

Abstract
Much evidence has gathered that nitric oxide (NO) signaling, via cGMP-dependent mechanisms, may activate pro-survival pathways in hippocampal neurons and inhibit apoptosis. Past research has revealed that the enhancement of monoaminergic neurotransmission via exercise or treatment with antidepressant medications leads to an enhanced expression of brain-derived neurotrophic factor (BDNF). In isolated hippocampal neurons, norepinephrine (NE) application also increases the immunoreactivity of BDNF and several pro-survival signaling molecules. The data herein support the possibility that NO signaling plays an important role in enhancing neurotrophin expression and activation of the pro-survival phosphatidylinositol 3′ kinase (PI-3K) pathway stimulated by NE. In isolated hippocampal neurons, the NO donor, sodium nitroprusside, increases BDNF, PI-3K, and phospho-ERK1 immunoreactivity. Specific inhibitors of the NO system suggest that NE-induced increases in hippocampal BDNF and the PI-3K pathway, but not stimulation of the MAPK pathway, depend upon NO signaling. In addition, inhibiting cGMP suggest that the effects of NE on BDNF immunoreactivity and Akt phosphorylation are also cGMP- dependent. Finally, the application of l-NAME to hippocampal neurons increases cell death. This is the first study of its kind demonstrating the involvement of NE-induced pro-survival signaling in three distinct signaling pathways: PI-3K, MAPK, and NO/cGMP. Possible mechanisms are discussed in light of the results.
Keywords: BDNF, nitric oxide, hippocampus, Akt, MAPK
INTRODUCTION
Nitric oxide (NO) is a volatile gas that has been attributed with the ability to promote both cell survival and neuronal death, depending upon the levels of NO present, as well as the specific cellular system involved (Araujo and Carvalho, 2005). Although high levels of NO can lead to tissue damage via the rapid formation of free radical species (Maher and Schubert 2000), this molecule can also play a prominent role in neural plasticity (Contestabile, 2000; Garthwaite and Boulton, 1995; Holscher, 1997) through highly specific signaling mechanisms Gross and Wolin, 1995; Bredt, 1999) that are capable of directing cellular activities via the activation of protein kinases (Holscher, 1997). This highly important biological action of NO involves its binding to the heme moiety in the heterodimeric enzyme, soluble guanylate cyclase. Activation of this enzyme by NO results in the production of the second messenger molecule, cGMP, which can regulate numerous physiological events such as vasodilatation and neurotransmission (Krumenacker et al., 2004). As NO is closely associated with the NMDA receptor complex (Contestabile, 2000), via this interaction, glutamatergic activation following sensory stimulation, learning or heightened neuronal activity activates the synthesis of NO, which in turn activates cGMP (Garthwaite and Boulton, 1995). As noted above, via the activation of protein kinases, cGMP activation may lead to growth or regenerative activities within the cell (Hindley et al., 1997). Therefore, NO signaling could be one means by which environmental enrichment, learning and/or physical activity lead to enhanced neuronal survival or growth.
In recent years, it has been observed that chronic treatment with antidepressant medications enhances the expression of a key neurotrophin, brain-derived neurotrophic factor (BDNF), in the hippocampus and neocortex, and that increase in BDNF may be an important aspect of the therapeutic response (Duman et al., 1997). General physical activity (exercise, voluntary wheel running) also increases BDNF (Neeper et al., 1996). This effect is more rapid than antidepressants (enhanced BDNF transcription is evident within hours rather than weeks), and voluntary wheel running combined with antidepressant treatment potentiates BDNF mRNA expression in an additive manner (Oliff et al., 1998; Russo-Neustadt et al., 1999). Other evidence exists that antidepressant-induced regulation and activity-induced changes in BDNF transcription may occur through convergent intracellular mechanisms (Russo-Neustadt et al., 2000).
Both antidepressent treatment and voluntary exercise enhance BDNF transcription through increased activation of monoaminergic neurotransmitter signaling, and evidence is particularly strong that exercise-induced changes in BDNF are dependent upon norepinephrine (NE) activation (Garcia et al., 2003; Ivy et al., 2003; Russo-Neustadt et al., 2005). This exercise-induced NE release activates the protein kinase A/cAMP pathway, just as increased intrasynaptic NE and/or 5-HT following antidepressant treatment is thought to do (Duman et al., 2001; Russo-Neustadt and Chen, 2005) leading to CREB activation and enhanced BDNF synthesis. In addition to this mechanism, one means by which exercise can rapidly and robustly increase BDNF is via glutamatergic activation and NO signaling. General physical activity enhances NE content (Dishman et al., 2000) and metabolism (Dunn et al., 1996) and glutamate (Leung et al., 2006; Richter-Levin et al., 1998) release, as well as NMDA receptor levels (Farmer et al., 2004; Molteni et al., 2002) within the hippocampus. These stimuli activate NO synthesis (Fedele et al., 2001), which is capable of enhancing NE release through increased glutamate release and rapid amplification of the signal (Lonart et al., 1992; Stout and Woodward, 1994, 1995). In addition, NO is a mediator of calcium-dependent activation of the G-protein p21 Ras (Ras) through NMDA receptor stimulation (Deora et al., 1998; Koo et al., 2005). By this means, the pro-survival phosphatidylinositol 3′ kinase (PI-3K) pathway is activated, leading to the subsequent transcription of many pro-survival genes (Kang et al., 2004).
The hypothesis that exercise-induced increases in BDNF may be dependent upon NO synthesis was recently tested by treating animals chronically with the NO synthase inhibitor, l-NAME (Chen et al., 2006). The results supported this hypothesis, as exercising l-NAME treated animals did not show increased hippocampal BDNF mRNA, unlike their vehicle-treated counterparts. On the other hand, increases in BDNF transcription resulting from an exercise/antidepressant combination were not prevented with concurrent l-NAME treatment (Chen et al., 2006). It is possible that only the exercise-induced increases in BDNF, and not those stimulated by antidepressant treatment, are NO-dependent.
In both the whole animal and in cell culture models, exercise or NE application respectively increase cell survival signaling via activation of the PI-3K pathway (Chen and Russo-Neustadt, 2005; Chen et al., 2007). In the current study, we wished to assess whether the phenomenon of enhanced BDNF expression following the application of NE is also dependent upon NO signaling. In addition, we have investigated whether cGMP activation is necessary and vital for the observed increase in BDNF and/or PI-3K within hippocampal neurons.
MATERIALS AND METHODS
Antibodies
Antibodies were purchased from the following sources: anti-BDNF (Santa Cruz Biotech, Santa Cruz, CA); anti-phospho-T308-Akt and anti-Akt (Cell Signaling, Beverly, MA); anti-phospho- ERK1/2, anti-ERK1/2, anti-PI-3K, and anti-α-tubulin (Upstate Biotech.Inc., Charlottesville, VA).
Hippocampal Dissection at Embryonic Day 18 (E18) and Tissue Culture
Hippocampi of each embryo were excised and cultured according to the method of Banker and Cowan (Banker and Cowan, 1977). Briefly, on their 18th day of pregnancy, female rats (Sprague-Dawley) were sacrificed using an overdose of isofluorane, followed by rapid decapitation. E18 embryos were then removed by cesarean section and immediately placed on ice. Neurons were dissected in Ca-Mg-free Medium (CMF) and rinsed in CMF twice in 15-ml conical tube (5-10 ml/rinse). 0.125% trypsin (in CMF, ~ 4-5 ml) was then added for 5-10 min in a water bath (37°C), and gently inverted every 2-3 min. Neurons were quenched in 10% FBS/DMEM (warmed to 37°C, 1-2 volumes; 2 volumes ~ 8 ml) to stop the reaction and centrifuged at 200 g for 5 min. The supernatant was then carefully removed and the pellet resuspended in ~2 ml serum-free (SF) DMEM. Neurons were then triturated with a constricted glass pipette, filtered through a 40-μm cell strainer (Falcon) into a new 50-ml conical tube, and the volume supplemented to 5 ml with serum-free DMEM/1 % N2 supplements. Ten μl trypan blue was then added to 10 μl cells and then counted in a hemocytometer. Neurons were plated in 12-well, poly-d-lysine-coated plates (each well 3.8 cm2) at a density of 50,000 per cm2. Treatment of hippocampal neuronal cultures (below) was conducted in duplicate.
Application of Compounds
Sources and handling
Norepinephrine, hemoglobin (Hb), Nω-nitro-l-arginine methyl ester (l-NAME), sodium nitroferricyanide (III) dihydrate (sodium nitroprusside, SNP), hemoglobin (Hb), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), and 8-bromoguanosine 3′:5′-cyclic monophosphate (8-Br-cGMP) were purchased from Sigma (St. Louis, MO). All compounds were dissolved in DMEM/SF, except for ODQ, which was initially dissolved in DMSO. At no time, did the DMSO concentration exceed 0.5% of the total volume in the tissue culture well.
Pre-treatment and treatment procedures
One day following plating, l-NAME (500 μM), SNP (500 μM), and/or Hb (380 μM) were added to the designated cells. Cells were then returned to the incubator (37°C, 5% CO2). On the third day following plating, NE (100 nM) was added to the appropriate treatment conditions and allowed to incubate for 2 hr; 8-Br-cGMP (25 μM) or ODQ (50 μM) were added to the appropriate treatment conditions allowed to incubate for 1 hr. The design was implemented so that all cells were of the same age when they were lysed and harvested for subsequent SDS-PAGE and Western blotting. All inhibitors and drugs were prepared in semi-darkness and made fresh just before each use. Cells used as controls were treated with the same vehicle as that used to dissolve the inhibitor (SF DMEM or DMSO).
Harvesting Cultured Hippocampal Neurons
Cultured hippocampal neurons from 2 pooled tissue culture wells were suspended in hot lysis buffer (~ 80°C), boiled for 5 min, pushed through a 26-gauge syringe 10 times to sheer genomic DNA and decrease sample viscosity, centrifuged for 5 min at 14,000 rpm, and the supernatant saved at -20°C until ready for protein analyses and immunoblotting. Protein concentrations were determined using the Lowry method (Lowry et al., 1951) with bovine serum albumin as the standard.
SDS-PAGE/Western blotting
Equal amounts of protein were applied to each well on a 10% polyacrylamide gel and electrophoresed according to established procedures (Laemmli, 1970). Proteins were then electrotransfered to nitrocellulose membranes (Amersham, Pharmacia-Biotech, Piscataway, NJ) and immunoblotted with the appropriate antibody according to the respective manufacturer’s specifications. Blots were then treated with the reagents for enhanced chemuminescence (ECL. Amersham, Pharmacia-Biotech, Piscataway, NJ) and then apposed to hyperfilm (Amersham, Pharmacia-Biotech, Piscataway, NJ). In the determination of phosphorylated protein levels, each blot was then stripped (100 mM 2-mercapto-ethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7, 55°C, 10 min, with agitation) and re-probed using the total form of the respective antibody, and Western blotting repeated according to the respective manufacturer’s instructions, followed by ECL. To control for inadvertent differences in protein loading and variability in transfer efficiency, all blots were stripped once again and re-probed with anti-α-tubulin in accordance with the manufacturer’s instructions, again followed by ECL. Specific dilution of the primary and secondary antibodies during western blotting was conducted in accordance with the respective manufacturer’s recommendations. Western blotting for each treatment condition was performed 2-3 times.
Quantification of immunoreactive bands was performed using computer-assisted densitometry (MCID image processing system, St. Catherine’s, Ontario, Canada). Lightly exposed bands were quantified within the linear range of a standard curve. The optical density of each band was divided by that of its respective α-tubulin band to yield the corrected band intensity. Data were then statistically normalized with respect to values obtained with vehicle–treated control neurons (controls). All data analyses entailed using a one-way ANOVA, followed by Fisher’s post-hoc least significant difference (PLSD) multiple comparisons test with a significance level of p < 0.05.
Cell Death Assay
All experiments were conducted concurrently with those used for the Western blotting procedures (above), but in separate tissue culture plates. Neuronal cell death was evaluated using the Live/Dead Cytotoxicity Kit (Molecular Probes, Eugene, OR). Two-three fields within each tissue culture well were counted for numbers of live and dead cells, determined by fluorescent FITC and Texas Red staining, respectively. As for the Western blotting experiments, statistical analyses of data entailed using a one-way ANOVA, followed by Fisher’s PLSD multiple comparisons test with a significance level of p < 0.05.
RESULTS
NE increases BDNF, PI-3K, P-Akt and P-ERK1 Immunoreactivity
Application of NE to E18 hippocampal neurons led to a time-and concentration-dependent increase in BDNF, PI-3K and P-ERK. Previous studies revealed that exposure to 100 nM NE for 2 hours led to peak levels of BDNF immunoreactivity in these cultures (Chen et al. 2007). These conditions were therefore used for our subsequent experiments. Under these conditions, NE exposure led to a robust increase in BDNF expression [F(15,32) = 16.46, p < .0001] (Figure 1). Similarly, NE also increased the expression of PI-3K [F(15,32) = 7.27, p < .0001] (Figure 2), P-Akt [F(15,32) = 5.42, p < 0.0001] (Figure 3) and P-ERK1 [F(15,32) = 13.68, p < .0001] (Figure 4).
Figure 1.
NE-induced increases in BDNF expression in cultured hippocampal neurons are NO-dependent. Relative levels of BDNF immunoreactivity, as determined by Western blotting, reveal that NE and/or SNP increased BDNF immunoreactivity. These results were reversed when neurons were co-incubated with the NO synthase inhibitor, l-NAME. Western blotting experiments were conducted and analyzed as specified in Experimental Procedures. Asterisks denote a treatment that is significantly different from vehicle-treated controls (p < .05). Lower-case “a” indicates that a treatment is significantly different from NE-treated cells (p < .05). Other relevant treatments that are significantly different from each other at p < .05 are indicated by the bracket. Data are the mean + SEM. Each experiment was conducted 2-3 times, with each sample measured in duplicate.
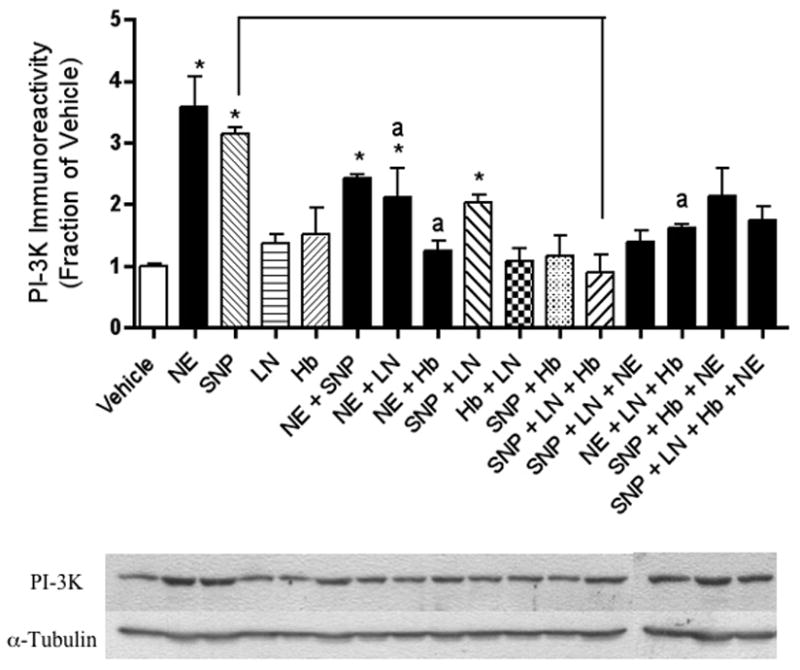
Figure 2.
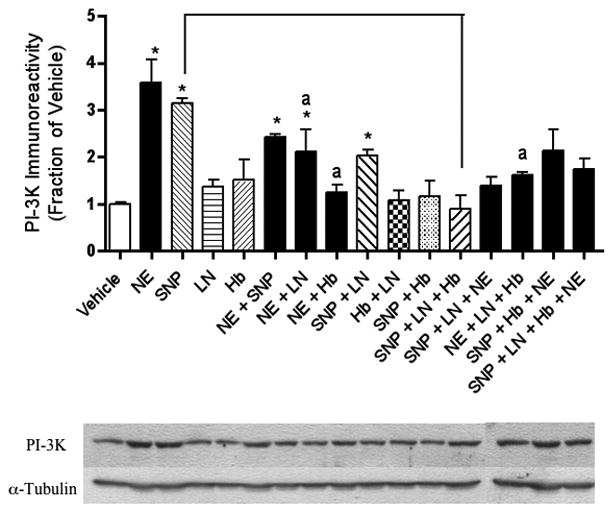
The NE-stimulated increase in PI-3K immunoreactivity observed in hippocampal neurons is NO-dependent. Relative levels of PI-3K immunoreactivity, as determined by Western blotting, reveal that NE and/or SNP increased PI-3K immunoreactivity. These results were reversed when neurons were co-incubated with the NO synthase inhibitor, l-NAME. Western blotting experiments were conducted and analyzed as specified in Experimental Procedures. Asterisks denote a treatment that is significantly different from vehicle-treated controls (p < .05). Lower-case “a” indicates that a treatment is significantly different from NE-treated cells (p < .05). Other relevant treatments that are significantly different from each other at p < .05 are indicated by brackets. Data are the mean + SEM. Each experiment was conducted 2-3 times, with each sample measured in duplicate.
Figure 3.
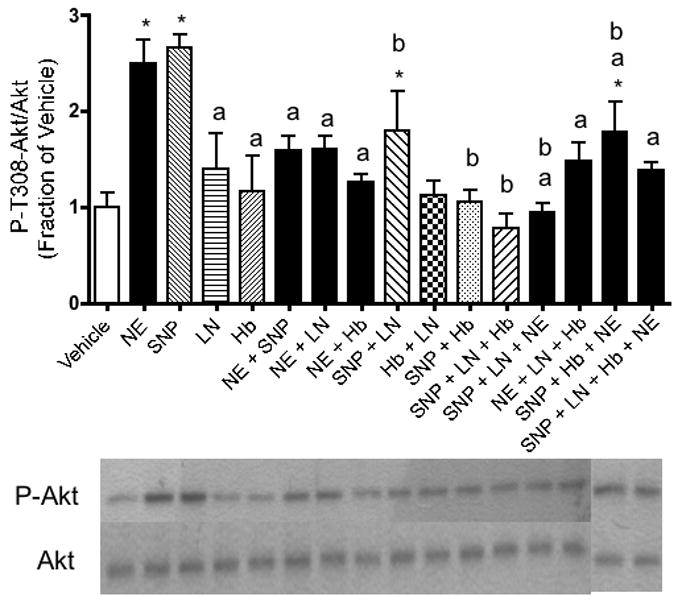
The NE-stimulated increase in P-T308-Akt immunoreactivity observed in hippocampal neurons is NO-dependent. Relative levels of P-T308-Akt immunoreactivity, as determined by Western blotting, reveal that NE and/or SNP increased P-T308-Akt immunoreactivity. These results were reversed when neurons were co-incubated with the NO synthase inhibitor, l-NAME. Western blotting experiments were conducted and analyzed as specified in Experimental Procedures. Asterisks denote a treatment that is significantly different from vehicle-treated controls (p < .05). Lower-case “a” indicates that a treatment is significantly different from NE-treated cells (p < .05). Lower-case “b” indicates that a treatment is significantly different from SNP-treated cells (p < .05). Other relevant treatments that are significantly different from each other at p < .05 are indicated by brackets. Data are the mean + SEM. Each experiment was conducted 2-3 times, with each sample measured in duplicate.
Figure 4.
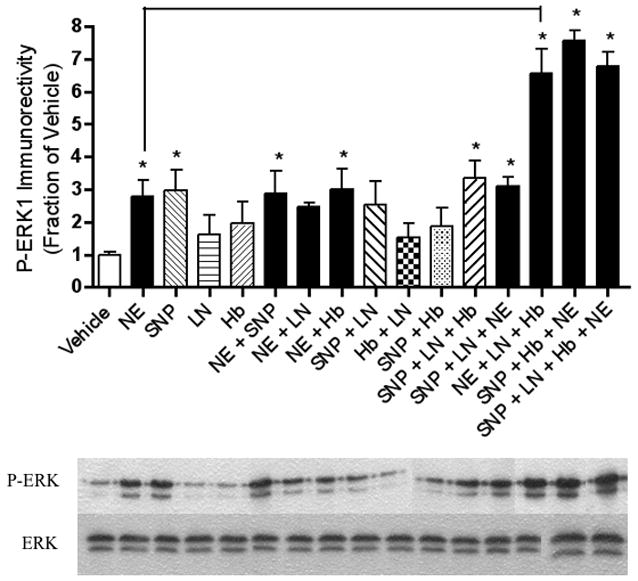
NE-induced increases in phospho-ERK1 expression are not NO-dependent. Relative levels of P-ERK1 immunoreactivity as determined by Western blotting reveal that NE and/or SNP increase P-ERK1 immunoreactivity, and these results are unaffected by co-incubation with l-NAME. Western blotting experiments were conducted and analyzed as specified in Experimental Procedures. Asterisks denote a treatment that is significantly different from vehicle-treated controls (p < .05). Treatments that are significantly different from each other at p < .05 are indicated by brackets. Data are the mean + SEM. Each experiment was conducted 2-3 times, with each sample measured in duplicate.
SNP increases BDNF, PI-3K, and P-ERK1 Immunoreactivity
Similar to NE, application of the NO donor SNP also led to a robust increase in BDNF (Figure 1), PI-3K (Figure 2), P-Akt (Figure 3), and P-ERK1 (Figure 4) expression.
The effects of NE on BDNF protein levels were inhibited when NE was applied in the presence of an NO scavenger or an NO synthase inhibitor
Adding the extracellular NO scavenger, Hb, to our NE treatment condition removed the increasing effects of NE on BDNF (Figure 1), PI-3K (Figure 2) and P-Akt (Figure 3) immunoreactivity. Likewise, the NO synthase (NOS) inhibitor, l-NAME, inhibited the effects of NE on BDNF (Figure 1), P-Akt (Figure 3) and PI-3K (Figure 2). The increased P-ERK1 immunoreactivity elicited by NE, however, was inhibited by neither Hb nor l-NAME (Figure 4). Curiously, however, when NE, SNP, and Hb were incubated together, P-ERK1 immunoreactivity was robustly and significantly increased above that of NE alone, SNP alone, and Hb alone (Figure 4). Additionally, the combination of SNP, NE, and l-NAME significantly increased P-ERK1 immunoreactivity (Figure 4), but not that of BDNF (Figure 1), PI-3K (Figure 2) nor P-Akt (Figure 3).
L-NAME potently increases neuronal death
In our experiment, l-NAME potently increased hippocampal neuronal death significantly above that of vehicle-treated controls [F(15,32) = 184.27, p < .0001] as well as above every other treatment (Figure 5). Neither NE nor SNP reversed these effects of l-NAME (Figure 5). It should also be noted that NE applied alone did not alter baseline cell survival.
Figure 5.
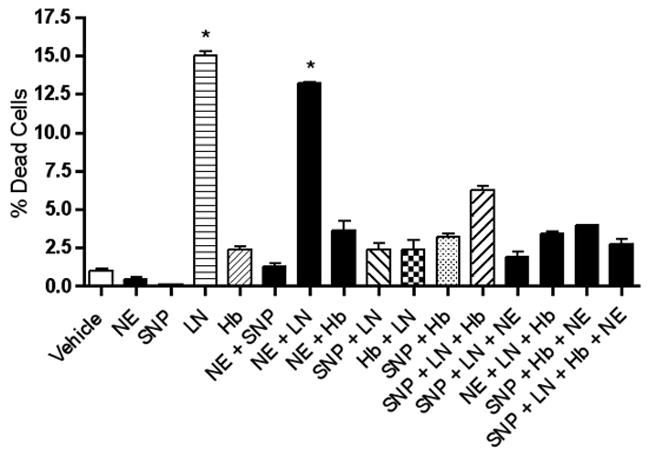
L-NAME potently increases hippocampal neuronal death significantly above that of vehicle-treated controls. Asterisks denote results that are significantly different from vehicle-treated controls (p < .05). Data are the mean + SEM.
ODQ inhibits the BDNF-enhancing effects of NE, but not the increasing effects of NE on PI-3K P-Akt, and P-ERK1 immunoreactivity
When the guanylate cyclase inhibitor, ODQ, was applied to hippocampal neurons in the presence of NE, the effects of NE on BDNF [F(5,12) = 5.57, p = .04] (Figure 6) and P-Akt [F(5,14) = 2.80, p = .007] (Figure 8) immunoreactivity were completely reversed. In fact, ODQ significantly reduced BDNF immunoreactivity below baseline levels (p = .0234) (Figure 6). The PI-3K- and P-ERK1-enhancing effects of NE, on the other hand, were not inhibited by ODQ. In the presence of NE, ODQ did not decrease PI-3K [F(5,12) = 2.58, p = .722] (Figure 7) nor P-ERK1 [F(5,12) = 6.60, p = .065] (Figure 9) immunoreactivity, relative to that of NE alone. Like NE, the cGMP analog, 8-Br-cGMP, increased BDNF (p = .05, Figure 6), PI-3K (p = .048, Figure 7), and P-ERK1 (p = .018, Figure 9), but not P-T308-Akt (p = .972, Figure 8) immunoreactivity significantly above that of vehicle-administered neurons.
Figure 6.
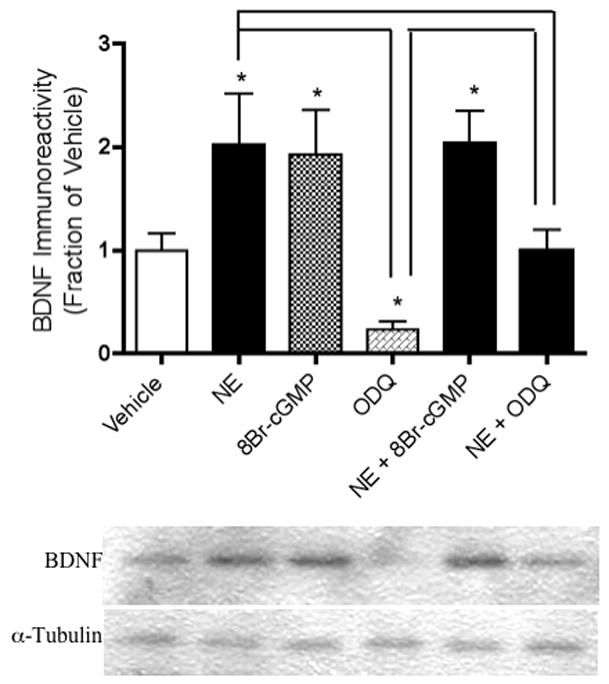
NE and/or 8-Br-cGMP increase BDNF immunoreactivity, which is reversed by treatment with ODQ. Western blotting experiments were conducted and analyzed as specified in Experimental Procedures. Asterisks denote treatments that are significantly different from vehicle-treated controls (p < .05). Treatments that are significantly different from each other at p < .05 are indicated by brackets. Data are the mean + SEM. Each experiment was conducted 2-3 times, with each sample measured in duplicate.
Figure 8.
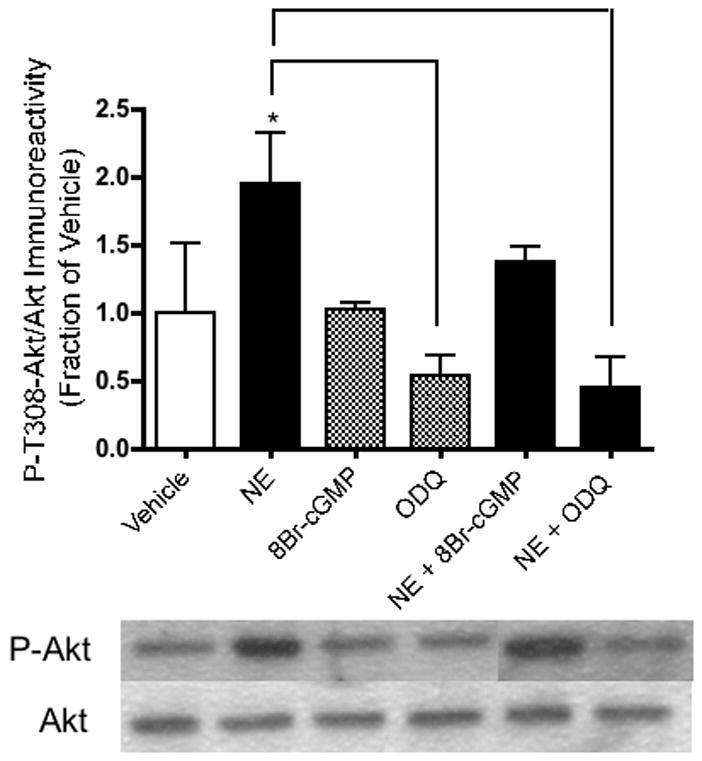
NE increases P-T308-Akt immunoreactivity in hippocampal neurons, but is reversed by ODQ. Western blotting experiments were conducted and analyzed as specified in Experimental Procedures. Asterisks denote treatments that are significantly different from vehicle-treated controls (p < .05). Treatments that are significantly different from each other at p < .05 are indicated by brackets. Data are the mean + SEM. Each experiment was conducted 2-3 times, with each sample measured in duplicate.
Figure 7.

NE and/or 8-Br-cGMP increase PI-3K immunoreactivity in hippocampal neurons. This result is unaffected by treatment with ODQ. Western blotting experiments were conducted and analyzed as specified in Experimental Procedures. Asterisks denote a treatment that are significantly different from vehicle-treated controls (p < .05). Treatments that are significantly different from each other at p < .05 are indicated by brackets. Data are the mean + SEM. Each experiment was conducted 2-3 times, with each sample measured in duplicate.
Figure 9.

NE and or 8-Br-cGMP increase P-ERK1 immunoreactivity, and this result is decreased by ODQ. Western blotting experiments were conducted and analyzed as specified in Experimental Procedures. Asterisks denote treatments that are significantly different from vehicle-treated controls (p < .05). Treatments that are significantly different from each other at p < .05 are indicated by brackets. Data are the mean + SEM. Each experiment was conducted at 2-3 times, with each sample measured in duplicate.
NE decreases cell death brought about by guanylate cyclase inhibition
Although NE on its own did not significantly affect survival of E18 hippocampal neurons, it led to a significant increase in cell survival compared to the level resulting from treatment with ODQ when co-incubated with NE [F(5,17) = 5.196, p = .03] (Figure 10). The combination of NE-plus-ODQ also led to levels of cell death that were lower than vehicle conditions (p = .0068) (Figure 10).
Figure 10.
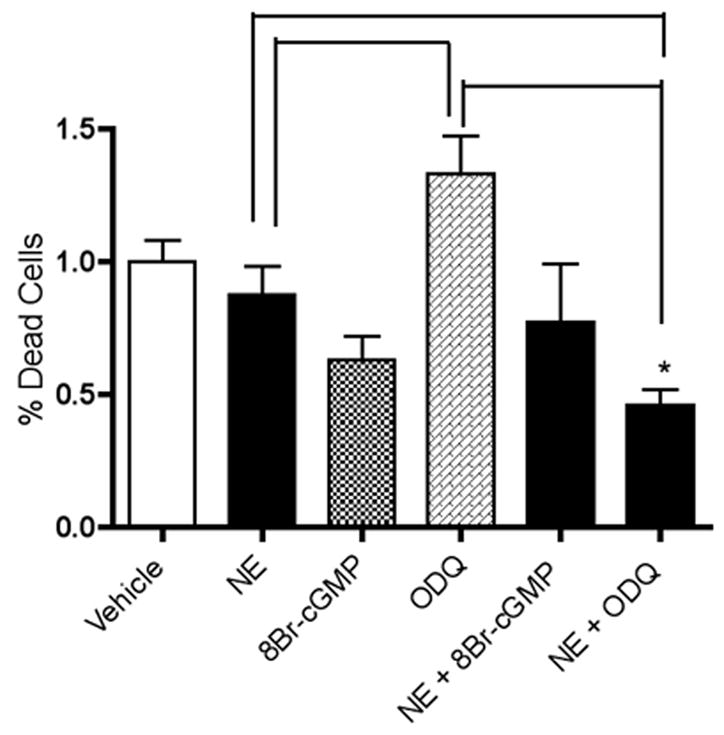
NE decreases cell death brought about by guanylate cyclase inhibition. NE-plus-ODQ treatment led to a significantly lower level of cell death in hippocampal neurons than treatment with ODQ (p = .0003). Treatment with ODQ also resulted in significantly higher cell death than NE-treated (p = .0275) and 8-Br-cGMP-treated (p = .0018) cells. Asterisks denote results that are significantly different from vehicle-treated controls (p < .05). Treatments that are significantly different from each other at p < .05 are indicated by brackets. Data are the mean + SEM. Each experiment was conducted at 2-3 times, with each sample measured in duplicate.
DISCUSSION
Our experimental evidence suggests that the BDNF-enhancing effects of NE, as applied to primary hippocampal neurons, are dependent upon NO signaling. Increased NE neurotransmission is thought to be an important component of the BDNF-enhancing response to exercise or antidepressant treatment in vivo. (Duman, 1998; Ivy et al., 2003). Our results support the hypothesis that NO signaling is an important part of this NE-mediated mechanism in vitro. The results of our current study are consistent with a recent study conducted in vivo, in which increased neuronal activation brought about by exercise led to a robust increase in BDNF mRNA and protein expression, both of which were prevented by concomitant treatment with the NOS inhibitor, l-NAME (Chen et al., 2006). In the current study, we demonstrated that an increase in BDNF immunoreactivity could be elicited not only by NE, but also by an NO donor, SNP. In addition, the BDNF-enhancing effects of NE were prevented by compounds removing NO from the culture medium (Hb) or inhibiting its synthesis within the cell (l-NAME).
Since the discovery of guanylate cyclase and the role of the labile NO molecule in promoting its activation, it is known that NO is a highly specific signaling molecule with important pro-survival (and pro-differentiation) activities. NO signaling is highly active in the hippocampus. Both cellular NOS (Dinerman et al., 1994) and cGMP signaling molecules such as protein kinase G I and II (Fiscus, 2002) are plentiful in this brain region. Evidence indicates that NO synthesis and subsequent signaling promotes growth and survival responses in hippocampal neurons (for review, see (Schuman and Madison, 1994). NO production is regulated in an activity-dependent manner in the hippocampus during the period of developmental synaptogenesis (Contestabile, 2000; Ogilvie et al., 1995) and participates in the regulation of synaptic plasticity both during development and throughout the lifespan (Bohme et al., 1991; Shibuki and Okada, 1991). In the adult brain, NO promotes neurite outgrowth (Hindley et al., 1997; Yamazaki et al., 2005) neurogenesis-mediated functional repair (Zhang et al., 2001), neuroplasticity (Bredt, 1999; Puzzo et al., 2005) neuronal protection (Lin et al., 2004) and cellular survival (Ciani et al., 2002; Culmsee et al., 2005; Estevez et al., 1998a,b; Wirtz-Brugger and Giovanni, 2000). NO is also thought to serve as a retrograde messenger secreted from hippocampal pyramidal cells to stimulate release of excitatory neurotransmitters such as glutamate and NE from the Schaeffer collaterals (Dinerman et al., 1994). Thus, NO induces NE release from hippocampal slices (Lonart et al., 1992) both directly from noradrenergic nerve terminals and indirectly through glutamate release (Lonart and Johnson, 1995). Under certain experimental conditions using NO donors, increased NE and glutamate release were reported to occur in a cGMP-independent manner (Lei et al., 2000; Satoh et al., 1996a,b).
As noted above, several studies have demonstrated that NO has important neuroprotective properties that may involve cGMP signaling. Consistently, cGMP analogs have been shown to protect neurons against NO deprivation or ODQ-induced neuronal death (Estevez et al., 1998a). In line with this, our results indicate that NE-mediated increases in BDNF, as well as Akt activation, require cGMP signaling (Figure 5). Additionally, inhibition of cGMP signaling appears to decrease neuronal survival in the presence of NE (Figure 8). In previous studies, inhibition of NO synthesis or NO availability has been shown to decrease neuroprotection (Hashiguchi et al., 2004) and neuronal survival (Estevez et al., 1998b). In our current study, cell death was significantly increased with l-NAME, supporting a pro-survival role for NO in hippocampal neurons. Inasmuch as NO is known to be synthesized in significant quantities upon NMDA receptor activation (Garthwaite, 1991; Garthwaite and Boulton, 1995; Luo and Vincent, 1994), NO signaling may be an important means by which globally-activating interventions, such as exercise or environmental enrichment, can enhance BDNF expression and survival signaling. It must be acknowledged, on the other hand, that contradictory evidence exists for NO signaling promoting survival in neurons. In primary cortical cultures, the administration of NOS inhibitors has been shown to prevent NMDA-induced cell death, thus suggesting that NO mediates glutamate-induced neurotoxicity (Dawson et al., 1991). This discrepancy could possibly be explained by the relative dose of NO involved: NMDA neurotoxicity in vitro may involve the production and release of larger amounts of NO, mediating apoptosis through separate mechanisms. Nevertheless, the administration of NOS inhibitors increased BDNF secretion in cultured hippocampal cells (Canossa et al., 2002) and enhanced hippocampal neurogenesis in adult animals (Packer et al., 2003; Park et al., 2004). It is possible that NO-mediated survival may be a feature of the hippocampus, but possibly not all other brain regions.
Because BDNF is important for survival, as well as synaptic plasticity within the hippocampus, the promotion of BDNF expression in hippocampal neurons has been regarded as vital for cellular maintenance, as well as protection from neurotoxic insults. Our studies have demonstrated that NE, when directly applied to hippocampal neurons, increases BDNF concentration within these cells. These experiments, as well as previous studies, have shown that this NE-induced enhancement in BDNF is associated with increased survival signaling along the PI-3K and MAPK pathways. Previous evidence indicates that BDNF and NO may be co-regulated at the cellular level in the CNS (Xiong et al., 1999) and that BDNF regulates synaptic plasticity in the hippocampus (Figurov et al., 1996; Kang and Schuman, 1995) in a manner similar to that of NO (Bohme et al., 1991; Shibuki and Okada, 1991). Supporting the possibility that NO signaling may also regulate neurogenesis, in a recent study, l-NAME and Hb blocked the stimulatory effect of BDNF on neuronal differentiation of neuroprogenitor cells (Cheng et al., 2003).
Via the activation of guanylate cyclase, the synthesis of cGMP and the subsequent activation of protein kinase G, NO has been demonstrated to activate the transcription of CREB and several other proteins leading to growth and survival responses within the cell (Ciani et al., 2002; Ha et al., 2003). NO is also known to interact more directly with cell-signaling machinery at the plasma membrane to set cell-survival signaling into motion. eNOS, the predominant NOS isoform within hippocampal pyramidal neurons (Dinerman et al., 1994) is most active as a membrane-associated molecule, and is found localized to particulate fractions of the cell, presumably plasma (Busconi and Michel, 1993). As such, rapid eNOS regulation can be elicited by activating physiological stimuli (Dimmeler et al., 1999). Recent evidence indicates that endogenous NO within hippocampal neurons is a major mediator of rapid, calcium-dependent activation of Ras (Deora et al., 1998; Koo et al., 2005). This mechanism could lead to a more rapid, direct activation of the PI-3K pathway via NO. Consistently, our experimental results indicate that activation of hippocampal NO may be an important part of the intracellular mechanism leading to increased PI-3K, as well as downstream activation of P-Akt, within hippocampal neurons following NE application, and that this mechanism may be only partially dependent upon cGMP.
All of the increased measures of survival signaling observed in this study following NE application were mimicked with the use of an NO donor, SNP. Enhanced BDNF expression following NE application appears to be dependent upon both intracellular NO synthesis and extracellular NO release, as these effects were reversed in the presence of both an NOS inhibitor and an NO scavenger. It is likely that enhanced BDNF synthesis results from a cooperative activation of multiple signaling pathways following NE stimulation, as well as positive feedback resulting from events such as increased Trk receptor activation and increased NE, glutamate release following NO synthesis (Figure 10). This could explain why the application of the NOS inhibitor, l-NAME, reversed SNP-mediated changes, even though SNP directly increases extracellular NO in a manner independent of NOS. As illustrated in Figure 11, it is possible that the transcription of BDNF undergoes a rapid feedback cycle (Kovalchuk et al., 2004) that NO synthesis can drive (Cheng et al., 2003).
Figure 11.
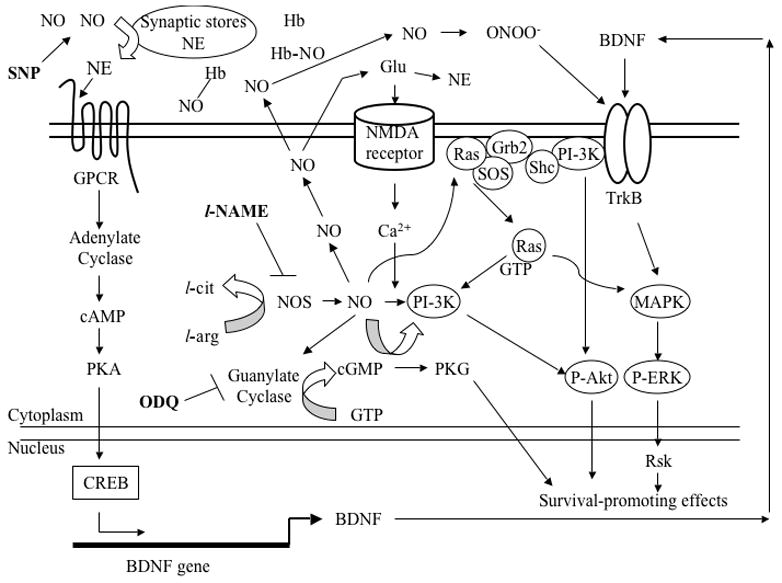
A hypothetical scheme illustrates the putative intracellular signaling cascades serving as the context for the data collected in the current study. SNP provides extracellular NO, which then stimlulates NE release from its synaptic stores. NE then binds its receptor (GPCR), thereby activating the cAMP-dependent protein kinase (PKA), which, in turn, phosphorylates CREB to transcribe BDNF. The neurotrophin is then packaged and secreted extracellularly to then bind its receptor, TrkB, in either an autocrine or paracrine fashion. Major signaling pathways (e.g., PI-3K and MAPK) are then activated, helping to promote neuronal survival through activation of survival-promoting genes, one of which, may be BDNF, which can then act in a positive feedback manner. But because neither l-NAME nor Hb decreased ERK1 phosphorylation, relative to that caused by NE alone, NE-induced ERK1 activation does not depend on NO signaling. Inside the cell, NOS produces NO, which, in our study, has a wide variety of possible fates: NO can diffuse out of the cell to activate NE release directly (as above) or through glutamate activation; NO can be sequestered by Hb, thereby decreasing its availability; NO can take part in what is thought to be its major cell survival signaling strategy by activating guanylate cyclase, producing cGMP and activating protein kinase G; NO can participate in the Ca2+-dependent activation of PI-3K, which can then phosphorylate Akt and/or cross-talk with the MAPK pathway via Ras activation, both leading to the promotion of neuronal survival; And, NO can be converted into a toxic intermediate, peroxynitrite (ONOO-), which can then bind TrkB to activate survival-promoting cascades. Alternatively, in removing NO, and therefore, ONOO-, it is possible that Hb and/or l-NAME take on anti-apoptotic roles via the increase in ERK1 phosphorylation. The NOS inhibitor, l-NAME, decreases intracellular NO, thereby decreasing PI-3K and subsequent cell survival-promoting effects (e.g., BDNF). The guanylate cyclase inhibitor, ODQ, prevented NE-simulated increases in BDNF expression in our experiments, suggesting that transcriptional activation via cGMP (e.g. through PKG or PI-3K) is an important source of BDNF. Our data suggests that ODQ also decreases P-ERK1 and subsequent cell survival-promoting effects, such as BDNF, possibly through the PI-3K-Ras-MAPK cross-talk pathway. ODQ did not decrease PI-3K, indicating that its activation is not cGMP-dependent and can be activated via the NE-GPCR-PKA-CREB-BDNF-TrkB route.
As noted above, NE activation also led to significant increases in the levels of PI-3K, P-Akt and P-ERK1 in primary hippocampal neurons. These effects were also imitated with the use of an NO donor. Our results suggest that increased PI-3K, as well as downstream Akt activation, is dependent on overall NO availability (both extracellular and intracellular) as these results were prevented with either Hb or with l-NAME. It is possible that increased PI-3K is an early event resulting from a more direct activation of its synthesis by NO (one that does not require transcriptional activation, protein synthesis or cellular feedback). One possible mechanism (indicated in Figure 11) is the known direct activation of p21Ras (Ras) by NO associated with the plasma membrane. Rapid activation of Ras-TrkB via NO release (Koo et al., 2005) is one means by which NE could lead to increased PI-3K levels. Further, it is possible that via PI-3K/MAPK pathway cross-talk (Opazo et al., 2003; Owada et al., 1999; Subramaniam et al., 2005; Tolbert et al., 2003), NO can more efficiently contribute to enhanced PI-3K pathway activation. Therefore, removing NO (using Hb) or preventing its synthesis (using l-NAME) could counteract any positive effects of NE.
Results from our current experiments suggest that the effects of NE on BDNF expression in hippocampal neurons are also dependent upon cGMP activity. The cGMP analog, 8-Br-cGMP, increased BDNF levels to a degree comparable to that achieved with NE alone, whereas the guanylate cyclase inhibitor, ODQ, inhibited the effects of NE when co-incubated with the latter. Consistently, ODQ has been shown to decrease CREB phosphorylation (Ciani et al., 2002) and survival of neurons treated with BDNF (Estevez et al., 1998b). It might be noted that ODQ also tended to decrease BDNF immunoreactivity below that of vehicle-administered controls when applied to neurons alone, suggesting that cGMP signaling may also be partially responsible for some proportion of basal BDNF synthesis in hippocampal neurons. Additional support for the possibility that enhanced BDNF synthesis requires transcriptional activation mechanisms (discussed above) is provided by the observation that NE-stimulated increases in BDNFwere dependent upon cGMP activity.
The NE-induced increases in ERK1 activation observed in our experiments do not appear to be dependent upon NO signaling at all. Although the MAPK pathway has been shown to be required for NOS induction (Schonhoff et al., 2001), ultimate CREB-dependent BDNF transcription does not depend on ERK signaling (Riccio et al., 2006). Similarly, the MAPK pathway can be activated by a wide variety of pathways other than those that are NO-mediated (e.g., cross-talk with PI-3K, growth factors (Huang and Reichardt, 2003; Segal, 2003). It is quite likely that in our current study, multiple pathways are operating when multiple compounds were added to the culture medium as revealed by the P-ERK1 immunoreactivity profile. Combination of three or more compounds that included NE, Hb, and l-NAME or SNP led to a compounded increase in P-ERK1 immunoreactivity (approximately twice the amount obtained with NE or SNP alone). It is possible that under these conditions, NO has the opposite effects from those we observe when these compounds are added individually to the culture medium. Among the many roles that NO plays a part, one of its most prominent is that of free radical, which, when combined with superoxide, produces the more potently toxic oxidant, peroxynitrite (Estevez et al., 1998a). Thus, the removal of NO (and therefore, the potential to produce peroxynitrite) from the culture medium by Hb or l-NAME may synergize with NE in which each of the former takes on an anti-apoptotic role (Estevez et al., 1998a). This could account for the observed increased P-ERK1 activation. Alternatively, with the combination of NE, Hb, and SNP, the similarly dramatic increase in P-ERK1 may represent anti-apoptotic effects of Hb (not observed when administered alone) combined with the pro-survival properties of NE and SNP. In contrast to the potentially toxic peroxynitrite that is produced as a result of NO reactions with superoxide, this product also has potential survival-promoting properties. Peroxynitrite has been shown to directly activate Trk receptors (Yuen et al., 2000), which can then activate the downstream MAPK pathway. When combined with NE, therefore, it is possible that this synergistic effect underlies the dramatic increase in P-ERK immunoreactivity we see with the combination of NE, SNP, and Hb. Consistently, BDNF immunoreactivity increased as a trend, and not significantly, in response to this three-way treatment, relative to that of controls.
CONCLUSIONS
In conclusion, in isolated hippocampal neurons, we have shown that BDNF and PI-3K immunoreactivity can be increased by application of NE (or an NO donor), and that these changes are NO-dependent. Similar to earlier results gleaned in vivo (Chen et al., 2006), NOS inhibition decreases BDNF and PI-3K immunoreactivity. Because BDNF and PI-3K are survival-associated proteins (Brunet et al., 1999; Huang and Reichardt, 2003; Rossler et al., 2004; Segal, 2003), it is logical to infer that the presence of NO is consistent with cellular survival-promoting strategies. In the preceding paragraph, we have broached the possibility that NO regulation and its beneficial vs. toxic potential may be exquisitely sensitive to the experimental conditions/treatments employed, including any other NO-involved compounds concurrently added to the culture medium. Thus, it makes logical sense that the effects of NO and the many NO-dependent signaling pathways be thoroughly characterized according to the experimental circumstances and conditions under which NO is survival-promoting vs. those under which NO is toxic (Araujo and Carvalho, 2005; Samdani et al., 1997). This way, one can devise an optimally appropriate therapeutic strategy that targets NO, considering the wide variety of disorders in which NO is theorized to participate (Bredt, 1999; Gross and Wolin, 1995).
Acknowledgments
Funded by PHS Grant MH 59776 to ARN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michael J. Chen, 5151 State University Dr., Department of Biological Sciences, California State University, Los Angeles, CA 90032 USA.
Amelia A. Russo-Neustadt, 5151 State University Dr., Department of Biological Sciences, California State University, Los Angeles, CA 90032 USA
References
- Araujo IM, Carvalho CM. Role of nitric oxide and calpain activation in neuronal death and survival. Curr Drug Targets CNS Neurology and Disorders. 2005;4(4):319–324. doi: 10.2174/1568007054546126. [DOI] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Research. 1977;126(3):397–342. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Bohme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. European Journal of Pharmacology. 1991;199(3):379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radical Research. 1999;31(6):577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Busconi L, Michel T. Endothelial nitric oxide synthase. N-terminal myristoylation determines subcellular localization. Journal of Biological Chemistry. 1993;268(12):8410–8413. [PubMed] [Google Scholar]
- Canossa M, Giordano E, Cappello S, Guarnieri C, Ferri S. Nitric oxide down-regulates brain-derived neurotrophic factor secretion in cultured hippocampal neurons. Proceedings of the National Academy of Sciences, U S A. 2002;99(5):3282–3287. doi: 10.1073/pnas.042504299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Ivy AS, Russo-Neustadt AA. Nitric oxide synthesis is required for exercise-induced increases in hippocampal BDNF and phosphatidylinositol 3′ kinase expression. Brain Research Bulletin. 2006;68(4):257–268. doi: 10.1016/j.brainresbull.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Chen MJ, Nguyen TV, Pike CJ, Russo-Neustadt AA. Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cellular Signalling. 2007;19(1):114–128. doi: 10.1016/j.cellsig.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Cheng A, Wang S, Cai J, Rao MS, Mattson MP. Nitric oxide acts in a positive feedback loop with BDNF to regulate neural progenitor cell proliferation and differentiation in the mammalian brain. Developmental Biology. 2003;258(2):319–333. doi: 10.1016/s0012-1606(03)00120-9. [DOI] [PubMed] [Google Scholar]
- Ciani E, Virgili M, Contestabile A. Akt pathway mediates a cGMP-dependent survival role of nitric oxide in cerebellar granule neurones. Journal of Neurochemistry. 2002;81(2):218–228. doi: 10.1046/j.1471-4159.2002.00857.x. [DOI] [PubMed] [Google Scholar]
- Contestabile A. Roles of NMDA receptor activity and nitric oxide production in brain development. Brain Research Reviews. 2000;32(23):476–509. doi: 10.1016/s0165-0173(00)00018-7. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proceedings of the National Academy of Sciences, USA. 1991;88(14):6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deora AA, Win T, Vanhaesebroeck B, Lander HM. A redox-triggered ras-effector interaction. Recruitment of phosphatidylinositol 3′-kinase to Ras by redox stress. Journal of Biological Chemistry. 1998;273(45):29923–29928. doi: 10.1074/jbc.273.45.29923. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Dinerman JL, Dawson TM, Schell MJ, Snowman A, Snyder SH. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proceedings of the National Academy of Sciences, USA. 1994;91(10):4214–4218. doi: 10.1073/pnas.91.10.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishman RK, Renner KJ, White-Welkley JE, Burke KA, Bunnell BN. Treadmill exercise training augments brain norepinephrine response to familiar and novel stress. Brain Research Bulletin. 2000;52(5):337–342. doi: 10.1016/s0361-9230(00)00271-9. [DOI] [PubMed] [Google Scholar]
- Duman RS. Novel therapeutic approaches beyond the serotonin receptor. Biological Psychiatry. 1998;44(5):324–335. doi: 10.1016/s0006-3223(98)00031-6. [DOI] [PubMed] [Google Scholar]
- Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Archives of General Psychiatry. 1997;54(7):597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- Duman RS, Malberg J, Nakagawa S. Regulation of adult neurogenesis by psychotropic drugs and stress. Journal of Pharmacology and Experimental Therapeutics. 2001;299(2):401–407. [PubMed] [Google Scholar]
- Dunn AL, Reigle TG, Youngstedt SD, Armstrong RB, Dishman RK. Brain norepineprhine and metabolites after treadmill training and wheel running. Medicine Science and Sports Exercise. 1996;28(2):204–209. doi: 10.1097/00005768-199602000-00008. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Spear N, Manuel SM, Radi R, Henderson CE, Barbeito L, Beckman JS. Nitric oxide and superoxide contribute to motor neuron apoptosis induced by trophic factor deprivation. Journal of Neuroscience. 1998a;18(3):923–931. doi: 10.1523/JNEUROSCI.18-03-00923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estevez AG, Spear N, Thompson JA, Cornwell TL, Radi R, Barbeito L, Beckman JS. Nitric oxide-dependent production of cGMP supports the survival of rat embryonic motor neurons cultured with brain-derived neurotrophic factor. Journal of Neuroscience. 1998b;18(10):3708–3714. doi: 10.1523/JNEUROSCI.18-10-03708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer J, Zhao X, van Praag H, Wodtke K, Gage FH, Christie BR. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience. 2004;124(1):71–79. doi: 10.1016/j.neuroscience.2003.09.029. [DOI] [PubMed] [Google Scholar]
- Fedele E, Marchi M, Raiteri M. In vivo NO/cGMP signalling in the hippocampus. Neurochemical Research. 2001;26(89):1069–1078. doi: 10.1023/a:1012309223236. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381(6584):706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Fiscus RR. Involvement of cyclic GMP and protein kinase G in the regulation of apoptosis and survival in neural cells. Neurosignals. 2002;11(4):175–190. doi: 10.1159/000065431. [DOI] [PubMed] [Google Scholar]
- Garcia C, Chen MJ, Garza AA, Cotman CW, Russo-Neustadt A. The influence of specific noradrenergic and serotonergic lesions on the expression of hippocampal brain-derived neurotrophic factor transcripts following voluntary physical activity. Neuroscience. 2003;119(3):721–732. doi: 10.1016/s0306-4522(03)00192-1. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends in Neuroscience. 1991;14(2):60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annual Review of Physiology. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Gross SS, Wolin MS. Nitric oxide: pathophysiological mechanisms. Annual Review of Physiology. 1995;57:737–769. doi: 10.1146/annurev.ph.57.030195.003513. [DOI] [PubMed] [Google Scholar]
- Ha KS, Kim KM, Kwon YG, Bai SK, Nam WD, Yoo YM, Kim PK, Chung HT, Billar TR, Kim YM. Nitric oxide prevents 6-hydroxydopamine-induced apoptosis in PC12 cells through cGMP-dependent PI3 kinase/Akt activation. FASEB Journal. 2003;17(9):1036–1047. doi: 10.1096/fj.02-0738com. [DOI] [PubMed] [Google Scholar]
- Hindley S, Juurlink BH, Gysbers JW, Middlemiss PJ, Herman MA, Rathbone MP. Nitric oxide donors enhance neurotrophin-induced neurite outgrowth through a cGMP-dependent mechanism. Journal of Neuroscience Research. 1997;47(4):427–439. [PubMed] [Google Scholar]
- Holscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends in Neuroscience. 1997;20(7):298–303. doi: 10.1016/s0166-2236(97)01065-5. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annual Review of Biochemistry. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Ivy AS, Rodriguez FG, Garcia C, Chen MJ, Russo-Neustadt AA. Noradrenergic and serotonergic blockade inhibits BDNF mRNA activation following exercise and antidepressant. Pharmacology, Biochemistry and Behavior. 2003;75(1):81–88. doi: 10.1016/s0091-3057(03)00044-3. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Schuman EM. Neurotrophin-induced modulation of synaptic transmission in the adult hippocampus. Journal of Physiology, Paris. 1995;89(1):11–22. doi: 10.1016/0928-4257(96)80547-x. [DOI] [PubMed] [Google Scholar]
- Kang YC, Kim PK, Choi BM, Chung HT, Ha KS, Kwon YG, Kim YM. Regulation of programmed cell death in neuronal cells by nitric oxide. In Vivo. 2004;18(3):367–376. [PubMed] [Google Scholar]
- Koo BS, Choi EG, Park JB, Cho CH, Chung KH, Kim CH. Neuroprotective effect of Chuk-Me-Sun-Dan on NMDA- and AMPA-evoked nitric oxide synthase activity in mouse brain. Immunopharmacology Immunotoxicology. 2005;27(3):499–514. doi: 10.1080/08923970500242319. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Holthoff K, Konnerth A. Neurotrophin action on a rapid timescale. Current Opinion in Neurobiology. 2004;14(5):558–563. doi: 10.1016/j.conb.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Krumenacker JS, Hanafy KA, Murad F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain Research Bulletin. 2004;62(6):505–515. doi: 10.1016/S0361-9230(03)00102-3. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lei S, Jackson MF, Jia Z, Roder J, Bai D, Orser BA, MacDonald JF. Cyclic GMP-dependent feedback inhibition of AMPA receptors is independent of PKG. Nature Neuroscience. 2000;3(6):559–565. doi: 10.1038/75729. [DOI] [PubMed] [Google Scholar]
- Leung LY, Tong KY, Zhang SM, Zeng XH, Zhang KP, Zheng XX. Neurochemical effects of exercise and neuromuscular electrical stimulation on brain after stroke: a microdialysis study using rat model. Neuroscience Letters. 2006;397(12):135–139. doi: 10.1016/j.neulet.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Lin YF, Raab-Graham K, Jan YN, Jan LY. NO stimulation of ATP-sensitive potassium channels: Involvement of Ras/mitogen-activated protein kinase pathway and contribution to neuroprotection. Proceedings of the National Academy of Sciences, USA. 2004;101(20):7799–7804. doi: 10.1073/pnas.0402496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Johnson KM. Characterization of nitric oxide generator-induced hippocampal [3H]norepinephrine release. II. The role of calcium, reverse norepinephrine transport and cyclic 3′,5′-guanosine monophosphate. Journal of Pharmacology and Experimental Therapeutics. 1995;275(1):14–22. [PubMed] [Google Scholar]
- Lonart G, Wang J, Johnson KM. Nitric oxide induces neurotransmitter release from hippocampal slices. European Journal of Pharmacology. 1992;220(23):271–272. doi: 10.1016/0014-2999(92)90759-w. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry. 1951;193(1):265–275. [PubMed] [Google Scholar]
- Luo D, Vincent SR. NMDA-dependent nitric oxide release in the hippocampus in vivo: interactions with noradrenaline. Neuropharmacology. 1994;33(11):1345–1350. doi: 10.1016/0028-3908(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Maher P, Schubert D. Signaling by reactive oxygen species in the nervous system. Cellular and Molecular Life Sciences. 2000;57(89):1287–1305. doi: 10.1007/PL00000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molteni R, Ying Z, Gomez-Pinilla F. Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. European Journal of Neuroscience. 2002;16(6):1107–1116. doi: 10.1046/j.1460-9568.2002.02158.x. [DOI] [PubMed] [Google Scholar]
- Neeper SA, Gómez-Pinilla F, Choi J, Cotman CW. Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Research. 1996;726(12):49–56. [PubMed] [Google Scholar]
- Ogilvie P, Schilling K, Billingsley ML, Schmidt HH. Induction and variants of neuronal nitric oxide synthase type I during synaptogenesis. FASEB Journal. 1995;9(9):799–806. doi: 10.1096/fasebj.9.9.7541381. [DOI] [PubMed] [Google Scholar]
- Oliff HS, Berchtold NC, Isackson P, Cotman CW. Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Brain Research Molecular Brain Research. 1998;61(12):147–153. doi: 10.1016/s0169-328x(98)00222-8. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O’Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. Journal of Neuroscience. 2003;23(9):3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, Michikawa M. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. Journal of Neurochemistry. 1999;73(5):2084–2092. [PubMed] [Google Scholar]
- Packer MA, Stasiv Y, Benraiss A, Chmielnicki E, Grinberg A, Westphal H, Goldman SA, Enikolopov G. Nitric oxide negatively regulates mammalian adult neurogenesis. Proceedings of the National Academy of Sciences, USA. 2003;100(16):9566–9571. doi: 10.1073/pnas.1633579100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Cho K, Ryu JH, Shin KS, Kim J, Ahn H, Huh Y. 7-Nitroindazole upregulates phosphorylated cAMP response element binding protein, polysialylated-neural cell adhesion molecule and tryptophan hydroxylase expression in the adult rat hippocampus. Brain Research. 2004;1008(1):120–125. doi: 10.1016/j.brainres.2004.01.055. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O. Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. Journal of Neuroscience. 2005;25(29):6887–6897. doi: 10.1523/JNEUROSCI.5291-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Alvania RS, Lonze BE, Ramanan N, Kim T, Huang Y, Dawson TM, Snyder SH, Ginty DD. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Molecular Cell. 2006;21(2):283–294. doi: 10.1016/j.molcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Richter-Levin G, Canevari L, Bliss TV. Spatial training and high-frequency stimulation engage a common pathway to enhance glutamate release in the hippocampus. Learning and Memory. 1998;4(6):445–450. doi: 10.1101/lm.4.6.445. [DOI] [PubMed] [Google Scholar]
- Rossler OG, Giehl KM, Thiel G. Neuroprotection of immortalized hippocampal neurones by brain-derived neurotrophic factor and Raf-1 protein kinase: role of extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase. Journal of Neurochemistry. 2004;88(5):1240–1252. doi: 10.1046/j.1471-4159.2003.02255.x. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A, Beard RC, Cotman CW. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology. 1999;21(5):679–682. doi: 10.1016/S0893-133X(99)00059-7. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt AA, Beard RC, Huang YM, Cotman CW. Physical activity and antidepressant treatment potentiate the expression of specific brain-derived neurotrophic factor transcripts in the rat hippocampus. Neuroscience. 2000;101(2):305–312. doi: 10.1016/s0306-4522(00)00349-3. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt AA, Chen MJ. Brain-derived neurotrophic factor and antidepressant activity. Current Pharmaceutical Design. 2005;11(12):1495–1510. doi: 10.2174/1381612053764788. [DOI] [PubMed] [Google Scholar]
- Samdani AF, Newcamp C, Resink A, Facchinetti F, Hoffman BE, Dawson VL, Dawson TM. Differential susceptibility to neurotoxicity mediated by neurotrophins and neuronal nitric oxide synthase. Journal of Neuroscience. 1997;17(12):4633–4641. doi: 10.1523/JNEUROSCI.17-12-04633.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Kimura T, Toda M, Miyazaki H, Ono S, Narita H, Murayama T, Nomura Y. NO donors stimulate noradrenaline release from rat hippocampus in a calmodulin-dependent manner in the presence of L-cysteine. Journal of Cell Physiology. 1996a;169(1):87–96. doi: 10.1002/(SICI)1097-4652(199610)169:1<87::AID-JCP9>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Satoh S, Murayama T, Nomura Y. Sodium nitroprusside stimulates noradrenaline release from rat hippocampal slices in the presence of dithiothreitol. Brain Research. 1996b;733(2):167–174. doi: 10.1016/0006-8993(96)00295-8. [DOI] [PubMed] [Google Scholar]
- Schonhoff CM, Bulseco DA, Brancho DM, Parada LF, Ross AH. The Ras-ERK pathway is required for the induction of neuronal nitric oxide synthase in differentiating PC12 cells. Journal of Neurochemistry. 2001;78(3):631–639. doi: 10.1046/j.1471-4159.2001.00432.x. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. Nitric oxide and synaptic function. Annual Review of Neuroscience. 1994;17:153–183. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- Segal RA. Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neuroscience. 2003;26:299–330. doi: 10.1146/annurev.neuro.26.041002.131421. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Okada D. Endogenous nitric oxide release required for long-term synaptic depression in the cerebellum. Nature. 1991;349(6307):326–328. doi: 10.1038/349326a0. [DOI] [PubMed] [Google Scholar]
- Stout AK, Woodward JJ. Differential effects of nitric oxide gas and nitric oxide donors on depolarization-induced release of [3H]norepinephrine from rat hippocampal slices. Neuropharmacology. 1994;33(11):1367–1374. doi: 10.1016/0028-3908(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Stout AK, Woodward JJ. Mechanism for nitric oxide’s enhancement of NMDA-stimulated [3H]norepinephrine release from rat hippocampal slices. Neuropharmacology. 1995;34(7):723–729. doi: 10.1016/0028-3908(95)00058-e. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Shahani N, Strelau J, Laliberte C, Brandt R, Kaplan D, Unsicker K. Insulin-like growth factor 1 inhibits extracellular signal-regulated kinase to promote neuronal survival via the phosphatidylinositol 3-kinase/protein kinase A/c-Raf pathway. Journal of Neurosci,ence. 2005;25(11):2838–2852. doi: 10.1523/JNEUROSCI.5060-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert LM, Russell DS, Duman RS. Norepinephrine activates extracellular-regulated kinase in cortical neurons. Biological Psychiatry. 2003;54(10):983–993. doi: 10.1016/s0006-3223(03)00346-9. [DOI] [PubMed] [Google Scholar]
- Wirtz-Brugger F, Giovanni A. Guanosine 3′,5′-cyclic monophosphate mediated inhibition of cell death induced by nerve growth factor withdrawal and beta-amyloid: protective effects of propentofylline. Neuroscience. 2000;99(4):737–750. doi: 10.1016/s0306-4522(00)00243-8. [DOI] [PubMed] [Google Scholar]
- Xiong H, Yamada K, Han D, Nabeshima T, Enikolopov G, Carnahan J, Nawa H. Mutual regulation between the intercellular messengers nitric oxide and brain-derived neurotrophic factor in rodent neocortical neurons. European Journal of Neuroscience. 1999;11(5):1567–1576. doi: 10.1046/j.1460-9568.1999.00567.x. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Chiba K, Mohri T. Fundamental role of nitric oxide in neuritogenesis of PC12h cells. British Journal of Pharmacology. 2005;146(5):662–669. doi: 10.1038/sj.bjp.0706370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EC, Gunther EC, Bothwell M. Nitric oxide activation of TrkB through peroxynitrite. Neuroreport. 2000;11(16):3593–3597. doi: 10.1097/00001756-200011090-00038. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhang L, Zhang Z, Wang Y, Lu M, Lapointe M, Chopp M. A nitric oxide donor induces neurogenesis and reduces functional deficits after stroke in rats. Annals of Neurology. 2001;50(5):602–611. doi: 10.1002/ana.1249. [DOI] [PubMed] [Google Scholar]