

Abstract
We investigated the role of NF-κB activation by the bacterial product lipopolysaccharide (LPS) in inducing caveolin-1 (Cav-1) expression and its consequence in contributing to the leakiness of the endothelial barrier. We observed that LPS challenge of human lung microvascular endothelial cells induced concentration- and time-dependent increases in expression of Cav-1 mRNA and protein. The NEMO (NF-κB essential modifier binding domain)-binding domain peptide (IkB kinase (IKK)-NEMO-binding domain (NBD) peptide), which prevents NF-κB activation by inhibiting the interaction of IKKγ with the IKK complex, blocked LPS-induced Cav-1 mRNA and protein expression. Knockdown of NF-κB subunit p65/RelA expression with small interfering RNA also prevented LPS-induced Cav-1 expression. Caveolae open to the apical and basal plasmalemma of endothelial cells increased 2–4-fold within 4 h of LPS exposure. IKK-NBD peptide markedly reduced the LPS-induced increase in the number of caveolae as well as transendothelial albumin permeability. These observations were recapitulated in mouse studies in which IKK-NBD peptide prevented Cav-1 expression and interfered with the increase in lung microvessel permeability induced by LPS. Thus, LPS mediates NF-κB-dependent Cav-1 expression that results in increased caveolae number and thereby contributes to the mechanism of increased transendothelial albumin permeability.
The interaction between bacteria and endothelial cell plasma membrane is mediated by components of the bacterial wall outer membrane, the most important being LPS.3 LPS binds to CD14 (1–3) and Toll-like receptor 4 (TLR4) (1–4) expressed in the membrane. NF-κB, the transcription factor activated by LPS-CD14-TLR4 signaling (5), results in the transcriptional induction of cytokines (interleukin-1 (IL-1), IL-6, IL-8), tissue factor, and adhesion molecules (E- and P-selectins, VCAM-1 (vascular cell adhesion molecule), and ICAM-1) (6).
Cav-1, the structural protein of caveolae in endothelial cells and other cell types, regulates the formation of caveolae, the vesicle carriers involved in the transcytosis of albumin across the endothelial barrier (7). Studies showed that caveolae-mediated transcytosis contributes to the regulation of microvascular permeability (7) secondary to the activation of Src kinase (8). Cav-1-null mice, lacking caveolae (9), showed defective albumin transcytosis (10). In an experimental model of diabetes, increased Cav-1 expression in endothelial cells was associated with increased transcytosis of albumin (11). LPS was shown to induce the expression of Cav-1 in endothelial cells (12) and murine macrophages (13, 14); however, the mechanisms of the response and its consequences in regulating endothelial barrier function are not clear.
NF-κB is composed of dimers of five different proteins (p50, p52, p65/RelA, RelB, c-Rel) (15). These dimers exist in the cytoplasm in inactive forms bound to the inhibitory protein I-κB (IκB) (15). A variety of agonists activate IκB kinases α and β (15), which in turn phosphorylate serines 32 and 36 of IκBα and serines 19 and 23 of IκBβ, respectively (15). Phosphorylation of IκBα and IκBβ leads to the proteolytic degradation of IκB and dissociation of NF-κB, and NF-κB translocates to the nucleus to induce gene transcription (15). The IκB kinase complex consists of two catalytic IKKα and IKKβ, and a regulatory subunit, IKKγ (or NF-κB essential modulator (NEMO)) (16). NEMO interaction with IKKα and IKKβ is required for IκB kinase catalytic activity. Based on our observation that the intronic region of Cav-1 contains NF-κB consensus sites, we addressed the possibility that LPS mediates Cav-1 expression by an NF-κB-dependent mechanism. We surmised that this pathway thereby contributes to the mechanism of increased transendothelial albumin permeability seen with LPS. We demonstrate here that LPS activation of endothelial cells increased Cav-1 protein expression as well as caveolae number and that both were dependent on activation of NF-κB. Moreover, inhibiting NF-κB activation pharmacologically, knockdown of p65/RelA expression and knockdown of Cav-1 expression each interfered with the increase in transendothelial albumin permeability induced by LPS. Thus, these findings suggest a critical role of increased caveolar trafficking of albumin in promoting the LPS-mediated albumin leakiness of the endothelial barrier.
EXPERIMENTAL PROCEDURES
Materials
Human lung microvascular endothelial cells (HLMVECs) and microvascular endothelial growth medium (EGM-2 MV) were obtained from Cambrex Bio Science (Walkersville, MD). Fetal bovine serum (FBS) was from Hyclone (Logan, UT). LPS (Escherichia coli 0111:B4) was obtained from Calbiochem. Cell-permeable NEMO binding domain (NBD) synthetic peptides (wild type, drqikiwfqnrrmkwkkTALD-WSWLQTE (IKK-NBD); mutant, drqikiwfqnrrmkwkkTAL-DASALQTE (mutant-IKK-NBD)) were obtained from Biomol (Plymouth Meeting, PA). Anti-Cav-1 polyclonal antibody (pAb) and anti-VE-cadherin monoclonal antibody (mAb) were from BD Biosciences. Anti-tubulin mAb was obtained from Cytoskeleton Inc. (Denver, CO). Human p65/RelA small interference RNA (siRNA; Validated siRNA, catalog #SI00301672) and scrambled siRNA were from Qiagen (Valencia, CA). NF-κB protein (p65/RelA)-specific antibody and siRNA transfection reagent (catalog #sc-29528) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Cav-1-specific NF-κB oligonucleotides and PCR primers were custom-synthesized from IDT (Coralville, IA).
Cell Culture
HLMVECs grown in EGM-2 MV supplemented with 15% FBS were used between passages 2 and 4. To study LPS effect, HLMVECs were exposed to LPS in the presence of 2% FBS-containing medium.
Immunoblot Analysis
HLMVECs exposed to LPS were lysed with lysis buffer (50 mM Tris-HCl buffer, pH 7.4, continuing 150 mM NaCl, 1 mM EGTA, 1%Triton X-100, 0.25% sodium deoxycholate, 0.1% SDS, and protease inhibitors), and lysates were immunoblotted with anti-Cav-1 pAb (17). For loading control, membranes were re-probed with anti-tubulin mAb.
Real-time Quantitative-PCR
Total RNA, extracted according to manufacturer’s recommendations with RNeasy kit (Qiagen, CA), was used to generate first-strand complementary DNA by reverse transcriptase (Invitrogen). cDNA (10 ng), mixed with SYBR green PCR master mix (Applied Biosystems, CA), was used for real-time quantitative-PCR with the ABI prism 7000 sequence detection system (Applied Biosystems). Cav-1 mRNA was normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA. Primer sequences were: Cav-1, forward 5′-GAGCTGAGCGAGAAGCAAGT-3′, and reverse 5′-TCC-CTTCTGGTTCTGCAATC-3′; glyceraldehyde-3-phosphate dehydrogenase, forward 5′-GTGAAGGTCGGAGTCAACG-3′, and reverse 5′-TGAGGTCAATGAAGGGGTC-3′.
Nuclear Protein Extraction
Nuclear extracts were prepared from HLMVECs after LPS treatment as described (18). Cells grown in 100-mm cell culture dishes were washed twice with ice-cold Tris-buffered saline, scraped, and resuspended in 400 μl of buffer A (10 mM KCl, 10 mM HEPES, pH 7.9, 0.1 mM EDTA, pH 8.0, 0.1 mM EGTA, 1 mM dithiothreitol, and 0.5 mM phenylmethylsulfonyl fluoride). The suspension was homogenized with 10 strokes using Glass Dounce homogenizer and centrifuging at 3000 × g for 30 s. Nuclear pellets were then resuspended in 100 μl of solution B (20 mM HEPES, 1 mM EDTA, 0.4 M NaCl, 1 mM EGTA, 1 mM dithiothreitol, and 0.5 mM phenylmethylsulfonyl fluoride and incubated on ice for 20 min. The nuclei were then pelleted by centrifugation at 25,000 × g for 1 min. Supernatants containing nuclear proteins were used for electrophoretic mobility shift assay.
Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed as described (18). Using the TFSEARCH program, we identified two putative NF-κB binding sites within intron 1, +435 to +460 relative to the translation start site in the human Cav-1 gene (Fig. 3A). The 26-bp sequence of the Cav-1 gene encompassing the predicted NF-κB binding sites was used for the wild type Cav-1 NF-κB sequence, 5′-GGGGACAGTC-CCCGGGACTCTCCGCC-3′, and mutant Cav-1 NF-κB-sequence, 5′-GGGGACAGTATCCGATACTCTCCGCC-3′(Fig. 3A). Wild type Cav-1 sequence contains two putative NF-κB sites which scores 88.5 and 87 points. Altering two bases in each site reduced the scores to 67.7 and 66.2 and abolished the NF-κB binding to DNA (see Fig. 3B, middle panel). As a positive control, the ICAM-1 promoter-specific NF-κB sequence (5′-AGCTTGGAAAATTCCGGAGCTG-3′) was used. End labeling was performed by T4 kinase in the presence of [α-32P]ATP. Labeled oligonucleotides were purified on a Sephadex G-50 column. An aliquot of 10 μg of nuclear protein extract was incubated with the labeled double-stranded probe (~80,000 cpm) in the presence of 2.5 μl of binding buffer (Pro-mega). The binding reactions were incubated at 25 °C for 20 min. After adding non-denaturing sample buffer, the DNA-protein complexes were resolved by 6% native PAGE in low ionic strength buffer (0.5× Tris borate-EDTA). To study the effect of antibodies on DNA-protein binding, nuclear extracts were first incubated with NF-κB protein-specific antibodies (2 μg/assay) for 15 min at 25 °C, and then labeled double-stranded probe was added, and the incubation was continued for an additional 20 min. After this incubation, non-denaturing sample buffer added, and the DNA-protein complexes were separated as described above.
FIGURE 3. LPS induces NF-κB binding to Cav-1 gene intronic NF-κB DNA element.

A, schematic of human Cav-1 gene indicates two NF-κB binding sequence in the intron 1 between +435 and +460. In B, left panel, HLMVECs grown to confluence were exposed to LPS (4 μg/ml) for up to 6 h. After LPS treatment, nuclear extracts prepared were used for EMSAs as described under “Experimental Procedures.” LPS treatment induced two complexes (Bands 1 and 2). Mutation in the NF-κB binding sequence prevented DNA binding activity of NF-κB (middle panel). LPS challenge also induced ICAM-1 promoter-specific NF-κB oligos binding to NF-κB (right panel). WT, wild type. In C, lanes 1–3, competition studies using a 70-fold excess of unlabeled consensus NF-κB double-stranded oligonucleotide (cold probe) and labeled oligonucleotide (hot probe) were performed (LPS 4 μg/ml, 1 h); lane 1, control (untreated cell) nuclear extract and hot probe; lane 2, nuclear extract and hot probe (1X); lane 3, nuclear extract and hot probe (1X) and cold probe (70X). In D, immunodepletion was demonstrated by preincubating the nuclear extract with anti-p65/Rel A Ab or anti-p50 Ab. Lane 1, nuclear extract and control Ab (preimmune IgG) and hot probe; lane 2, nuclear extract and anti-p65 Ab and hot probe; lane 3, nuclear extract and anti-p50 Ab and hot probe. Band 1 was diminished upon the addition of Abs specific for p65 and p50 as indicated. Note the p65 complex (band 1) present in lane 1 is absent from lane 2. Results are representative of three independent experiments. E, p65/RelA knockdown using siRNA prevents p65/RelA expression in endothelial cells. HLMVECs grown to ~80% confluence were transfected with control-siRNA or p65-siRNA as described details under “Experimental Procedures.” At 48 h after transfection, cells were lysed and immunoblotted (IB) with anti-p65 Ab. The membrane was stripped and probed with anti-tubulin mAb for loading control. The experiment was repeated three times with similar results. F, p65/RelA knockdown prevents LPS-induced Cav-1 expression in endothelial cells. HLMVECs were transfected p65-siRNA as described above. At 48 h after transfection, cells were exposed to LPS (4 μg/ml) for up to 6 h. After LPS stimulation, cells were lysed and immunoblotted with anti-Cav-1 Ab. The membrane was stripped and probed with anti-tubulin mAb for loading control. Results are representative of >4 experiments.
p65/RelA-siRNA Transfection
HLMVECs grown to ~80% confluence were washed with transfection medium before transfection. The cells were then transfected with indicated concentrations of human p65/RelA siRNA or scrambled siRNA using siRNA transfection reagents from Santa Cruz Biotechnology. At 48 h after transfection the cells were washed with serum-free medium and incubated with 2% FBS-containing medium for 2 h. After this period, the cells were used for either immunoblotting with anti-p65 Ab or treated with LPS for different time intervals and immunoblotted anti-Cav-1 Ab.
Electron Microscopy and Morphometric Analysis
HLM-VECs were washed 3× at 2 min each with phosphate-buffered saline, 1× with washing buffer (0.1 M sodium cacodylate buffer, pH 7.4, containing 5% sucrose), fixed (1.5% glutaraldehyde in washing buffer) for 30 min at room temperature, washed 3 × 15 min each, post-fixed in 1% OsO4, pH 6.0 (45 min in the dark on ice), and then in 7.5% uranyl magnesium acetate (1 h in the dark at room temperature). Specimens dehydrated in increasing concentrations of ethanol and embedded in Epon were sectioned (60 mm thick) and viewed using a JEOL 1220 transmission electron microscope (19).
Randomly chosen Epon blocks (2 or 3) were used to obtain 6–8 grids/block and 15–20 sections/grid for 95 micrographs per condition at 84,000× final magnification. Vesicle number was determined using the Morphometrix program, and counts were expressed as the average number of vesicle per experimental condition. Vesicles associated with the apical or baso-lateral surface of an endothelial cell were considered open, and those >100 nm away from the plasma membrane were considered inside the cell (19).
Confocal Microscopy
Confocal imaging was performed to identify Cav-1-positive vesicles in HLMVECs as described (8). HLMVECs grown on glass coverslips were incubated with 2% FBS containing medium for 2 h, and then cells were exposed to LPS for up to 4 h. Cells were washed 3 times, fixed with 4% paraformaldehyde in Hanks’ balanced salt solution (HBSS) for 30 min at 22 °C, and blocked with 5% goat serum in HBSS containing 0.1% Triton X-100 (blocking buffer) for 30 min at 22 °C. After washing, the cells were incubated with either anti-Cav-1 pAb diluted (1:3000) or anti-VE-cadherin mAb diluted (1:200) in blocking buffer at 4 °C overnight. After washing 2 times, cells were incubated with Alexa 488-labeled secondary goat anti-rabbit or goat anti-mouse Ab in blocking buffer for 60 min at 4 °C. Confocal images were acquired with the Zeiss LSM 510 confocal microscope (8).
Transendothelial Electrical Resistance Measurement
The real-time change in endothelial monolayer electrical resistance was measured as described by us (20). In brief, HLMVECs were grown to confluence on small gold electrode (4.9 × 10−4 cm2). The small electrode and the larger counter electrode were connected to a phase-sensitive lock-in amplifier. An approximate constant current 1 μA was supplied by a 1-V, 4000-Hz AC signal connected serially to 1-megaohm resistor between the small electrode and the larger counter electrode. The voltage between small electrode and large electrode was monitored by lock-in amplifier, stored, and processed by a personal computer. The same computer controlled the output of the amplifier and switched the measurement to different electrodes in the course of an experiment. Before the experiment a confluent endothelial monolayer was kept in 2% FBS containing medium for 2 h, and then cells were challenged with LPS, IKK-NBD, or mutant IKK-NBD. The change in monolayer resistance was monitored up to 4 h. As a positive control, HMLVECs were challenged with thrombin. The data are presented in resistance normalized to its value at time 0 as described (20).
Transendothelial 125I-Labeled Albumin Permeability in Monolayers
Permeability of 125I-labeled albumin in confluent HLMVECs on polycarbonate Transwell membranes was determined (21). Single-stranded siRNA (90 pmol/well) corresponding to human Cav-1 (5′-GAGAAGCAAGUGUACGACG-3′) and control siRNA (4 bases altered, 5′-GAGAAGCAGU-GAUACGACG-3′) together with FuGENE 6 reagent at a ratio of siRNA:lipid (1:3, w/v) were transfected into monolayers. After 72 h, monolayers were treated with LPS alone and either IKK-NBD (100 μM) plus LPS or mutant IKK-NBD (100 μM) peptides in Me2SO. IKK-NBD or mutant IKK-NBD peptides were preincubated with the monolayers for 1 h before the addition of LPS. Data were analyzed by Student’s t test or analysis of variance (Bonferroni) with values p < 0.05 considered as significant.
Determination of Lung Microvessel Permeability of 125I-Labeled Albumin in Mice
C57BL6J mice were obtained from Jackson Laboratory, housed in the University of Illinois Animal Care Facility, and used according to approved animal protocols. Female mice weighing 25–30 g were anesthetized (2.5% sevoflurane in room air) for insertion of an indwelling jugular catheter and then were permitted to recover for 30 min. At 30 min before LPS challenge, mice received the vehicle (100 μl/mouse, 20% Me2SO), IKK-mutant peptide (100 μl/mouse, 2.0 mM in 20% Me2SO), or IKK-NBD peptide (100 μl/mouse, 2.0 mM in 20% Me2SO) via the jugular catheter. Mice were challenged with LPS (10 mg/kg, i.p) at zero time. Control animals received the same volume of vehicle. After LPS challenge or sham injection, at time points of 1, 3, and 5 h mice received repeat injections of vehicle, IKK-mutant peptide, or IKK-NBD peptide. At 340 min after LPS, mice received ~1 μCi of 125I-labeled albumin via the jugular catheter. At 360 min animals were again anesthetized (2.5% sevoflurane in air), and a blood sample of 100 μl was withdrawn by puncture of the inferior vena cava to determine vascular tracer counts. Lungs were flushed with RPMI solution containing 3% unlabeled albumin to remove the vascular albumin tracer. The lung tissue was removed, weighed, and counted for γ radioactivity. Albumin permeability-surface area product was calculated as described from vascular counts, tissue counts, and tracer exposure time (22). In another set of experiments lungs were harvested for immunoblotting to determine Cav-1 expression.
RESULTS
LPS Induces Cav-1 Protein Expression
We exposed HLM-VECs to varying concentrations of LPS to for up to 4 h and determined Cav-1 protein expression. Fig. 1A shows that LPS induced Cav-1 protein expression in a concentration-dependent manner. Cav-1 protein expression reached a maximum between 2 and 4 μg/ml LPS concentration, and levels decreased significantly at 8 μg/ml. To assess the time course, HLMVECs were exposed to fixed LPS concentration for up to 6 h. Cav-1 expression reached a maximum with LPS concentration at 4 μg/ml after 4 h of exposure (Fig. 1B). To address whether the LPS-induced Cav-1 expression is dependent on transcriptional activation of Cav-1, we determined the effect of transcriptional inhibitor actinomycin D. Actinomycin D treatment prevented LPS-induced Cav-1 protein expression (Fig. 1C). The protein synthesis inhibitor cycloheximide also prevented LPS-induced Cav-1 expression in HLMVECs (data not shown). As a positive control we studied the effect of LPS on ICAM-1 expression in HLMVECs. LPS stimulation also increased ICAM-1 expression (Fig. 1D). Likewise, TNF-α exposure also increased Cav-1 expression (Fig. 1E).
FIGURE 1.
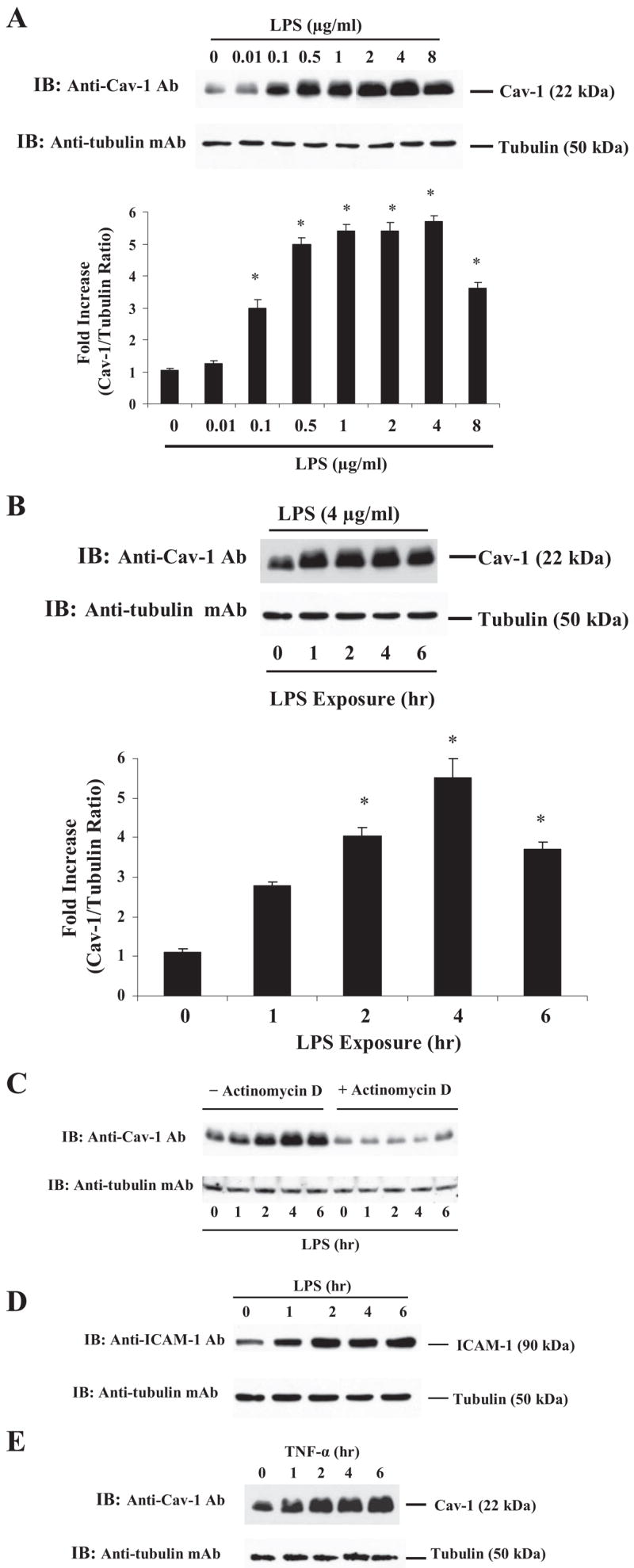
A, LPS induces Cav-1 expression in endothelial cells. HLMVECs grown to confluence were incubated with 2% FBS containing medium for 2 h and challenged with the indicated concentrations of LPS for 4 h. After this treatment, cells were washed and lysed using lysis buffer. Total cell lysate proteins were subjected to immunoblot (IB) using anti-Cav-1 pAb (see details under “Experimental Procedures”). For the loading control, the membrane was probed with anti-tubulin-mAb. Cav-1 induction -fold was calculated by measuring the ratio of Cav-1 to tubulin (bottom panel). Values are the means ± S.E. from four experiments. *, p < 0.05, different from control. B, time-dependent increase in LPS-induces Cav-1 expression. HLMVECs were exposed to LPS from 0–6 h and immunoblotted as described above. *, p < 0.05, different from the control. C, actinomycin D prevents LPS-induced Cav-1 expression. HMLVECs, incubated with 2% FBS containing medium, were pre-treated with actinomycin D (1 μM) 1 h and then exposed to LPS (4 μg/ml) for up to 6 h. After LPS treatment, cells were lysed and immunoblotted with anti-Cav-1 pAb as described in Fig. 1A. D, LPS induces ICAM-1 expression. HLMVECs, exposed to LPS (4 μg/ml; 0 – 6 h) were subjected to immunoblot using anti-ICAM-1 pAb (upper panel). For loading control, the membrane was re-probed with anti-tubulin mAb (bottom panel). E, TNF-α induces Cav-1 expression. HLMVECs were exposed to TNF-α (20 ng/ml; 0 – 6 h) and immunoblotted with anti-Cav-1 pAb as described in Fig. 1A. Results are representative of >3 experiments.
IKK-NBD Peptide Inhibits LPS-induced Cav-1 mRNA and Protein Expression
Expression of Cav-1 mRNA after LPS stimulation was elevated within 2 h after LPS stimulation and was maximal at 4 h (Fig. 2A). The membrane-permeant IKK-NBD peptide, which blocks the interaction of NEMO with the IKK, complex preventing its phosphorylation of IκB and degradation of IκB and activation of NF-κB (16), was used to assess the role of NF-κB in the mechanism of Cav-1 expression in cell and in vivo studies (described below). We treated HLMVECs with 100 μM IKK-NBD or mutant IKK-NBD peptide 1 h before LPS challenge. IKK-NBD blocked LPS induction of Cav-1 mRNA, whereas mutant IKK-NBD had no significant effect (Fig. 2B). IKK-NBD treatment also markedly reduced LPS-induced Cav-1 protein expression (Fig. 2C).
FIGURE 2.
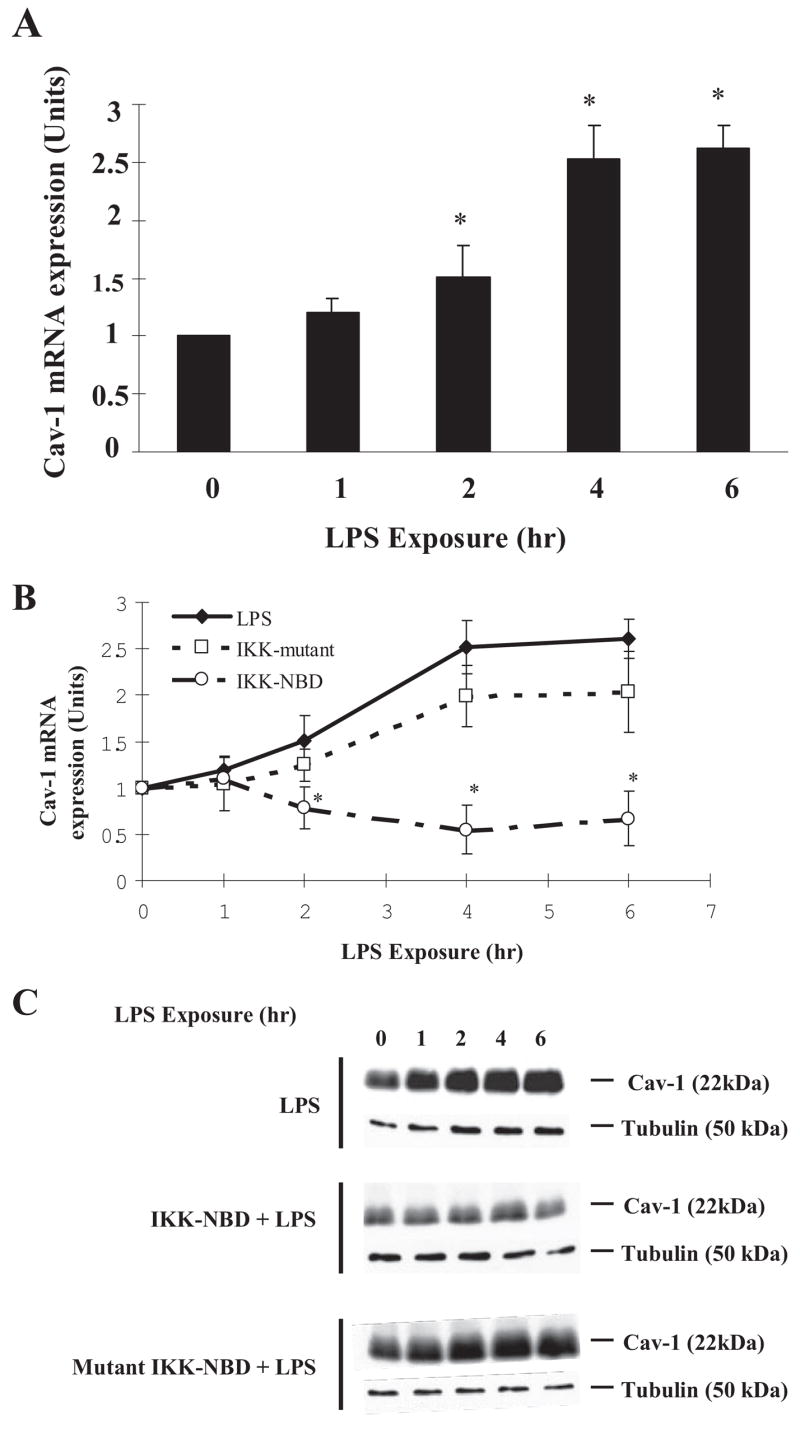
A, LPS induces Cav-1 mRNA expression. HLMVECs grown to confluence were exposed to LPS (4 μg/ml) for up to 6 h. After LPS treatment total RNA was isolated, and real-time quantitative-PCR was performed (see the details under “Experimental Procedures”). The graph shows -fold induction of Cav-1 mRNA normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA levels. Data are the mean ± S.E., n = 3; *, p < 0.05 compared with control (not stimulated with LPS). Results are representative of three experiments. B, IKK-NBD peptide prevents Cav-1 mRNA expression in LPS-activated endothelial cells. HLMVECs were pretreated with vehicle (Me2SO), IKK-NBD peptide (100 μM), or mutant-IKK-NBD (100 μM) for 1 h and then exposed to LPS (4 μg/ml) for up to 6 h. After LPS treatment, total RNA was isolated, and real-time quantitative-PCR was performed as described above. Data are the mean ± S.E., n = 3; *, p < 0.05 compared with LPS alone. C, IKK-NBD inhibits Cav-1 protein expression in LPS-activated endothelial cells. HLMVECs, pre-treated with either IKK-NBD peptide (100 μM) or mutant IKK-NBD peptide (100 μM), were challenged with LPS (0 – 6 h). Cell lysates were immunoblotted as in Fig. 1A. Results are representative of three independent experiments.
LPS Promotes NF-κB Binding to Intronic NF-κB Consensus Sites
We identified two putative NF-κB binding sites within intron 1, +435 to +460 relative to the translation start site in the human Cav-1 gene (Fig. 3A). To test whether LPS induces NF-κB binding to the introinc NF-κB consensus sequence, we incubated nuclear extracts from control and LPS-stimulated cells with 32P-labeled double-stranded Cav-1 gene intronic NF-κB oligonucleotides (Fig. 3A) and performed EMSA (see details under “Experimental Procedures”). Binding of nucleoproteins to DNA fragments generated two labeled complexes at slower (Band 1) and faster (Band 2) mobilities (Fig. 3B, left panel). DNA binding activity was absent when LPS-stimulated nuclear extract was incubated with 32P-labeled double-stranded mutant Cav-1 NF-κB oligonucleotide (Fig. 3B, middle panel). Using the ICAM-1 promoter-specific NF-κB oligonucleotide as a positive control for EMSA, we observed that LPS increased the NF-κB-DNA complex formation (Fig. 3B, right panel). Competition studies (70-fold excess unlabeled NF-κB oligonucleotide (cold probe) with labeled oligonucleotide (hot probe) in nuclear extracts from cells stimulated with LPS (Fig. 3C)) demonstrated specificity of NF-κB interaction with Cav-1 gene intronic NF-κB DNA sequence. To identify NF-κB subunits binding to the DNA, nuclear extracts were immunodepleted by preincubation with anti-p65 Ab or anti-p50 Ab. Band 1 was markedly reduced by anti-p65 Ab (Fig. 3D), indicating that LPS mediates p65/RelA binding to Cav-1 gene to transcription.
p65/RelA Knockdown Prevents LPS-induced to Cav-1 Expression
We transfected HLMVECs with siRNA specific to p65/RelA. At 48 h after siRNA transfection, HLMVECs were immunoblotted with anti-p65 Ab (see details under “Experimental Procedures”). p65/RelA expression was markedly reduced in p65-siRNA-transfected cells (Fig. 3E), whereas the scrambled (Control)-siRNA had no effect (Fig. 3F). LPS-induced Cav-1 protein expression was also prevented in p65-siRNA transfected cells compared with control (Fig. 3F), indicating that p65/RelA signaling plays an important role in the mechanism of LPS-induced Cav-1 expression in HLMVECs.
LPS Induces NF-κB-dependent Increase in Caveolae Number
Next we addressed whether the increased Cav-1 protein expression in response to LPS challenge was coupled to the formation of caveolae by electron microscopy. The mean number of vesicles per cell increased 2.6-fold compared with control after 4 h of LPS challenge (Table 1 and Fig. 4A). LPS resulted in greater number of membrane-associated vesicles communicating with the media and vesicles within the cytosol itself (Table 1). Treatment with IKK-NBD or IKK-NBD mutant failed to alter significantly the number of vesicles in HLMVECs under basal conditions, whereas IKK-NBD, but not mutant IKK-NBD, prevented the LPS-induced increase in vesicle number (Table 1 and Fig. 4A). Confocal microscopy also showed that Cav-1-positive vesicles were significantly increased after LPS challenge compared with control cells (Fig. 4B).
TABLE 1. NF-κB-dependent increase in vesicle number in endothelial cells induced by LPS challenge.
The number of vesicles in cultured HLMVECs was quantified from electron micrographs; 215 micrographs were used for each of the first three experimental conditions, and 117 micrographs were used for each of the last three conditions. In column 1, the mean number refers to the number of endothelial cells with complete profiles quantified per micrograph. In columns 2 and 3, the mean numbers refer to the number of vesicles counted per micrograph. Column 4 refers to the total vesicle number normalized to number of cells counted. Data in first three columns are shown as mean ± S.E.
| No. cell profiles counted | No. vesicles communicating with media | No. cytosolic vesicles | No. vesicles/cell | |
|---|---|---|---|---|
| Control | 62 ± 9 | 4677 ± 112 | 4398 ± 92 | 146 |
| LPS | 58 ± 12 | 8944 ± 322a | 13105 ± 446a | 380a |
| IKK-NBD | 44 ± 7 | 3348 ± 191 | 2961 ± 96 | 143 |
| IKK-NBD + LPS | 69 ± 11 | 4235 ± 205b | 4957 ± 133b | 133b |
| Mutant-IKK-NBD | 35 ± 8 | 2268 ± 191 | 2359 ± 221 | 132 |
| Mutant-IKK-NBD + LPS | 32 ± 4 | 4922 ± 177 | 4209 ± 325 | 285 |
p < 0.05 versus control.
p < 0.05 versus LPS challenge.
FIGURE 4.
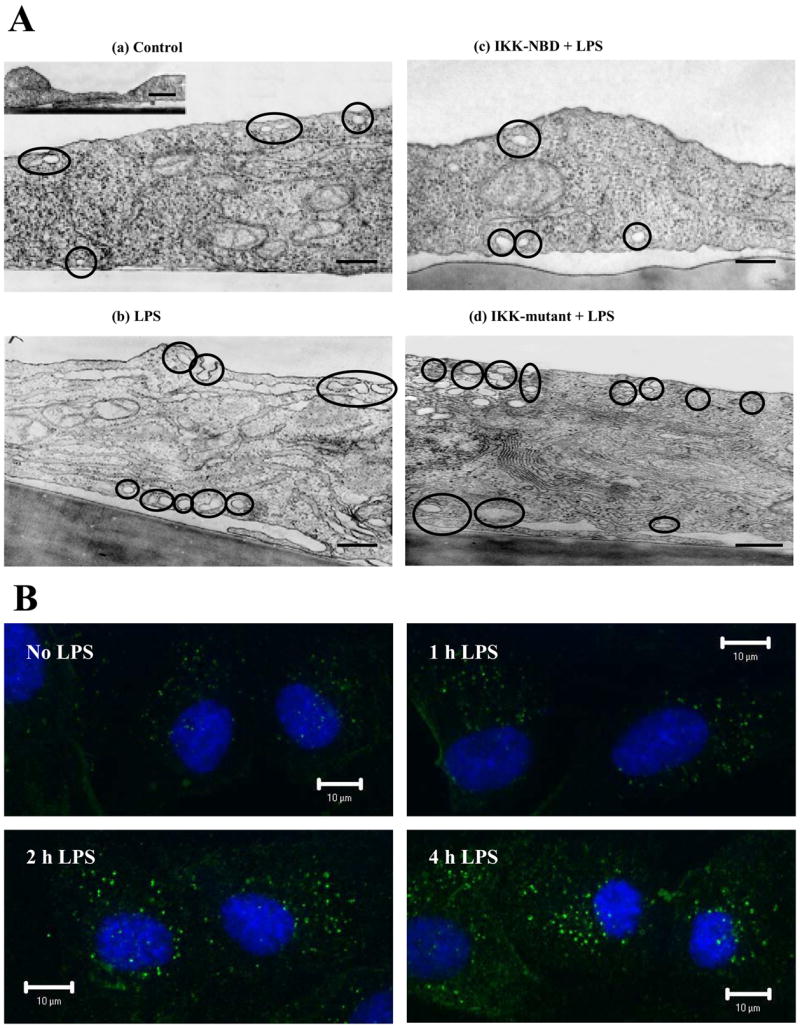
A, LPS challenge of endothelial cells increases caveolae number and its dependence on NF-κB. Shown are electron micrographs of control HLM-VECs (a), LPS-treated HLMVECs, LPS concentration of 4 μg/ml, 4 h (b), IKK-NBD peptide (100 μM) plus LPS (4 μg/ml, 4 h)-treated HLMVECs (c), and mutant IKK-NBD peptide (100 μM) plus LPS ((4 μg/ml, 4 h)-treated HLMVECs (d). Data are representative of Table 1 Data. Scale bar, 200 nm. B, LPS-induced increase in number of Cav-1-positive vesicles is dependent on NF-κB. HLMVECs were challenged with 4 μg/ml LPS for up to 4 h, fixed, and stained with anti-Cav-1 pAb plus Alexa 488-labeled secondary Ab. Confocal fluorescence imaging was used to detect the presence of Cav-1-positive vesicles. Representative images from three experiments are shown.
A NF-κB- and Cav-1-dependent Increase in Endothelial Permeability Induced by LPS
To assess whether increased endothelial permeability induced by LPS occurred via opening of the inter-endothelial junctional pathway, we measured TER (20). LPS had no significant effect of TER (see supplemental data). Also, addition of treatment of cells with either IKK-NBD or mutant IKK-NBD had no significant effect on TER (see the supplemental data). We challenged HLMVECs with LPS and stained with anti-VE-cadherin mAb to assess adherens junction integrity (see the supplemental data). LPS exposure for up to 4 h did not induce junctional disassembly (see the supplemental data).
We next addressed the effects of LPS on transendothelial permeability by measuring 125I-labeled albumin flux. LPS exposure (4 h at 4 μg/ml) increased transendothelial 125I-labeled albumin permeability by 80% from control (8.6 ± 1.9 to 17.1 ± 2.0 nl/min/cm2) (Fig. 5A). IKK-NBD prevented the response (7.9 ± 2.7 nl/min/cm2), whereas mutant IKK-NBD had no significant effect (14.5 ± 3.0 nl/min/cm2). Cav-1-specific siRNA was used to determine whether increased endothelial permeability induced by LPS was the result of increased Cav-1 expression. Immunoblotting showed that 72 h treatment of HLM-VECs with Cav-1 siRNA significantly reduced Cav-1 expression (Fig. 5B). Basal endothelial albumin permeability also decreased by 60% in Cav-1-depleted monolayers compared with control siRNA-treated cells (from 8.6 ± 1.9 to 3.5 ± 1.3 nl/min/cm2) (Fig. 5A). In addition, Cav-1 siRNA prevented the LPS-induced increase in transendothelial albumin permeability, whereas control siRNA treatment had no effect (Fig. 5A).
FIGURE 5. Contribution of NF-κB in mediating increased transendothelial 125I-labeled-albumin permeability induced by LPS.
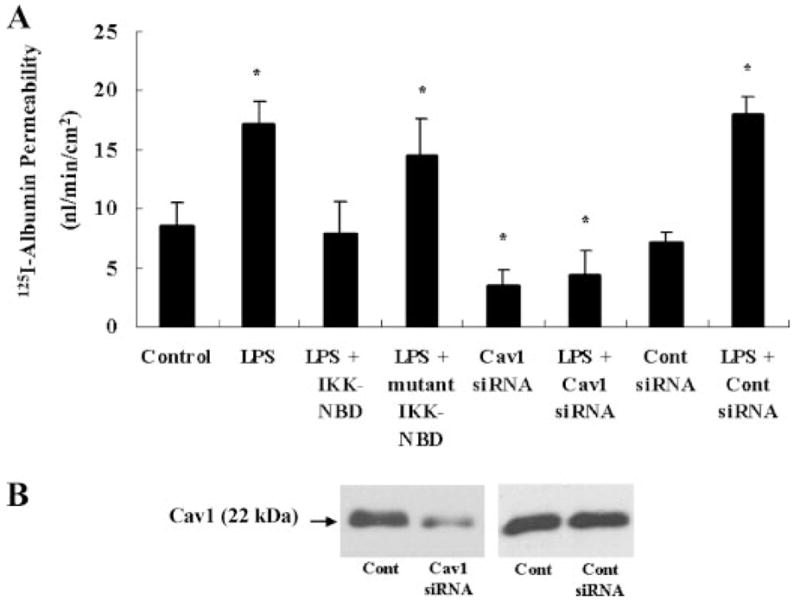
A, 125I-labeled albumin transport was determined in HLMVECs grown to confluence in Transwell inserts (21). Transcellular flux across endothelial monolayers treated with IKK-NBD, mutant IKK-NBD, Cav-1 siRNA, or control siRNA was measured. Data are expressed as nl/min/cm2, mean ± S.E., n = 3; *, p < 0.05 versus control. B, immunoblot analysis of HLMVECs transfected with Cav-1 siRNA or control siRNA for 72 h.
Inhibition of NF-κB Activation Prevents LPS-induced Increase in Vascular Permeability in Mouse Lungs
To address the in vivo relevance of LPS-induced Cav-1 expression, we prevented NF-κB-mediated Cav-1 expression by treating mice with IKK-NBD. Fig. 6A depicts the protocol of the experiments. LPS challenge (6 h) increased lung Cav-1 expression >3-fold compared with control (Fig. 6B). IKK-NBD peptide treatment prevented the LPS-induced increase in Cav-1 expression in mouse lung tissue, whereas IKK-mutant peptide was ineffective as expected (Fig. 6B). LPS challenge increased lung microvascular permeability to 125I-labeled albumin >2-fold (Fig. 6C), and this increase was prevented by IKK-NBD peptide but not IKK-mutant peptide (Fig. 6C), indicating that NF-κB-dependent Cav-1 expression contributes to LPS-induced increase in vascular permeability.
FIGURE 6. NF-κB inhibitor prevents LPS-induced Cav-1 expression and increased lung vascular permeability in mice.
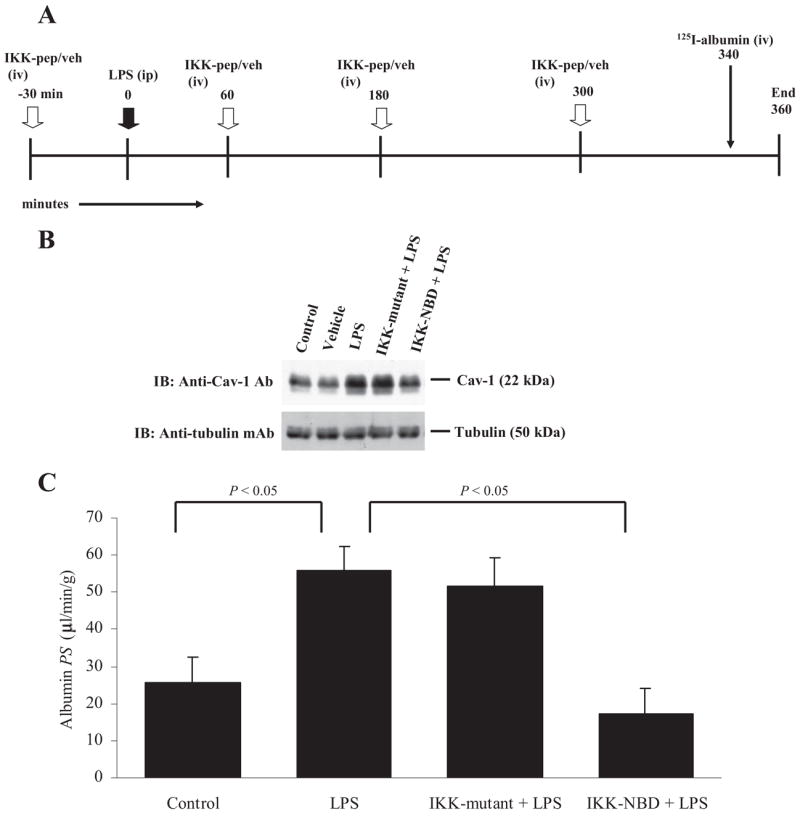
Panel A shows the timeline for in vivo experiments in mice challenged with LPS (intraperitoneal) at zero time. In the experimental group multiple injections of IKK-NBD peptide were made before (at 30 min) and after LPS (at 60, 180, and 300 min). In control experiments we replaced the active peptide with an inactive mutant peptide or with the vehicle. At 340 min, tracer 125I-labeled albumin was injected intravenously. At 360 min blood samples were taken, and lungs were flushed with unlabeled albumin solution to remove the vascular 125I-labeled albumin tracer; lungs were excised, weighed, and counted for γ radioactivity. In separate experiments employing identical steps, tissue homogenate was made from excised lungs for Western blot analysis. In B, lung tissue from each group mice was used for immunoblotting with anti-Cav-1 Ab to determine Cav-1 expression. The membrane was stripped and probed with anti-tubulin mAb for loading control. Note that Cav-1 expression increased >3-fold after LPS challenge, and the increase was prevented by treating with IKK-NBD peptide but not with IKK-mutant peptide. In C, LPS challenge increases lung microvessel 125I-labeled albumin permeability-surface area product 2-fold, and this increase was also prevented by IKK-NBD peptide but not the inactive IKK-mutant peptide (n = 4 in each group).
DISCUSSION
The bacterial endotoxin LPS contributes to the mechanism of inflammation in acute lung injury and other inflammatory diseases (23, 24). LPS stimulation results in increased lung microvascular endothelial permeability and pulmonary edema formation (25), both characteristic features of the acute lung injury syndrome (6). It has been shown that LPS exposure of endothelial cells induces the expression of a broad array of proteins involved in the host defense and inflammatory responses (12). Expression of many of these proteins (e.g. ICAM-1) is dependent on activation of the transcription factor NF-κB (6). We followed the lead of previous observations that Cav-1, the 22-kDa primary constituent of caveolae, the invaginated plasma membrane structures abundant in endothelial cells, adipocytes, and vascular smooth muscle cells (26), may also be upregulated by LPS (12). Here, we observed that LPS induced a significant 5-fold increase in Cav-1 expression at pathophysiologically relevant LPS concentration of ~100 ng/ml (27). Cav-1 protein expression increased at 1 h after LPS exposure, whereas the mRNA increase was somewhat delayed. This delay may be ascribed to the stability of Cav-1 mRNA as shown for a number of genes (28). RNA-binding proteins (e.g. human antigen R) stabilize mRNA by binding to the AU-rich elements in the 3′-untranslated region (28). Antigen R present in the nucleus is translocated to the cytosol upon LPS challenge where it stabilizes mRNA (29). Knockdown of antigen R using siRNA in human aortic smooth muscle cells inhibited LPS-induced TLR4 mRNA expression (29). The human Cav-1 mRNA sequence shows similar AU-rich elements in the 3′-untranslated region (28), suggesting that RNA-binding proteins may control Cav-1 mRNA stability in a similar manner.
Studies have identified sterol-responsive elements in the human Cav-1 gene 5′-regulatory region between −781 and −106 (i.e. upstream of ATG) that are required for induction of Cav-1 expression in response to cholesterol (30). In the present study we identified two putative NF-κB binding sites within intron 1 of the human Cav-1 gene that also transcriptionally regulate Cav-1. Similar intronic NF-κB binding sites have been identified in other genes (e.g. MHC class I-related chain A and polymeric Ig receptor) in mucosal epithelial cells and T lymphocytes (31, 32). We observed by EMSA that LPS induced NF-κB binding to the intronic NF-κB sites in Cav-1 gene, suggesting that these sites are functionally competent in the induction of Cav-1 transcription.
LPS is known to activate the IKK-mediated IκB phosphorylation that leads to p65/p50 heterodimer translocation to the nucleus to induce transcription (15). Thus, to address the role of IKK in mediating Cav-1 expression, we used the cell-permeant IKK-NBD peptide that prevents NEMO/IKKγ association with IKKα and IKKβ required for NF-κB activation (16). We showed that IKK-NBD, but importantly not its mutant form, prevented LPS-induced Cav-1 mRNA and protein expression. Knockdown of p65/RelA expression utilizing siRNA also prevented LPS-induced Cav-1 expression. Together, these observations show that NF-κB signaling is required for Cav-1 expression in endothelial cells.
We carried out an extensive electron microscopic analysis of endothelial cell alterations occurring as the result of Cav-1 expression induced by LPS. The most notable finding was a marked increase in the number of caveolae. The increase was evident both in the number of plasmalemmal-associated vesicles and free cytosolic vesicles. Previous studies have shown that exogenous expression of Cav-1 in Cav-1-null cells induced the formation of caveolae (33, 34); thus, our findings are in accord with the requirement of Cav-1 in the formation of caveolae. Because increased number of vesicles could be both caveolae and endosomes, we used confocal microscopy to address the relative number of each population. We observed an increase in the Cav-1-positive vesicle number induced by LPS, indicating that they were caveolae. Importantly, treatment of endothelial cells with IKK-NBD to inhibit NF-κB activation did not significantly change the basal caveolae number, but it did prevent the LPS-induced increase number. Thus, LPS activation of NF-κB increases caveolae number secondary to the up-regulation of Cav-1 expression. In a previous study (35), a 2-fold increase in endothelial-specific Cav-1 expression in mice failed to increase caveolae number. This difference may be attributed to the significantly greater (5-fold) increase in Cav-1 protein expression seen in the present study. In addition there was an important difference in that we used LPS to increase Cav-1 expression as compared with the overexpression of Cav-1 studied previously (35).
Because caveolae are vesicle carriers responsible for transendothelial permeability of albumin (7), we addressed the possibility that the LPS-induced Cav-1 expression and increased caveolae number contributed to the mechanism of LPS-mediated increased permeability. We observed that indeed LPS increased transendothelial 125I-labeled albumin permeability and that the response was significantly reduced by pretreating cells with IKK-NBD to inhibit NF-κB activation as well as suppressing Cav-1 expression using siRNA. Thus, our findings demonstrate an important role of NF-κB-dependent Cav-1 expression and resultant increased caveolae-mediated transcytosis in contributing to increased transendothelial albumin permeability induced by LPS.
We ruled out the possibility that opening of inter-endothelial junctions was responsible for the increase in endothelial permeability. Real-time changes in TER, sensitive to increased endothelial permeability via the junctional pathway (20), were measured. LPS challenge of endothelial cells did not significantly decrease TER; however, in a positive-control experiment, thrombin induced a junctional opening reflected by the decreased TER. At the morphological level adherens junctions were also not altered, consistent with the absence of LPS-induced junction disruption.
We have shown previously that LPS-induced increase in endothelial permeability was significantly reduced in Cav-1-null mice (36). The endothelial barrier-protective effect of Cav-1 gene deletion was the result of high levels of eNOS-derived NO production generated after LPS exposure and, thereby, NO-dependent inhibition of neutrophil-mediated vascular lung injury. In the present study we have studied a different question; that is, the role of LPS in mediating Cav-1 expression through activation of NF-κB in endothelial cells. To address whether NF-κB-induced Cav-1 expression can also increase transendothelial albumin transport in vivo, we determined the effects of LPS on Cav-1 expression and lung microvessel permeability to 125I-labeled albumin in mice. We observed that intraperitoneal LPS challenge increased Cav-1 expression 3-fold and lung microvessel permeability to 125I-labeled albumin 2-fold in mice. Also, IKK-NBD peptide pre-treatment prevented the LPS-induced Cav-1 expression as well as the increase in lung microvessel albumin permeability, consistent with the endothelial monolayer data described above.
In summary, we show here that NF-κB signaling mediates the LPS-induced expression of Cav-1 and increases caveolae-mediated transendothelial albumin permeability. Our previous studies showed that increased albumin permeability through transcytosis results in increased transport of plasma proteins carried in the fluid phase of caveolae (37). Thus, a possible function of increased transcytosis induced by LPS may be the transport across of the endothelial barrier of acute phase plasma proteins and immunoglobulins involved in innate immunity.
Supplementary Material
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M703153200/DC1
Footnotes
This study was supported by National Institutes of Health Grants P01HL060678, P01HL077806, R01GM58531, and R01HL045638.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
The abbreviations used are: LPS, lipopolysaccharide; HLMVEC, human lung microvascular endothelial cell; Cav-1, caveolin-1; NEMO, NF-κB essential modifier binding domain; siRNA, small interfering RNA; ICAM-1, intercellular adhesion molecule 1; IκB, inhibitory protein I-κB; NBD, NEMO binding domain; FBS, fetal bovine serum; Ab, antibody; pAb, polyclonal Ab; mAb, monoclonal Ab; EMSA, electrophoretic mobility shift assay; IKK, IkB kinase; TER, transendothelial electrical resistance.
References
- 1.Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, Kubes P. J Clin Invest. 2003;111:1011–1020. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faure E, Equils O, Sieling PA, Thomas L, Zhang FX, Kirschning CJ, Polentarutti N, Muzio M, Arditi M. J Biol Chem. 2000;275:11058–11063. doi: 10.1074/jbc.275.15.11058. [DOI] [PubMed] [Google Scholar]
- 3.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 4.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang G, Ghosh S. J Endotoxin Res. 2000;6:453–457. doi: 10.1179/096805100101532414. [DOI] [PubMed] [Google Scholar]
- 6.Bannerman DD, Goldblum SE. Lab Investig. 1999;79:1181–1199. [PubMed] [Google Scholar]
- 7.Mehta D, Malik AB. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 8.Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, Malik AB. J Cell Biol. 2000;150:1057–1070. doi: 10.1083/jcb.150.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 10.Schubert W, Frank PG, Razani B, Park DS, Chow CW, Lisanti MP. J Biol Chem. 2001;276:48619–48622. doi: 10.1074/jbc.C100613200. [DOI] [PubMed] [Google Scholar]
- 11.Pascariu M, Bendayan M, Ghitescu L. J Histochem Cytochem. 2004;52:65–76. doi: 10.1177/002215540405200107. [DOI] [PubMed] [Google Scholar]
- 12.Zhao B, Bowden RA, Stavchansky SA, Bowman PD. Am J Physiol Cell Physiol. 2001;281:C1587–C1595. doi: 10.1152/ajpcell.2001.281.5.C1587. [DOI] [PubMed] [Google Scholar]
- 13.Lei MG, Morrison DC. Infect Immun. 2000;68:5084–5089. doi: 10.1128/iai.68.9.5084-5089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lei MG, Tan X, Qureshi N, Morrison DC. Infect Immun. 2005;73:8136–8143. doi: 10.1128/IAI.73.12.8136-8143.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayden MS, Ghosh S. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 16.May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 17.Shajahan AN, Tiruppathi C, Smrcka AV, Malik AB, Minshall RD. J Biol Chem. 2004;279:48055–48062. doi: 10.1074/jbc.M405837200. [DOI] [PubMed] [Google Scholar]
- 18.Rahman A, Kefer J, Bando M, Niles WD, Malik AB. Am J Physiol. 1998;275:L533–L544. doi: 10.1152/ajplung.1998.275.3.L533. [DOI] [PubMed] [Google Scholar]
- 19.Miyawaki-Shimizu K, Predescu D, Shimizu J, Broman M, Predescu S, Malik AB. Am J Physiol Lung Cell Mol Physiol. 2006;290:L405–L413. doi: 10.1152/ajplung.00292.2005. [DOI] [PubMed] [Google Scholar]
- 20.Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Proc Natl Acad Sci U S A. 1992;89:7919–7923. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.John TA, Vogel SM, Tiruppathi C, Malik AB, Minshall RD. Am J Physiol Lung Cell Mol Physiol. 2003;284:L187–L196. doi: 10.1152/ajplung.00152.2002. [DOI] [PubMed] [Google Scholar]
- 22.Vogel SM, Gao X, Mehta D, Ye RD, John TA, Andrade-Gordon P, Tiruppathi C, Malik AB. Physiol Genomics. 2000;4:137–145. doi: 10.1152/physiolgenomics.2000.4.2.137. [DOI] [PubMed] [Google Scholar]
- 23.Stephens KE, Ishizaka A, Larrick JW, Raffin TA. Am Rev Respir Dis. 1988;137:1364–1370. doi: 10.1164/ajrccm/137.6.1364. [DOI] [PubMed] [Google Scholar]
- 24.Ware LB, Matthay MA. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 25.Lantz RC, Keller GE, 3rd, Burrell R. Am Rev Respir Dis. 1991;144:167–172. doi: 10.1164/ajrccm/144.1.167. [DOI] [PubMed] [Google Scholar]
- 26.Razani B, Woodman SE, Lisanti MP. Pharmacol Rev. 2002;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Tupper JC, Bannerman DD, Winn RK, Rhodes CJ, Harlan JM. Infect Immun. 2003;71:4414–4420. doi: 10.1128/IAI.71.8.4414-4420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misquitta CM, Chen T, Grover AK. Cell Calcium. 2006;40:329–346. doi: 10.1016/j.ceca.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Lin FY, Chen YH, Lin YW, Tsai JS, Chen JW, Wang HJ, Chen YL, Li CY, Lin SJ. Arterioscler Thromb Vasc Biol. 2006;26:2622–2629. doi: 10.1161/01.ATV.0000246779.78003.cf. [DOI] [PubMed] [Google Scholar]
- 30.Bist A, Fielding PE, Fielding CJ. Proc Natl Acad Sci U S A. 1997;94:10693–10698. doi: 10.1073/pnas.94.20.10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molinero LL, Fuertes MB, Girart MV, Fainboim L, Rabinovich GA, Costas MA, Zwirner NW. J Immunol. 2004;173:5583–5590. doi: 10.4049/jimmunol.173.9.5583. [DOI] [PubMed] [Google Scholar]
- 32.Schjerven H, Tran TN, Brandtzaeg P, Johansen FE. J Immunol. 2004;173:1849–1857. doi: 10.4049/jimmunol.173.3.1849. [DOI] [PubMed] [Google Scholar]
- 33.Fra AM, Williamson E, Simons K, Parton RG. Proc Natl Acad Sci U S A. 1995;92:8655–8659. doi: 10.1073/pnas.92.19.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engelman JA, Wykoff CC, Yasuhara S, Song KS, Okamoto T, Lisanti MP. J Biol Chem. 1997;272:16374–16381. doi: 10.1074/jbc.272.26.16374. [DOI] [PubMed] [Google Scholar]
- 35.Bauer PM, Yu J, Chen Y, Hickey R, Bernatchez PN, Looft-Wilson R, Huang Y, Giordano F, Stan RV, Sessa WC. Proc Natl Acad Sci U S A. 2005;102:204–209. doi: 10.1073/pnas.0406092102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garrean S, Gao XP, Brovkovych V, Shimizu J, Zhao YY, Vogel SM, Malik AB. J Immunol. 2006;177:4853–4860. doi: 10.4049/jimmunol.177.7.4853. [DOI] [PubMed] [Google Scholar]
- 37.Tiruppathi C, Song W, Bergenfeldt M, Sass P, Malik AB. J Biol Chem. 1997;272:25968–25975. doi: 10.1074/jbc.272.41.25968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M703153200/DC1