
Figure 5. CD4+ and CD8+ T cells differ in CD26 expression with age.
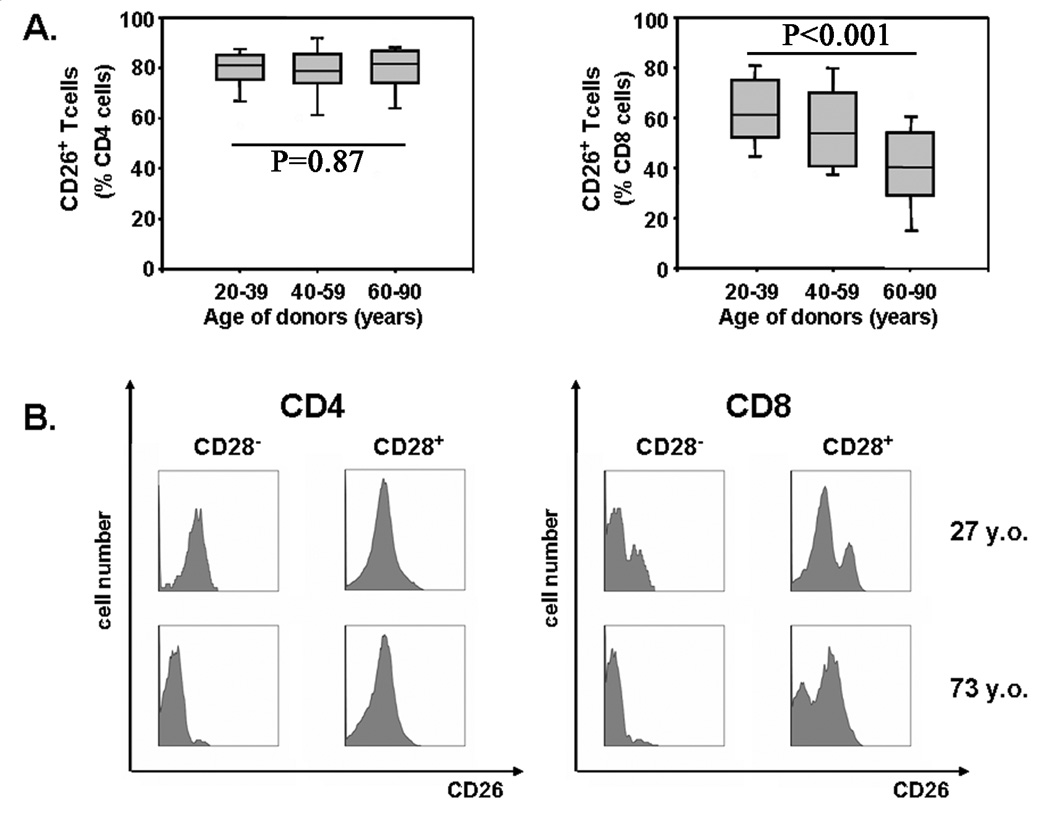
Results of CD26 expression within CD4 (left panel) and CD8 (right panel) T-cell populations in relation to age in cross-sectional studies are shown as box plots (A). Representative histograms of CD26 expression in CD28+ and CD28− CD4 and CD8 cells from a 27 (top row) and 73 year-old (bottom row) individual are shown in (B).
Genes of the MHC class II region were found to be differentially expressed in both series of gene arrays. Flow cytometry studies confirmed that the expression of HLA-DR is age dependent (Figure 6A). A small increase in the frequencies of HLA-DR+ cells was already seen between the ages of 20 and 60 years and then accelerated in the age group of 60 years and older who had significantly higher frequencies of HLA-DR-expressing CD4 and CD8 T cells compared to young adults (p<0.001). In contrast to KIRs, CD57 and CD85j, HLA-DR expression did not coincide with CD28 loss. Representative histograms of CD4 and CD8 T cells are shown in Figure 6B. In both CD4 and CD8 subsets, very few T cells expressed HLA-DR in a 27-year-old individual. Only CD8+ CD28− T cells had a slightly higher expression of 6%. In contrast, 30% of CD8+ CD28+ and 44% of CD8+ CD28− T cells in a 70-year-old individual were positive for HLA-DR. 10 and 13%, respectively, of CD4+ CD28+ and CD28− T cells in the same individual expressed the molecule. HLA-DR is known to be expressed on activated T cells, raising the possibility that elderly individuals have a larger fraction of activated cells. However, HLA-DR-positive cells had morphological characteristics similar to HLA-DR-negative cells in the forward-side scatter, and there was no correlation with other activation markers, such as CD69, which does not increase with age (data not shown). Increased HLA-DR expression can therefore not be explained as a result of constitutive activation of T cells in the elderly. Although the pattern of HLA-DR expression was strikingly different from those of CD57 and CD85j, the influence of age on HLA-DR expression was again more pronounced in CD8 than in CD4 T cells (Table 2). These findings suggest that CD8 T cells are more susceptible to phenotypic conversions with age independent of their preferred differentiation into effector cells.
Figure 6. Increased expression of HLA-DR on CD4 and CD8 T-cells with age.
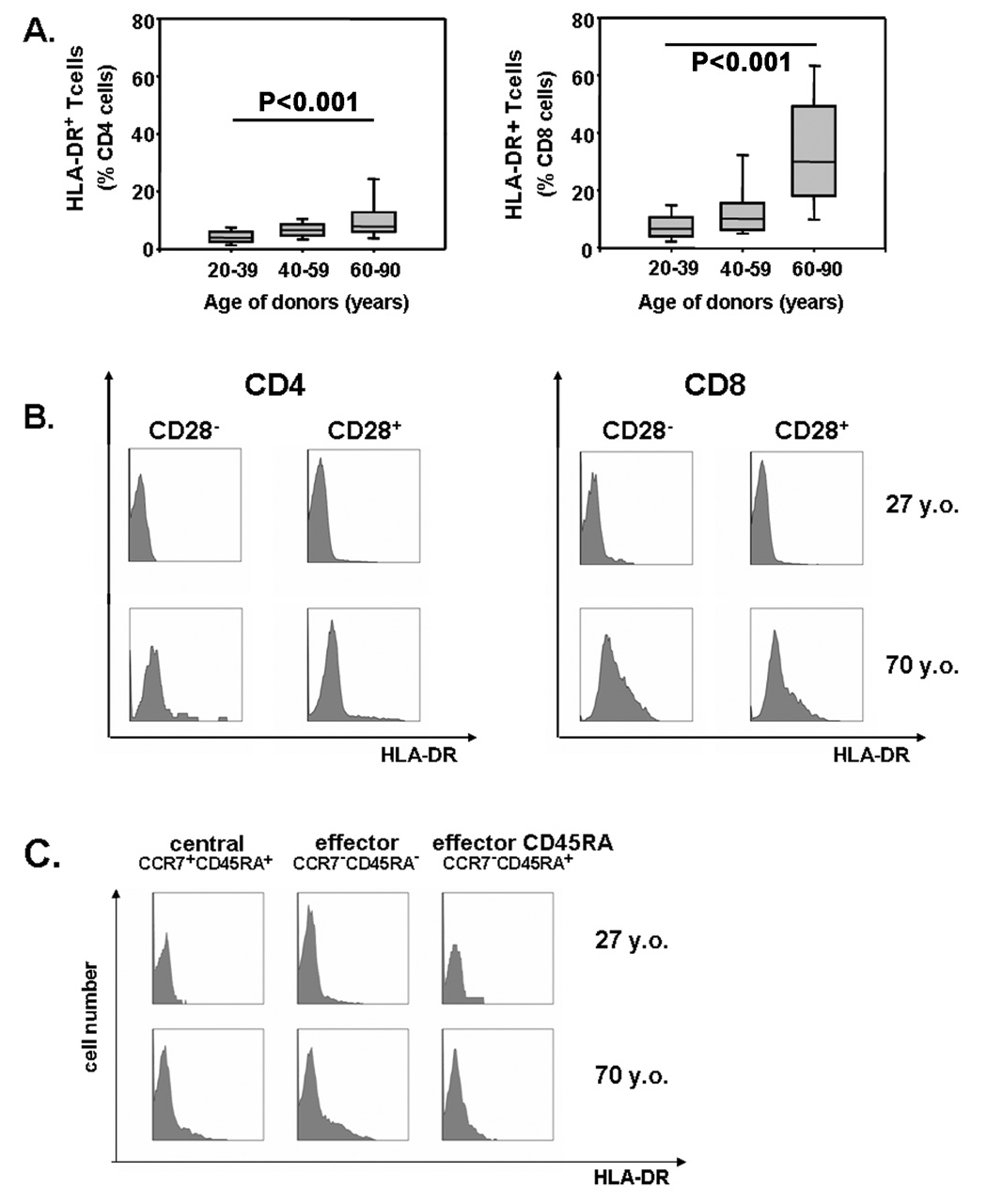
(A) The frequencies of HLA-DR+ cells within CD4 (left panel) and CD8 (right panel) T-cells are shown as box plots for different age groups. HLA-DR in elderly individuals is expressed on CD28+ as well as CD28− T cells (B) and on central as well as effector memory CD8 T cells (C). Representative histograms from a 27 (top row) and 70 year-old (bottom row) individual are shown.
Intrinsic increased susceptibility of CD8 T cells to undergo phenotypic conversions
To examine whether the phenotypic instability is intrinsic to CD8 T cells, we activated CD4 and CD8 T cells in vitro, expanded them in the presence of IL-2 and IL-15 and determine the expression of CD28, HLA-DR, and CD85j over time. To control for differences in population doublings, cells were labeled with CFSE, and CD4 and CD8 T cells with equal division numbers were compared. Representative histograms are shown in Figure 7. CD28 was more rapidly lost in CD8 than in CD4 T cells. On day 31, most CD8, but few CD4, T cells had lost CD28. Expression of CD85j emerged in a subset of CD8 T cells after 3 to 4 weeks of culture, but was nearly absent in CD4 T cells. Both T-cell subsets gained HLA-DR expression early during culture, and expression in CD8 T cells again preceded that in CD4 T cells. In general, phenotypic shifts were more pronounced and earlier in older individuals (data not shown).
Figure 7. Phenotypic changes of the surface molecules: CD28, CD85j, HLA-DR in CD8 and CD4 subsets caused by long-term culture of CD28 positive T cells.
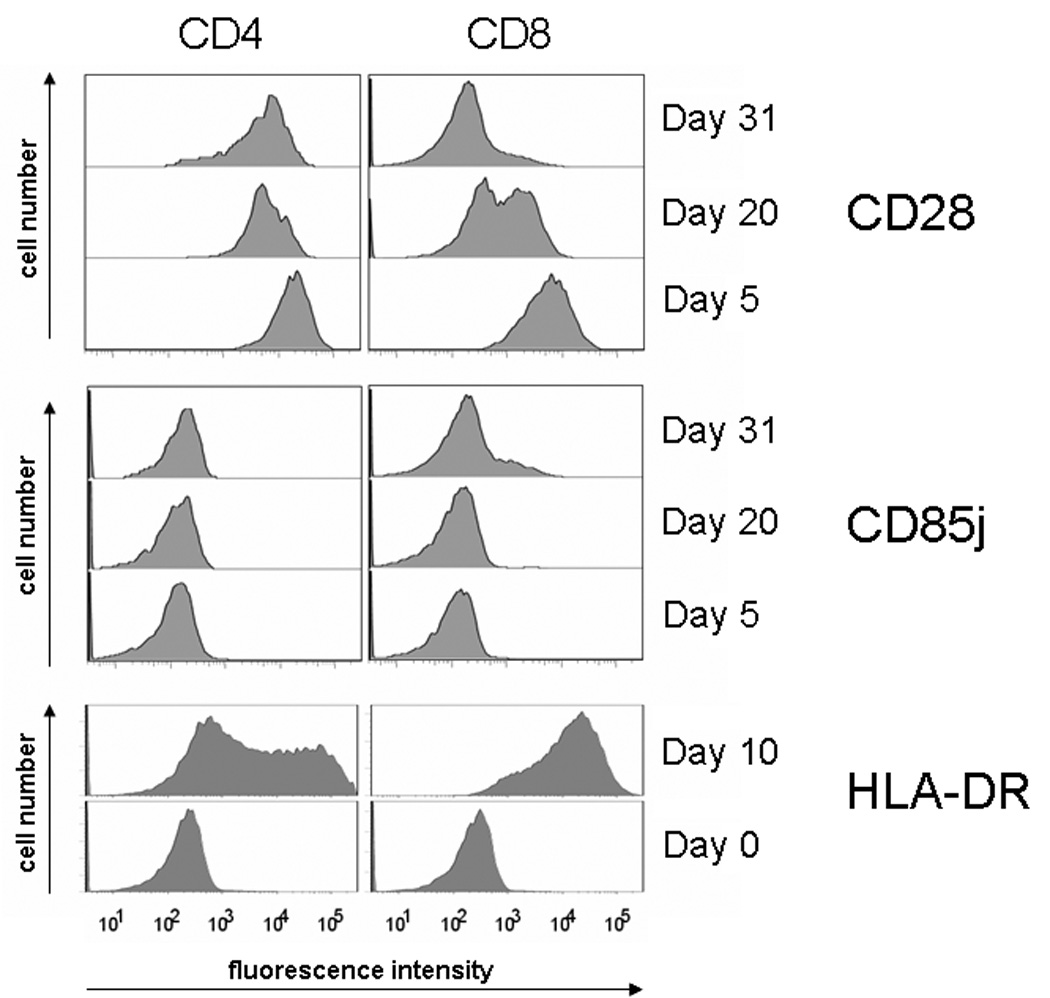
CD28 positive cells were sorted, labeled with CFSE, and stimulated with irradiated, EBV transformed PBMC and OKT3. FACS analysis was carried out every seventh day beginning from the first stimulation. CD28, HLA-DR, and CD85j molecules surface expression changes over time and is much more pronounced in CD8 T cells compared to the CD4 subset, suggesting that the phenotypic instability is intrinsic to CD8 T cells.
Discussion
In this manuscript, we describe that CD4 T cells are more resistant to phenotypic and functional changes with aging than CD8 T cells. CD4 and CD8 T cells undergo the same principal phenotypic shifts; however, the rate at which they occur or accumulate with age is vastly different. The increased susceptibility of CD8 T cells to age was seen for all functional subsets and for all cell surface markers tested. Diminution of naïve cells with age was drastic for CD8 T cells, but relatively minor for CD4 T cells. Similarly, the relative proportions of functional memory subsets were stable in the CD4 compartment, while for CD8 T cells, the aging central memory compartment contracted by more than 50%, and normal effector T cells were increasingly replaced by end-differentiated CD45RA effector T cells. Several cell surface marker profiles, such as the loss of CD28 and the gains of CD57, CD158, and CD85j expressions were typical for functional subsets, and these phenotypic changes with age were therefore in part explained by the accumulation of end-differentiated effector T cells. However, others, such as the expression of MHC class II molecules, were independent of T-cell differentiation, seen on central memory as well as effector T cells, and were still much more prevalent in CD8 than in CD4 T cells. In vitro culture studies where CD4 and CD8 T cells were driven into replicative stress reproduced this different susceptibility, with CD8 T cells more readily undergoing phenotypic changes. These data indicate a fundamental global difference between CD4 and CD8 T cells in their response to age that goes beyond the genetic regulation of a single cell surface molecule or a single differentiation pathway.
Why are homeostatic mechanisms to maintain the relative proportions of functional T-cell subsets more efficient for CD4 T cells? Thymic selection favors the production of CD4 T cells. Thymic production declines with age [7; 8; 9], and one could speculate that the bias towards producing naïve CD4 T cells is more pronounced with declining function. However, thymic production is quantitatively negligible after the age of 40 to 50 years and is therefore unlikely to sustain the naïve CD4 compartment while the naïve CD8 compartment shrinks [9; 37]. Most naïve T cells during adult life are produced by self-renewal of existing T cells, and differing kinetics between CD4 and CD8 T cells could lead to altered subset distributions. Studies using deuteriated glucose have estimated turnover rates of about 1 year for naïve T cells, 48 days for central memory T cells, and 15 days for effector T cells [5]. These studies have come to the conclusion that the overall kinetics are not different for CD4 and CD8 T cells and do not change with age; only the CD45RA effector T-cell population that accumulates with age in the CD8 compartment have higher survival rates [38]. There is therefore no evidence that an increased replicative history explains the higher susceptibility of CD8 T cells to undergo age-dependent changes. This interpretation is also consistent with our previous observation that CD8 T cells do not have more telomeric erosion than CD4 T cells [8] and the results from our in vitro system where we controlled for the number of generations (Figure 7).
Dysregulated T-cell homeostasis with the accumulation of end-stage effector cells can be the result of chronic viral infections; in particular, chronic CMV infection and the associated CMV-specific immune response have been implicated as detrimental for immune health and overall survival [26; 27]. Many end-differentiated effector CD8 cells in the elderly have been shown to be specific for dominant CMV peptides. These oligoclonal expansions compete for space, but also have indirect effects on the remaining repertoire [12]. We tested the hypothesis that chronic CMV infection would induce a preferential accumulation of CD8 and not CD4 effector T cells, explaining the preferential effect of age on CD8 T cells. However, we did not find support for the hypothesis that the higher susceptibility of CD8 T cells to age-dependent functional and phenotypic changes was related to CMV infection. Oligoclonal expansion of HLA-A2-restricted CMV pp65495–504 (NLVPMVATV)-specific clones was infrequent even in our 60–80 year-old population; also, the preferential accumulation of CD85j expression by CD8 T cells was equally seen in individuals that were HLA-A2-positive and did not have detectable CMV-specific CD8 T cells. Our interpretation that CMV is not responsible for the preference of changes to occur in the CD8 compartment is consistent with the finding that similar proportions of CD4 and CD8 T cells are involved in CMV responses [26].
Alternatively, CD8 T cells may be more susceptible to phenotypic and functional conversions than CD4 T cells. We initially thought that the preferential CD28 loss in CD8 T cells was a gene-specific effect. However, loss of CD28 expression in CD4 and CD8 T cells is correlated with the same disassembly of an initiator complex [30]. Our data here suggest that this increased susceptibility is not restricted to one particular gene but involves many genes. Results from the gene expression arrays show that the phenotypic changes in CD4 T cells are principally the same as have been reported for CD8 T cells; however, they occur much less frequently. Some of them, such as the expressions of CD85j and KIR, can be linked to T-cell differentiation [39] and indeed, they are mostly seen in CD28– effector T-cell population. Others, such as the expression of HLA-DR, are observed in central memory and in effector cells and appear to be more global markers. All of them have in common that they are preferentially expressed in CD8 T cells with age and that they can be more readily induced in CD8 T cells in vitro than in CD4 T cells.
It is striking that CD8 T cells age faster than CD4 T cells in every aspect studied although they principally undergo the same changes. It is tempting to postulate a common mechanism; however, the complexity and multitude of how CD4 and CD8 T cells differ in their susceptibility to age is difficult to reconcile in one overriding concept. Obviously, homeostatic control of the CD4 compartment is much more robust than that of CD8 T cells, as evidenced not only by the stability of the naïve and central memory compartments but also by the finding that the TCR diversity of naïve CD4 T cells is well maintained for decades after thymic function has ceased and that oligoclonal expansion of CD4 memory T cells is infrequent and, if it occurs, small in size [37]. Phenotypic conversions are only partially explained by the accumulation of CD8 effector T cells and thus a distorted equilibrium between functional T-cell subsets. Our in vitro studies provide further evidence for the interpretation that CD8 T cells are intrinsically more susceptible to phenotypic changes. If CD8 immunity is indeed more compromised in the elderly than the CD4 response, therapeutic or preventive interventions need to primarily target the CD8 T cell. Whether CD4 or CD8 T-cell aging is primarily responsible for increased morbidity and mortality due to immune-mediated diseases in the elderly is currently unclear. However, and in support of this interpretation, CD28 loss on CD8 and not on CD4 T cells is a strong predictor for the failure to produce antibodies to trivalent flu vaccination, an immune response that is clearly under the control of CD4 T cells [21; 22].
Acknowledgments
This work was funded in part by grants from the National Institutes of Health (RO1 AG 15043 and RO1 AI 57266), the General Clinical Research Center (MO1 RR00039), the Noble Foundation, and the Aging Registry. The authors thank Tamela Yeargin for manuscript editing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weng NP. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. 2006;24:495–499. doi: 10.1016/j.immuni.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grubeck-Loebenstein B, Wick G. The aging of the immune system. Adv Immunol. 2002;80:243–284. doi: 10.1016/s0065-2776(02)80017-7. [DOI] [PubMed] [Google Scholar]
- 3.Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature. 1999;402:255–262. doi: 10.1038/46218. [DOI] [PubMed] [Google Scholar]
- 4.Hellerstein MK. Measurement of T-cell kinetics: recent methodologic advances. Immunol Today. 1999;20:438–441. doi: 10.1016/s0167-5699(99)01529-7. [DOI] [PubMed] [Google Scholar]
- 5.Macallan DC, Wallace D, Zhang Y, De Lara C, Worth AT, Ghattas H, Griffin GE, Beverley PC, Tough DF. Rapid turnover of effector-memory CD4(+) T cells in healthy humans. J Exp Med. 2004;200:255–260. doi: 10.1084/jem.20040341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, Polis MA, Haase AT, Feinberg MB, Sullivan JL, Jamieson BD, Zack JA, Picker LJ, Koup RA. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 7.Haynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP. The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection. Annu Rev Immunol. 2000;18:529–560. doi: 10.1146/annurev.immunol.18.1.529. [DOI] [PubMed] [Google Scholar]
- 8.Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A. 2000;97:9203–9208. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hakim FT, Memon SA, Cepeda R, Jones EC, Chow CK, Kasten-Sportes C, Odom J, Vance BA, Christensen BL, Mackall CL, Gress RE. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J Clin Invest. 2005;115:930–939. doi: 10.1172/JCI22492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naylor K, Li G, Vallejo AN, Lee WW, Koetz K, Bryl E, Witkowski J, Fulbright J, Weyand CM, Goronzy JJ. The influence of age on T cell generation and TCR diversity. J Immunol. 2005;174:7446–7452. doi: 10.4049/jimmunol.174.11.7446. [DOI] [PubMed] [Google Scholar]
- 11.Goronzy JJ, Lee WW, Weyand CM. Aging and T-cell diversity. Exp Gerontol. 2007;42:400–406. doi: 10.1016/j.exger.2006.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Messaoudi I, Lemaoult J, Guevara-Patino JA, Metzner BM, Nikolich-Zugich J. Age-related CD8 T cell clonal expansions constrict CD8 T cell repertoire and have the potential to impair immune defense. J Exp Med. 2004;200:1347–1358. doi: 10.1084/jem.20040437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. 2004;4:123–132. doi: 10.1038/nri1292. [DOI] [PubMed] [Google Scholar]
- 14.Hadrup SR, Strindhall J, Kollgaard T, Seremet T, Johansson B, Pawelec G, thor Straten P, Wikby A. Longitudinal studies of clonally expanded CD8 T cells reveal a repertoire shrinkage predicting mortality and an increased number of dysfunctional cytomegalovirus-specific T cells in the very elderly. J Immunol. 2006;176:2645–2653. doi: 10.4049/jimmunol.176.4.2645. [DOI] [PubMed] [Google Scholar]
- 15.Akbar AN, Fletcher JM. Memory T cell homeostasis and senescence during aging. Curr Opin Immunol. 2005;17:480–485. doi: 10.1016/j.coi.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 16.Vallejo AN, Weyand CM, Goronzy JJ. T-cell senescence: a culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med. 2004;10:119–124. doi: 10.1016/j.molmed.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Effros RB, Boucher N, Porter V, Zhu X, Spaulding C, Walford RL, Kronenberg M, Cohen D, Schachter F. Decline in CD28+ T cells in centenarians and in long-term T cell cultures: a possible cause for both in vivo and in vitro immunosenescence. Exp Gerontol. 1994;29:601–609. doi: 10.1016/0531-5565(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 18.Posnett DN, Sinha R, Kabak S, Russo C. Clonal populations of T cells in normal elderly humans: the T cell equivalent to "benign monoclonal gammapathy". J Exp Med. 1994;179:609–618. doi: 10.1084/jem.179.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang I, Hong MS, Nolasco H, Park SH, Dan JM, Choi JY, Craft J. Age-associated change in the frequency of memory CD4+ T cells impairs long term CD4+ T cell responses to influenza vaccine. J Immunol. 2004;173:673–681. doi: 10.4049/jimmunol.173.1.673. [DOI] [PubMed] [Google Scholar]
- 20.Goodwin K, Viboud C, Simonsen L. Antibody response to influenza vaccination in the elderly: a quantitative review. Vaccine. 2006;24:1159–1169. doi: 10.1016/j.vaccine.2005.08.105. [DOI] [PubMed] [Google Scholar]
- 21.Goronzy JJ, Fulbright JW, Crowson CS, Poland GA, O'Fallon WM, Weyand CM. Value of immunological markers in predicting responsiveness to influenza vaccination in elderly individuals. J Virol. 2001;75:12182–12187. doi: 10.1128/JVI.75.24.12182-12187.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saurwein-Teissl M, Lung TL, Marx F, Gschosser C, Asch E, Blasko I, Parson W, Bock G, Schonitzer D, Trannoy E, Grubeck-Loebenstein B. Lack of antibody production following immunization in old age: association with CD8(+)CD28(−) T cell clonal expansions and an imbalance in the production of Th1 and Th2 cytokines. J Immunol. 2002;168:5893–5899. doi: 10.4049/jimmunol.168.11.5893. [DOI] [PubMed] [Google Scholar]
- 23.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. Jama. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 24.Gavazzi G, Krause KH. Ageing and infection. Lancet Infect Dis. 2002;2:659–666. doi: 10.1016/s1473-3099(02)00437-1. [DOI] [PubMed] [Google Scholar]
- 25.Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med. 1995;155:1605–1609. [PubMed] [Google Scholar]
- 26.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pawelec G, Akbar A, Caruso C, Solana R, Grubeck-Loebenstein B, Wikby A. Human immunosenescence: is it infectious? Immunol Rev. 2005;205:257–268. doi: 10.1111/j.0105-2896.2005.00271.x. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7− CD28− T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. 1996;97:2027–2037. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vallejo AN, Bryl E, Klarskov K, Naylor S, Weyand CM, Goronzy JJ. Molecular basis for the loss of CD28 expression in senescent T cells. J Biol Chem. 2002;277:46940–46949. doi: 10.1074/jbc.M207352200. [DOI] [PubMed] [Google Scholar]
- 30.Vallejo AN, Brandes JC, Weyand CM, Goronzy JJ. Modulation of CD28 expression: distinct regulatory pathways during activation and replicative senescence. J Immunol. 1999;162:6572–6579. [PubMed] [Google Scholar]
- 31.Baugh LR, Hill AA, Brown EL, Hunter CP. Quantitative analysis of mRNA amplification by in vitro transcription. Nucleic Acids Res. 2001;29:E29. doi: 10.1093/nar/29.5.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 33.Fann M, Chiu WK, Wood WH, 3rd, Levine BL, Becker KG, Weng NP. Gene expression characteristics of CD28null memory phenotype CD8+ T cells and its implication in T-cell aging. Immunol Rev. 2005;205:190–206. doi: 10.1111/j.0105-2896.2005.00262.x. [DOI] [PubMed] [Google Scholar]
- 34.Weyand CM, Brandes JC, Schmidt D, Fulbright JW, Goronzy JJ. Functional properties of CD4+ CD28− T cells in the aging immune system. Mech Ageing Dev. 1998;102:131–147. doi: 10.1016/s0047-6374(97)00161-9. [DOI] [PubMed] [Google Scholar]
- 35.Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. 2005;5:201–214. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- 36.Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, Casazza JP, Kuruppu J, Migueles SA, Connors M, Roederer M, Douek DC, Koup RA. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101:2711–2720. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 37.Goronzy JJ, Weyand CM. T cell development and receptor diversity during aging. Curr Opin Immunol. 2005;17:468–475. doi: 10.1016/j.coi.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 38.Wallace DL, Zhang Y, Ghattas H, Worth A, Irvine A, Bennett AR, Griffin GE, Beverley PC, Tough DF, Macallan DC. Direct measurement of T cell subset kinetics in vivo in elderly men and women. J Immunol. 2004;173:1787–1794. doi: 10.4049/jimmunol.173.3.1787. [DOI] [PubMed] [Google Scholar]
- 39.Fischer JC, Ottinger H, Ferencik S, Sribar M, Punzel M, Beelen DW, Schwan MA, Grosse-Wilde H, Wernet P, Uhrberg M. Relevance of C1 and C2 epitopes for hemopoietic stem cell transplantation: role for sequential acquisition of HLA-C-specific inhibitory killer Ig-like receptor. J Immunol. 2007;178:3918–3923. doi: 10.4049/jimmunol.178.6.3918. [DOI] [PubMed] [Google Scholar]