

Abstract
The tumor suppressor tuberin, encoded by the Tuberous Sclerosis Complex (TSC) gene TSC2, negatively regulates the mammalian target of rapamycin (mTOR) pathway, which plays a key role in the control of cell growth and proliferation. In addition to naturally occurring mutations, several kinases including Akt, RSK1, and ERK are known to phosphorylate and inactivate tuberin. We demonstrate a novel mechanism of tuberin inactivation through ubiquitination by Pam, a putative RING finger-containing E3 ubiquitin (Ub) ligase in mammalian cells. We show that Pam associates with E2 ubiquitin-conjugating enzymes, and tuberin can be ubiquitinated by Pam through its RING finger domain. Tuberin ubiquitination is independent of its phosphorylation by Akt, RSK1, and ERK kinases. Pam is also self-ubiquitinated through its RING finger domain. Moreover, the TSC1 protein hamartin, which forms a heterodimer with tuberin, protects tuberin from ubiquitination by Pam. However, TSC1 fails to protect a disease-associated missense mutant of TSC2 from ubiquitination by Pam. Furthermore, Pam knockdown by RNA interference (RNAi) in rat primary neurons elevates the level of tuberin, and subsequently inhibits the mTOR pathway. Our results provide novel evidence that Pam can function as an E3 Ub ligase toward tuberin and regulate mTOR signaling, suggesting that Pam can in turn regulate cell growth and proliferation as well as neuronal function through the TSC/mTOR pathway in mammalian cells.
Keywords: TSC, Tuberin, Hamartin, mammlian target of rapamycin, Ubiquitination, Pam, RING finger
1. Introduction
Inactivating mutations in either the TSC1 or TSC2 gene are responsible for tuberous sclerosis complex (TSC), an autosomal dominant disorder, which is characterized by formation of slow-growing, benign hamartomas in multiple organs [1]. The protein products of TSC1 and TSC2, hamartin and tuberin (also referred to as TSC1 and TSC2, respectively), form a tight complex and function together in the cell. Tuberin has GTPase activating protein activity toward the small GTP-binding protein Rheb. The TSC protein complex restrains cell growth and proliferation by acting as a focal point integrating a diverse range of signaling pathways with the mTOR pathway (for reviews [2, 3]). A growing body of evidence suggests that abnormal regulation of the TSC-mTOR pathway may be a widespread molecular mechanism for the pathological development of several hamartomatous syndromes including TSC (see review [3]).
We previously identified Pam (Protein associated with Myc) as a tuberin interactor and demonstrated a genetic interaction between the homologs of these proteins in Drosophila [4]. Pam is an extremely large protein which belongs to an evolutionally conserved family of proteins (PHR family) including Phr1 in mouse, Highwire (HIW) in Drosophila, Regulator of Presynaptic Morphology (RPM-1) in C. elegans, and Esrom in zebrafish. These proteins contain multiple domains including a Myc-binding region, regulator of chromosome condensation (RCC)-homology domains (RHD), and a C-terminal RING finger (RZF) domain which is present in a large family of E3 Ub ligases [5]. Several genetic studies demonstrate that Pam homologs regulate presynaptic growth [6–12]. Members of the PHR family have been suggested to control a variety of signaling pathways including JNK/p38 MAPK signaling in Drosophila and C. elegans [13, 14], TGF-β/BMP signaling in Drosophila [15], and cAMP signaling pathway in mammalian cells [16]. In addition, a very recent report suggests that RPM-1 positively regulates a Rab GTPase pathway to promote vesicular trafficking via late endosomes, thereby regulating synapse formation and axon termination [17]. The results obtained from both Drosophila and C. elegans suggest that the highly conserved RZF domain is critical for E3 Ub ligase activity of Pam homologs, particularly in regulation of synapse development [13, 14]. However, the function of Pam as an E3 Ub ligase in mammalian cells remains unknown.
In this study, we examine whether Pam targets tuberin for degradation through ubiquitination and thus can regulate TSC/mTOR signaling pathway. Our results demonstrate that Pam interacts with specific E2 enzymes and is capable of ubiquitinating tuberin. In addition, hamartin protects tuberin from ubiquitination by Pam. Furthermore, suppression of Pam in primary neurons results in stabilization of tuberin and downregulation of mTOR signaling. In addition to cell proliferation and growth, in neuronal cells, TSC1/2 and mTOR are implicated in many processes which are critical for neuronal development and long-term modification of synaptic strength [18–20]. Furthermore, Ub and ubiquitination enzymes have emerged as key regulators of synaptic development, function and plasticity [21]. Therefore, our findings suggest that Pam, as an E3 Ub ligase and a regulator of TSC/mTOR signaling, could play an essential role in synaptic development and function in mammalian neurons.
2. Materials and methods
2.1. Cell culture, antibodies, and reagents
Human embryonic kidney 293T (HEK293T) cells were maintained in DMEM with high glucose (4.5g/L glucose) (Gibco) containing 10% FBS (Gibco). Dissociated hippocampal or cortical neuronal cultures were prepared from E19 rats (Charles River Laboratories), plated either on coverslips coated with poly-D-lysine (PDL, 1mg/ml, Sigma) for transfection or on PDL (0.1mg/ml)-coated 60mm dish for lentiviral infection. Neuronal cultures were maintained in growth media containing Neurobasal Media (Gibco) supplemented with 2% B27 Supplement, 2mM L-glutamine, 50U/ml penicillin, and 50g/ml streptomycin, as described [20]. Primary antibodies used are anti-FLAG M2, anti-GAPDH (Sigma), anti-GST, anti-p53 (Santa Cruz), anti-myc 9E10 (Development Study Hybridoma Bank), anti-HA (Covance), anti-His (Qiagen), anti-phospho-S6 (S235/236), anti-S6, anti-phospho-S6K (T389) (Cell Signaling Technologies). Anti-Pam (PP1) and anti-TSC2 (TSDF) antibodies were described previously [4]. ALLN, MG132, and cycloheximide were obtained from Calbiochem.
2.2. Constructs
Generation of full-length Pam and truncated Pam fragments (Pam F1, Pam F2, and Pam F3) has been previously described [22]. Myc-tagged mutant Pam F3 (Pam F3-3A) and naturally occurring tuberin mutants R905Q and R611Q were generated using the QuickChange Site-Directed Mutagenesis kit (Stratagene). Myc-tagged Pam F3ΔRZF was generated by digestion of Pam F3 with Sap1 and Xho1 restriction enzymes to delete the C-terminus including the RZF domain. FLAG-tagged wild-type TSC2 and mutant TSC2 (S939A/T1462A) were a kind gift from B. D. Manning, and Ub-HA was a gift from Y. Jin. FLAG-tagged TSC2 (S1798A) was generously provided by J. Blenis, and Xpress-tagged TSC2 (S664/S540A) was kindly given by P. P. Pandolfi.
2.3. RNAi
To knock down expression of endogenous Pam in rat neurons, pSuper-rPam RNAi constructs were designed as described [23]. The highest efficiency of Pam suppression was observed when targeting bp 671–689 of rat Pam (5’-GGAGCCTCCAAGCCCTGCT-3’). The target sequence was not homologous to any other genes using a BLAST database search. A scrambled sequence (5’-CAGTCGCGTTTGCGACTGG-3’) and a modified sequence of rat Pam bp 671–689 containing two point mutations (5’-GGAGCCTCCGGGCCCTGCT-3’) were used as controls. These sequences were also used to generate the control and Pam RNAi constructs in the lentiviral pLKOpuro.1 vector kindly provided by Dr. Sheila Stewart of Washington University [24].
2.4. Transfection and infection
HEK293T cells (80–90% confluent) were transfected using lipofectamine 2000 (Invitrogen), as recommended by manufacturer’s instructions. For neuronal transfection, 4 days in vitro (DIV) rat hippocampal neuronal cells (8×104 cells/well of a 24-well plate) were transfected using lipofectamine 2000 (Invitrogen) with a 4:1 ratio of pSuper rPam-RNAi (1.6µg) or pSuper vector control (1.6µg) along with peGFP-N1 (0.4µg, Clontech) as a reporter of transfection. Cerebral cortical neurons (2 or 5 DIV, 1.7 ×106 cells/60mm dish) were also infected with the lentiviral constructs, and 4 days after infection cell lysates were prepared for western analysis. Lentiviral particles were produced by a NINDS funded Neuroscience Vector Core at Massachusetts General Hospital.
2.5. Ubiquitination assays and immunoprecipitation
At 20hr-post transfection, HEK293T cells were harvested and lysed in 0.5% NP-40 lysis buffer containing 150mM NaCl, 50mM Tris (pH7.5), 50mM NaF, 1mM Na orthovanadate, 2mM EDTA, and 1X Complete protease inhibitor cocktail (Roche). HEK293T cells were treated with proteasome inhibitor MG132 (50µM) or ALLN (10nM) for 1 hr prior to lysis. Lysates were then used for immunoprecipitation (IP). Briefly, protein lysates (1mg) were incubated with 2–3µg of anti-FLAG M2, anti-myc 9E10, anti-Xpress, anti-p53, or anti-TSC2 (TSDF) antibody for 15hr at 4°C. Subsequently, either protein-G Sepharose or protein-A Sepharose (Amersham-Pharmacia) was added and incubated for an additional 3hr. Precipitates were washed and resuspended in 2X SDS sample loading buffer. Lysates and immunoprecipitates were separated by 4–15% gradient or 5% SDS-PAGE (Bio-Rad), and transferred to nitrocellulose (Bio-Rad). Membranes were then blocked, detected with the appropriate primary antibody, followed by horseradish peroxidase-conjugated secondary antibodies (Chemicon), and then visualized using the ECL system (Amersham-Pharmacia).
2.6. In vitro binding assay
At 20hr-post transfection with Pam F3-Myc, HEK293T cells were harvested and lysed in 0.5% NP-40 lysis buffer. Lysates (500µg) were then incubated with 5µg of recombinant GST, UbcH2-GST (Calbiochem), UbcH5a-GST (EMD), UbcH5b-GST (Calbiochem), UbcH5c-GST (BIOMOL), Ubc6-His (EMD), or Ubc7-His (BIOMOL) proteins for 2hr at 4°C. Anti-Myc antibody 9E10 (2µg) was then added and incubated for 15hr at 4°C, followed by incubation with protein-G Sepharose beads for an additional 3hr. Bound proteins were then subjected to western analysis, and detected with anti-GST, anti-Myc, or anti-His antibody.
2.7. Quantification of immunofluorescence
Hippocampal cultures transfected with pSuper vector control or pSuper-rPam RNAi constructs at 4days post-transfection (DPT) were fixed in 3.7% paraformaldehyde/4% sucrose, permeablized with 0.1% Triton-X 100/phosphate-buffered saline (PBS) (Sigma), blocked in 1% goat serum/PBS (Jackson ImmunoLabs) for 1 hr, and incubated with anti-Pam (1:10) or anti-P-S6 (1:500) antibody, followed by Cy3-conjugated goat anti-rabbit (Jackson Immunoresearch) secondary antibody (1:500). Secondary antibody fluorescence was measured in GFP-positive neurons using an LSM510 confocal (Zeiss). Acquisitions of different experimental conditions were interleaved. Both image acquisition and image analysis were performed blinded to experimental conditions. To quantify somatic immunofluorescence, a binary mask was created using the fluorescence emitted from 500 nm to 530 nm (GFP). The somatic region was user-isolated, and the average fluorescence within this mask was calculated utilizing a MetaMorph custom journal (Molecular Devices). Multiple experiments were combined by normalizing each cell’s fluorescence to the average intensity of pSuper vector control.
2.8. Statistical Analysis
Statistical analysis of scanned images was determined using Student’s two-tailed t-test, and cumulative distributions of immunofluorescence intensities were compared using the non-parametric Kolmogorov-Smirnov test.
3. Results
3.1. Pam associates with E2 enzymes
Pam is an evolutionarily conserved, very large protein with several interesting motifs including two N-terminal RCC1 homology domains (RHD1 and RHD2), a central Myc-binding domain, and a RZF domain at the C- terminus (Figure 1A). To test the role of mammalian Pam in ubiquitination, we generated three different truncated Pam mutants along with full-length Pam [22]. The RZF domains in many E3 Ub ligases have been shown to directly bind to an E2 ubiquitin-conjugating (Ubc) enzyme [5]. We asked whether the C-terminal Pam (Pam F3), containing the RZF domain, could interact with a specific E2 enzyme. Myc-tagged Pam F3 was overexpressed in HEK293T cells, and its binding capacity to GST-, or His-tagged E2 recombinant proteins was examined by in vitro binding assays. Interestingly, Pam F3 was able to bind specifically to UbcH5a, UbcH5c and UbcH7, while no interaction was detected for the other E2 enzymes (Figure 1B). Another E3 Ub ligase, Parkin, was also shown to associate with two different E2 enzymes UbcH7 and UbcH8 [25], suggesting that certain E3 Ub ligases can interact with multiple E2 Ubc enzymes. Given our previous results showing that the C-terminal Pam region (residues 4312–4641), containing the RZF domain, was also able to bind tuberin [4], these results demonstrate that the Pam C-terminus has the capacity to interact with E2 Ubc enzymes as well as its putative substrate tuberin.
Fig. 1. Pam interacts with ubiquitin-conjugating E2 enzymes and ubiquitinates TSC2.
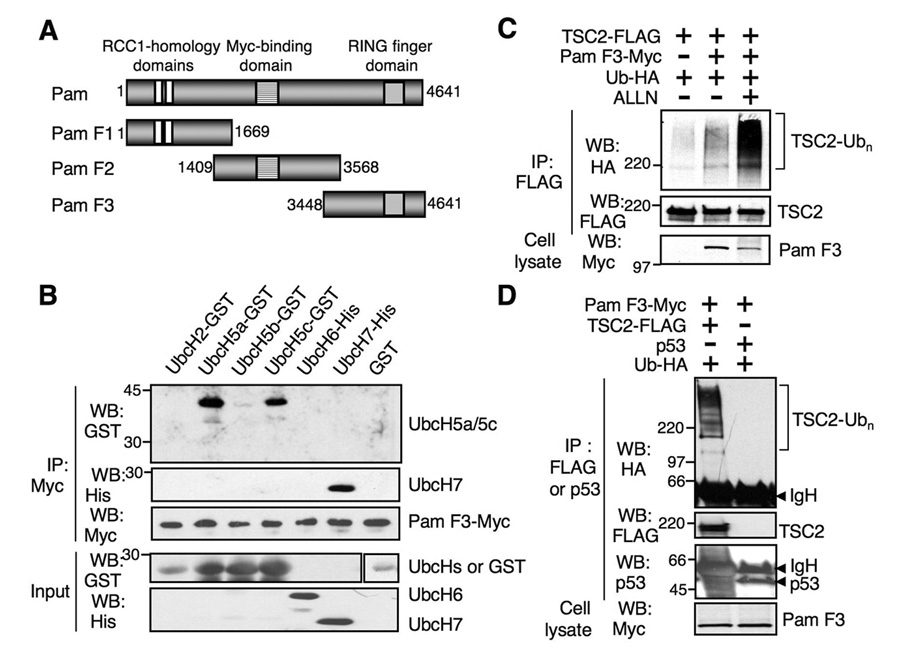
(A) Schematic diagram of Pam fragments. Pam F1 (aa 1–1669) contains two regulator of chromatin condensation (RCC)-homology domains (RHD1 and RHD2, residues 498–740 and 874–1065). Pam F2 (aa 1409–3568) contains a Myc-binding domain (aa 2413–2712) and Pam F3 (aa 3448–4641) possesses a RING finger domain (aa 4354–4440). (B) Carboxyl terminal Pam (Pam F3) binds to UbcH5a, UbcH5c and UbcH7 ubiquitin-conjugating E2 enzymes. Pull-down assays were carried out using recombinant E2 enzymes and HEK293T cell lysates transfected with Pam F3-Myc. (C) Pam F3 containing the RZF domain enhances ubiquitination of TSC2. HEK293T cells were transfected with TSC2-FLAG, Pam F3-Myc or Ub-HA. Immunoprecipitation (IP) performed with the anti-FLAG antibody was subjected to western analysis and ubiquitinated TSC2 was detected using the anti-HA antibody. (D) Tuberin, but not p53, is ubiquitinated by Pam. IPs were subjected to western analysis using anti-HA, anti-TSC2, or anti-p53 antibodies. Note the lack of ubiquitinated products in cells transfected with p53. However, ubiquitinated TSC2 was detected in cells transfected with Pam F3 and TSC2.
3.2. Pam enhances tuberin (TSC2) ubiquitination
We next examined whether Pam possesses E3 ubiquitin ligase activity toward tuberin. Pam C-terminus (Pam F3) containing the RZF domain promoted tuberin ubiquitination, and ALLN treatment significantly increased the accumulation of ubiquitinated tuberin (Figure 1C). The pattern of ubiquitination indicated both mono- and poly-ubiquitination of tuberin with variable lengths of poly-ubiquitin. The specificity of tuberin ubiquitination by Pam was next examined. Pam F3 failed to mediate ubiquitination of p53, which is known to be ubiquitinated by Mdm2 and E6-AP E3 Ub ligases [26, 27] (Figure 1D), indicating the specificity of tuberin ubiquitination by Pam. Neither Pam F1 nor Pam F2 showed any effect on ubiquitination of tuberin (data not shown), suggesting that the RZF domain of Pam is responsible for tuberin ubiquitination.
3.3. RZF domain of Pam C-terminus mediates self-ubiquitination as well as tuberin ubiquitination
Human Pam’s RZF domain has well-conserved cysteine and histidine residues, when compared with other well-known E3 Ub ligases, Mdm2 and Parkin [25, 28] (Fig. 2A). To determine the requirement of Pam’s RZF domain for ubiquitination, we generated a Pam F3-3A mutant in which three cysteine residues in the RZF domain were mutated to alanine residues (C4394A, C4409A, and C4417A). Since some RZF-containing proteins, including Mdm2, are known to be auto-ubiquitinated, we examined whether Pam could be self-ubiquitinated. Pam F3 was ubiquitinated when co-transfected with Ub-HA in HEK293T cells, whereas no accumulation of ubiquitinated Pam F3-3A mutant was observed, indicating that mutations in the RZF domain completely abolished self-ubiquitination (Fig. 2B). Most E3 Ub ligases are known to have a short half-life [21]. To determine the role of the RZF domain on stability, we chased the half-life of Pam F3 and Pam F3-3A mutant in the presence of the translation inhibitor cycloheximide up to 24 hr. Mutations in the RZF domain (PamF3-3A) resulted in significant prolongation of Pam’s half-life, whereas ~50% of wild type Pam F3 was degraded within 3–6 hr (Fig. 2C). In addition, the level of Pam F3 was consistently lower than the level of Pam F3-3A mutant in transfected cells (Fig. 2B, 2D and 2E), supporting that Pam is self-ubiquitinated, and intact RZF domain of Pam is essential for regulation of self-ubiquitination and stability. As is evident in Figure 2D, Pam F3-3A had significantly reduced tuberin ubiquitination, when compared to the wild type Pam F3. Furthermore, when Pam F3 and TSC2 were overexpressed, the TSC2 level was significantly reduced in HEK293T. However, Pam F3-3A was less efficient in downregulating TSC2, compared to Pam F3 (Figure 2E).
Fig. 2. The RZF domain of Pam is essential for self-ubiquitination and TSC2 ubiquitination.
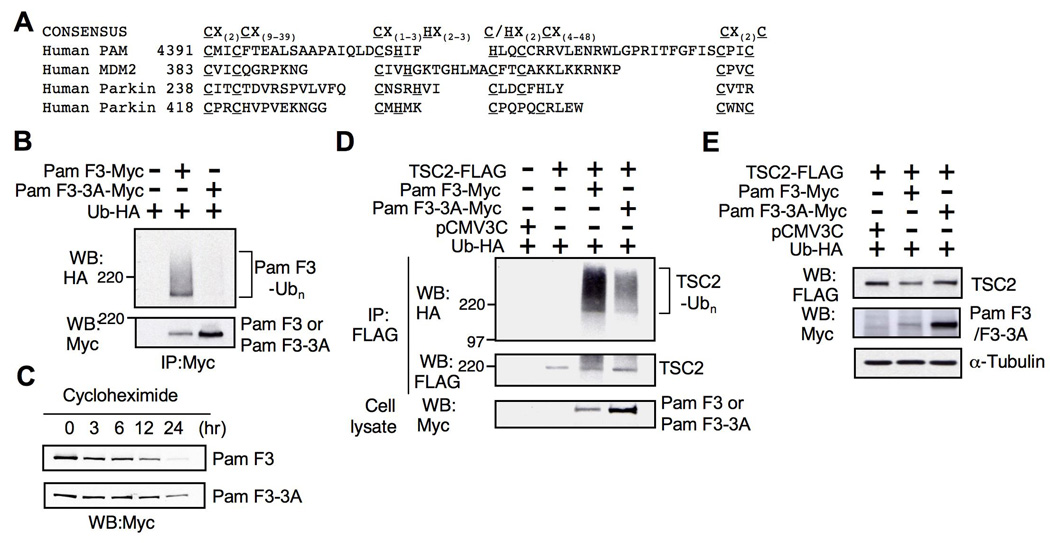
(A) Consensus sequence of RZF domain and sequence alignment of RZF domains from human Pam, Mdm2 and Parkin. C=cysteine, H=histidine, X=any amino acid. (B) Mutation of three different cysteine residues (Cys-4394, Cys-4409, and Cys-4417) in the RZF domain of Pam (PamF3-3A) resulted in significant loss of Pam self-ubiquitination compared to Pam F3. IPs performed with anti-Myc antibodies were analyzed to detect ubiquitinated Pam F3 using anti-HA antibody. (C) Increased half-life of Pam F3-3A mutant. Twenty hours post-transfection with either Pam F3-Myc or Pam F3-3A-Myc, HEK293T cells were treated with cycloheximide for up to 24 hr to inhibit protein synthesis. (D) The effect of the RZF domain mutations on TSC2 ubiquitination. Pam F3-3A displays a reduction in TSC2 ubiquitination when compared with Pam F3. (E) TSC2 is downregulated by Pam F3.
3.4. Phosphorylation-independent ubiquitination of TSC2 by Pam
Phosphorylation of a substrate is one of the initiation events for targeting a substrate for ubiquitination by serving as a recognition motif for an E3 ubiquitin ligase [29, 30]. Tuberin is known to be phosphorylated and inhibited by various kinases in the PI3K and MAPK signaling pathways, including Akt, ERK, and RSK1 kinases [31–34]. To determine whether tuberin phosphorylation is essential for initiation of tuberin ubiquitination by Pam, we tested the effect of mutations at several phosphorylation sites by using non-phosphorylatable tuberin mutants such as S939A/T1462A (mutant of Akt phosphorylation), S1798A (mutant of RSK1 phosphorylation) and S540A/S664A (mutant of ERK phosphorylation). We hypothesized that if phosphorylation is necessary for TSC2 ubiquitination, these non-phosphorylatable TSC2 mutants would not be ubiquitinated. Surprisingly, all mutants showed robust ubiquitination, similarly to the wild type tuberin (Figure 3A). These results suggest that tuberin ubiquitination by Pam is either mediated by other phosphorylation sites or is independent of its phosphorylation status.
Fig. 3. Regulation of TSC2 ubiquitination by Pam.
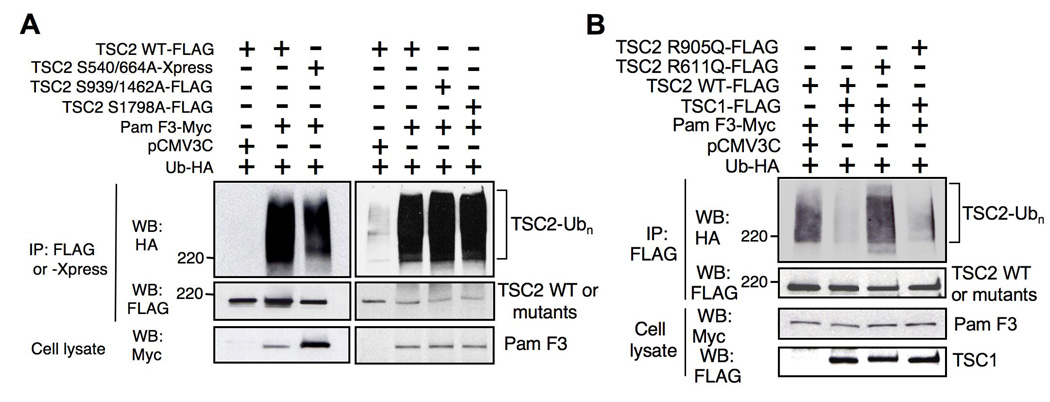
(A) Ubiquitination of TSC2 by Pam is independent of its phosphorylation status. HEK293T cells were transfected with either wild type or mutant TSC2 (S939A/T1462A, S1798A, and S540/664A), along with Pam F3-Myc and Ub-HA as indicated. IPs performed with anti-FLAG or anti-Xpress antibody were analyzed with anti-HA antibody for ubiquitinated TSC2. (B) TSC1 protects TSC2 from ubiquitination by Pam. HEK293T cells were transfected with either FLAG-TSC2 (wild type) or disease-associated TSC mutants (R611Q and R905Q), along with Pam F3-Myc, TSC1-FLAG, and Ub-HA. TSC1 inhibits Pam F3 induced ubiquitination of wild type and R905Q mutant, but not R611Q mutant.
3.5. A TSC disease-derived mutation (R611Q) enhances tuberin ubiquitination by Pam
Previous studies showed that hamartin inhibits tuberin ubiquitination in HEK293 and Cos-7 cells [35, 36]. Similarly, we found that tuberin ubiquitination by Pam was completely blocked by co-expression of TSC1 (Figure 3B, lane 2). These results support the prevailing idea that hamartin acts as a chaperone to stabilize tuberin. However, TSC1 was unable to inhibit TSC2 ubiquitination by Pam when a naturally occurring missense TSC2 mutant (TSC2 R611Q) was expressed along with TSC1 and Pam F3, compared to wild type TSC2 (Figure 3B, lane 3). Tuberin R611Q mutant has been shown to be incapable of binding hamartin [37]. In contrast, another pathogenic tuberin mutant (R905Q), which is capable of binding to hamartin [37], was not ubiquitinated by Pam when co-expressed with TSC1 (Figure 3B, lane 4). Therefore, these results suggest that a subset of disease-associated TSC2 mutations, which disrupt hamartin binding, are more susceptible to ubiquitination by Pam.
3.6. Pam regulates mTOR signaling through tuberin in neurons
Pam, similar to Drosophila and C. elegans homologs (HIW and RPM-1) is highly expressed in neurons. To determine whether Pam regulates tuberin levels in mammalian neurons, we examined whether Pam RNA interference (RNAi) results in stabilization of tuberin. We employed the lentiviral, vector-based pLKOpuro.1 RNAi system [24] to knock down Pam expression in primary cortical neurons derived from embryonic day 18 rat. At 4 days post-infection with lentiviral Pam RNAi, Pam expression was specifically knocked down, compared with neurons infected with control RNAi lentivirus expressing scrambled non-targeting sequence (Figure 4A). Furthermore, downregulation of Pam increased tuberin levels, when compared with control RNAi (Figure 4A and 4B). Since tuberin is known to inhibit mTOR activity, we tested whether the activity of components of the mTOR pathway were downregulated upon Pam knockdown. We observed that two targets of the mTOR signaling pathway, p70S6 kinase (S6K) and ribosomal S6, were downregulated, as monitored by the reduced levels of phospho-S6K (T389) and phospho-S6 (S235/236) (Figure 4A and 4C). These results suggest that Pam can regulate tuberin levels and thus the mTOR pathway in mammalian neurons.
Fig. 4. Pam regulates mTOR signaling through TSC2 downregulation in rat neurons.
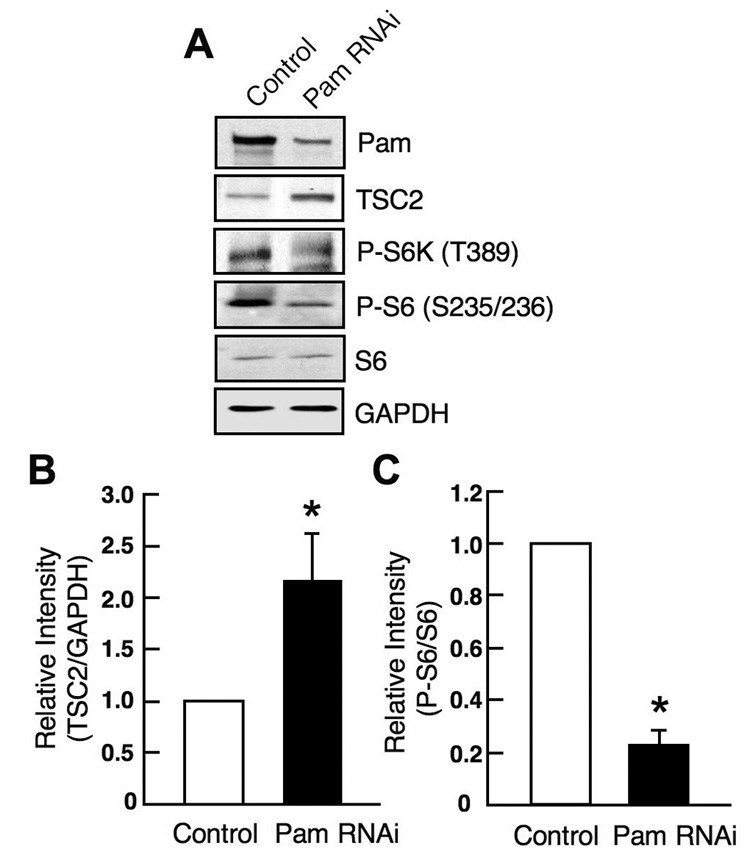
(A) Pam RNAi in rat cerebral cortical neurons resulted in an increase of TSC2 levels and subsequent downregulation of P-S6 and P-S6K levels. Dissociated cortical neuronal cells (5 DIV) from E18 rat were infected with lentiviruses expressing either control or Pam RNAi. At 9 DIV, cell lysates were prepared and analyzed by western analysis. (B) Quantitation of TSC2 levels in control and Pam RNAi groups (mean ± SEM; n=3, t-test, *p<0.05). Band intensities from western blots were analyzed using scanned images by densitometer (Bio-Rad). TSC2 levels were normalized against GAPDH levels. (C) Quantitation of P-S6 levels in control and Pam RNAi groups (mean ± SEM; n=3, t-test, *p<0.05). Band intensities were analyzed as described in B. P-S6 levels were normalized against total S6 levels.
To further determine the influence of Pam on the mTOR pathway in neurons, immunofluorescence was performed using dissociated hippocampal neuron cultures (4 DIV) to assess the effect of Pam RNAi on phospho-S6 (S235/236) levels, using a vector-based pSuper RNAi system [23]. Neurons were co-transfected with pSuper or pSuper-Pam RNAi, along with the pEGFP vector as a reporter of transfected cells. At 4 DPT, cells were fixed and subjected to immunofluorescence analysis using the anti-P-S6 (S235/236) antibody. Quantitative analysis revealed that Pam RNAi not only resulted in a significant reduction in Pam levels (supplemental Fig. 1.), but that it also resulted in a significant decrease of P-S6 level, when compared with vector control (Figure 5A and 5B). Additionally, neurons expressing a mutant Pam RNAi, having two point mutations at target recognition sequences, showed no decrease in P-S6 levels (supplemental Fig. 2.). These results indicate that Pam may play a role in regulating the mTOR signaling pathway in neurons.
Fig. 5. Pam RNAi results in a reduction of P-S6 levels.
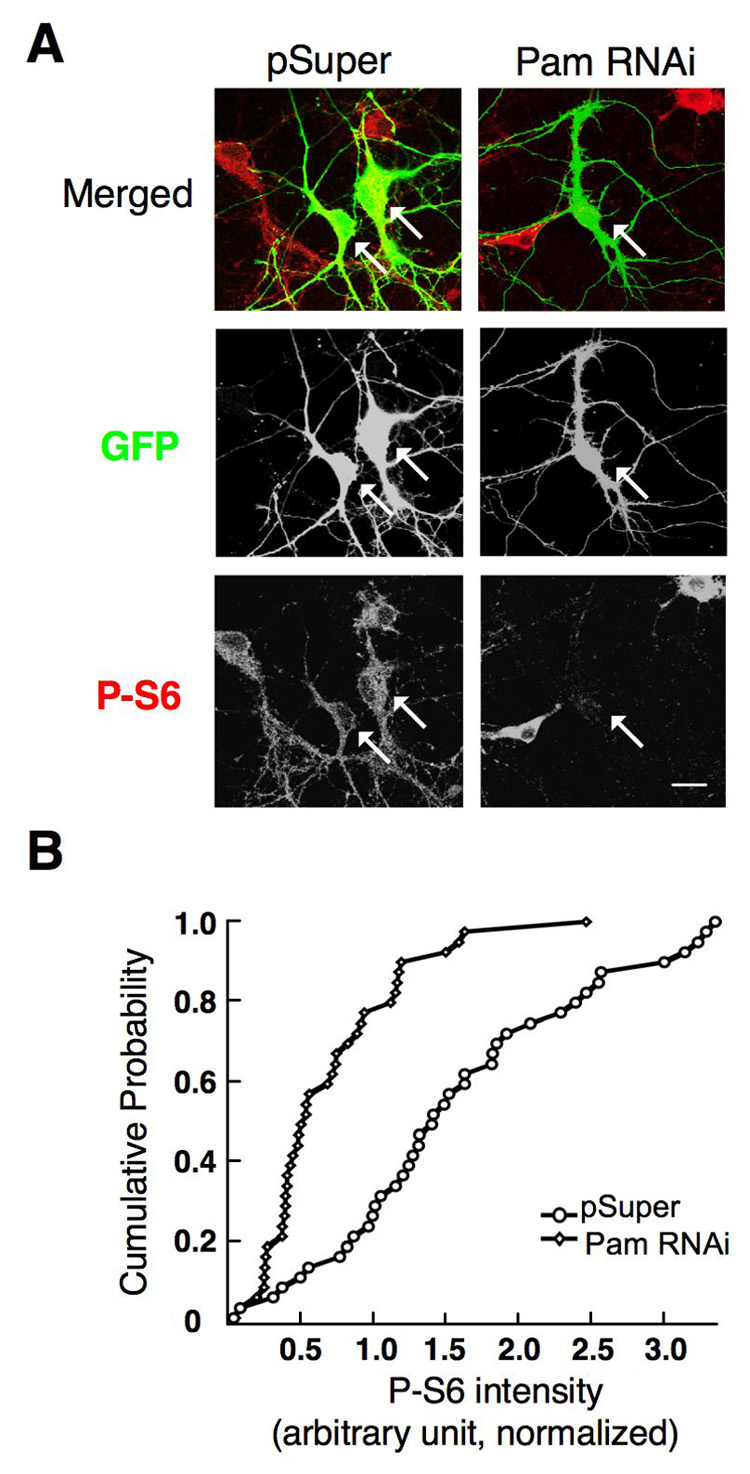
(A) Co-transfection of E19 rat hippocampal cultures four days post-dissociation with both eGFP-N1 and an RNAi against PAM caused a reduction in somatic phosphorylated S6 ribosomal protein (P-S6) levels, compared to co-transfection of eGFP-N1/pSuper backbone at 4 DPT. Arrows indicate transfected neurons. Representative images. Scale Bar, 20µm. (C) Cumulative distribution of P-S6 immunostaining in eGFP-N1/pSuper-transfected or eGFP-N1/Pam RNAi-transfected hippocampal neurons at 4 DPT (two independent experiments, pSuper (n=40), Pam RNAi (n=40), p<0.01
4. Discussion
In this study, we document for the first time that mammalian Pam functions as an E3 Ub ligase, and Pam, as an interactor of tuberin, can induce the ubiquitination of tuberin. RNAi mediated knockdown of Pam in rat primary neurons results in stabilization of tuberin and downregulation of mTOR signaling, which establishes a physiological role for Pam in regulating TSC/mTOR signaling in mammalian neurons. In addition to growth control in various organs, TSC proteins via mTOR signaling play an important role in neurodevelopment. Rapamycin-sensitive mTOR function is known to regulate neuronal activity-dependent local protein synthesis, long-term potentiation, and neuronal morphology [19, 20, 38]. A crucial role for mTOR signaling in neuronal translation has been established providing a functional role for this pathway in long-lasting forms of synaptic plasticity and memory [18, 39, 40]. Furthermore, a functional interplay between MAPK and mTOR-dependent signaling appears to determine the translational efficiency during the establishment of long-term synaptic plasticity [18, 41]. Two most recent publications employing knock out approaches for Pam (Phr1) in mouse implicate a role for Pam in axon tract formation and regulation of axon outgrowth [42, 43]. In the future, it is essential to examine whether the defects observed in the Phr1 deficient mice could be explained in part by aberrant regulation of TSC/mTOR signaling.
Tuberin is also ubiquitinated by other putative E3 Ub ligases including human papillomavirus E6 oncoprotein and the recently identified HERC1 protein. E6 oncoprotein from human papillomavirus 16 associates with and degrade tuberin, thus leading to upregulation of S6K and S6 activities, suggesting a potential pathogenic mechanism of E6-induced tumorigenesis [44]. Intriguingly, HERC1, similar to Pam, is a very large protein (532 kDa) with multiple domains including a HECT domain (homologous to E6-AP carboxyl-terminus) and a RCC1-like domain (RHD) [45]. A recent report demonstrates that HERC1 associates with TSC2, and TSC1 inhibits the interaction between TSC2 and HERC1. The authors further demonstrate that TSC1 stabilizes TSC2 by preventing the association between tuberin and HERC1 ubiquitin ligase [36]. The physiological functions of HERC1 in regulating the TSC-mTOR pathway are yet to be determined. However, it is important to note that in addition to phosphorylation by multiple kinases, tuberin stability is also determined by ubiquitination mediated by many Ub ligases, which adds another layer of complexity to TSC-mTOR regulation.
Protein phosphorylation plays an important role in the initiation of protein ubiquitination by providing a recognition motif for an E3 ligase and it was shown previously that activation of Akt could trigger the proteasomal degradation of tuberin [46]. Our results show that tuberin ubiquitination by Pam, in HEK293 cells, can occur independently of tuberin phosphorylation by Akt, RSK1, and ERK kinases. However, our results suggest that abolishing hamartin binding may enhance tuberin ubiquitination by Pam. While phosphorylation is known to trigger substrate ubiquitination by some E3 Ub ligases, it is also becoming evident that phosphorylation of E3 Ub ligase itself or interaction with other regulatory proteins can modulate the accessibility of an E3 ligase to its substrate, thus regulating the ubiquitination process [47, 48]. Further identification of upstream signal(s) or interacting proteins, which could regulate Pam’s E3 Ub ligase, may be necessary to understand precisely how Pam ubiquitinates its substrates.
Tuberin ubiquitination by these putative E3 Ub ligases and subsequent degradation may occur in a context or tissue-dependent manner. For example, Pam is expressed predominantly in the nervous system, while HERC1 is expressed ubiquitously. Given the specific role of Pam homologs in the nervous system, mammalian Pam may function primarily in the nervous system as an E3 Ub ligase toward tuberin. Furthermore, it will be interesting to identify other downstream targets that may serve as substrates for the E3 Ub ligase activity of Pam and to define whether Pam integrates mTOR signaling with other signaling pathways in neurons.
Supplementary Material
Acknowledgements
This work was supported in part by National Institutes of Health grants NS24279 (V. Ramesh), NS52707 (B. L. Sabatini), a Quan predoctoral fellowship (R. M. Witt), and a grant support from Autism Speaks (V. Ramesh). Lentiviral particles were produced Neuroscience Vector Core supported by NS45776. We thank members of our laboratory for helpful comments on the manuscript.
Abbreviations
- TSC
tuberous sclerosis complex
- mTOR
mammalian target of rapamycin
- Ub
ubiquitin
- RNAi
RNA interference
- Pam
Protein-associated with Myc
- HIW
highwire
- RPM-1
regulator of presynaptic morphology
- RHD
regulator of chromosome condensation-homolgy domain
- RZF
RING finger
- HEK293T
human embryonic kidney 293T cell
- IP
immunoprecipitation
- WB
western blotting
- DIV
days in vitro
- DPT
days post-transfection
- PBS
phosphate-buffered saline
- Ubc
ubiquitin-conjugating enzyme
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gomez M, Sampson J, Holtes-Whittemore V. Tuberous Sclerosis Complex. Oxford University Press; 1999. [Google Scholar]
- 2.Kwiatkowski DJ, Manning BD. Hum. Mol. Genet. 2005;14:251–258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 3.Inoki K, Guan KL. Trends Cell. Biol. 2006;16(4):206–212. doi: 10.1016/j.tcb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Murthy V, Han S, Beauchamp RL, Smith N, Haddad LA, Ito N, Ramesh V. J. Biol. Chem. 2004;279(2):1351–1358. doi: 10.1074/jbc.M310208200. [DOI] [PubMed] [Google Scholar]
- 5.Joazeiro CA, Weissman AM. Cell. 2000;102(5):549–552. doi: 10.1016/s0092-8674(00)00077-5. [DOI] [PubMed] [Google Scholar]
- 6.Burgess RW, Peterson KA, Johnson MJ, Roix JJ, Welsh IC, O'Brien TP. Mol. Cell. Biol. 2004;24(3):1096–1105. doi: 10.1128/MCB.24.3.1096-1105.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Souza J, Hendricks M, Le Guyader S, Subburaju S, Grunewald B, Scholich K, Jesuthasan S. Development. 2005;132(2):247–256. doi: 10.1242/dev.01578. [DOI] [PubMed] [Google Scholar]
- 8.DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS. Nature. 2001;412(6845):449–452. doi: 10.1038/35086595. [DOI] [PubMed] [Google Scholar]
- 9.Schaefer AM, Hadwiger GD, Nonet ML. Neuron. 2000;26(2):345–356. doi: 10.1016/s0896-6273(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 10.Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. Neuron. 2000;26(2):313–329. doi: 10.1016/s0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 11.Wu C, Wairkar YP, Collins CA, DiAntonio A. J. Neurosci. 2005;25:9557–9566. doi: 10.1523/JNEUROSCI.2532-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhen M, Huang X, Bamber B, Jin Y. Neuron. 2000;26(2):331–343. doi: 10.1016/s0896-6273(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 13.Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Neuron. 2006;51(1):57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 14.Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. Cell. 2005;120(3):407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 15.McCabe BD, Hom S, Aberle H, Fetter RD, Marques G, Haerry TE, Wan H, O'Connor MB, Goodman CS, Haghighi AP. Neuron. 2004;41(6):891–905. doi: 10.1016/s0896-6273(04)00073-x. [DOI] [PubMed] [Google Scholar]
- 16.Pierre SC, Hausler J, Birod K, Geisslinger G, Scholich K. EMBO J. 2004;23:3031–3040. doi: 10.1038/sj.emboj.7600321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grill B, Bienvenut WV, Brown HM, Ackley BD, Quadroni M, Jin Y. Neuron. 2007;55(4):587–601. doi: 10.1016/j.neuron.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Neuron. 2004;44(1):59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Parsons RG, Gafford GM, Helmstetter FJ. J. Neurosci. 2006;26(50):12977–12983. doi: 10.1523/JNEUROSCI.4209-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tavazoie S, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Nat. Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 21.DiAntonio A, Hicke L. Annu. Rev. Neurosci. 2004;27:223–246. doi: 10.1146/annurev.neuro.27.070203.144317. [DOI] [PubMed] [Google Scholar]
- 22.Santos TM, Han S, Bowser M, Sazani K, Beauchamp RL, Murthy V, Bhide PG, Ramesh V. J. Neurosci. Res. 2006;83:222–232. doi: 10.1002/jnr.20723. [DOI] [PubMed] [Google Scholar]
- 23.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296(5567):550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 24.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA, Novina CD. RNA. 2003;9(4):493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Nat. Genet. 2000;25(3):302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 26.Fuchs SY, Fried VA, Ronai Z. Oncogene. 1998;17:1483–1490. doi: 10.1038/sj.onc.1202184. [DOI] [PubMed] [Google Scholar]
- 27.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. Cell. 1993;75(3):495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 28.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. J. Biol. Chem. 2000;275(12):8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 29.Carrano AC, Eytan E, Hershko A, Pagano M. Nat. Cell Biol. 1999;1(4):193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 30.Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A. Nat. Cell Biol. 2001;3(3):321–324. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 31.Inoki K, Li Y, Zhu T, Wu J, Guan KL. Nat. Cell Biol. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 32.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Mol. Cell. 2002;10(1):151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 33.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Proc. Natl. Acad. Sci. U. S. A. 2004;101(37):13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 35.Benvenuto G, Li S, Brown SJ, Braverman R, Vass WC, Cheadle JP, Halley D, Sampson JR, Wienecke R, DeClue JE. Oncogene. 2001;19:6306–6316. doi: 10.1038/sj.onc.1204009. [DOI] [PubMed] [Google Scholar]
- 36.Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, Guan KL. J. Biol. Chem. 2006;281(13):8313–8316. doi: 10.1074/jbc.C500451200. [DOI] [PubMed] [Google Scholar]
- 37.Nellist M, Verhaaf B, Goedbloed MA, Reuser AJ, van den Ouweland AM, Halley DJ. Hum. Mol. Genet. 2001;10(25):2889–2898. doi: 10.1093/hmg/10.25.2889. [DOI] [PubMed] [Google Scholar]
- 38.Jaworski J, Sheng M. Mol. Neurobiol. 2006;34(3):205–219. doi: 10.1385/MN:34:3:205. [DOI] [PubMed] [Google Scholar]
- 39.Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Cell. 2004;116(3):467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 40.Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. Proc. Natl. Acad. Sci. U. S. A. 2002;99(1):467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. J. Neurosci. 2007;27(22):5885–5894. doi: 10.1523/JNEUROSCI.4548-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bloom AJ, Miller BR, Sanes JR, DiAntonio A. Genes Dev. 2007;21(20):2593–2606. doi: 10.1101/gad.1592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewcock JW, Genoud N, Lettieri K, Pfaff SL. Neuron. 2007;56(4):604–620. doi: 10.1016/j.neuron.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 44.Lu Z, Hu X, Li Y, Zheng L, Zhou Y, Jiang H, Ning T, Basang Z, Zhang C, Ke Y. J. Biol. Chem. 2004;279(34):35664–35670. doi: 10.1074/jbc.M403385200. [DOI] [PubMed] [Google Scholar]
- 45.Rosa JL, Casaroli-Marano RP, Buckler AJ, Vilaro S, Barbacid M. EMBO J. 1996;15(16):4262–4273. [PMC free article] [PubMed] [Google Scholar]
- 46.Plas DR, Thompson CB. J. Biol. Chem. 2003;278:12361–12366. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- 47.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. Cell. 2005;121(7):1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 48.Gallagher E, Gao M, Liu YC, Karin M. Proc. Natl. Acad. Sci. U. S. A. 2006;103(6):1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.