

Abstract
To control the rate of release of methylprednisolone (MP) in lysosomes, new dextran-MP conjugates with peptide linkers were synthesized and characterized. Methylprednisolone succinate (MPS) was attached to dextran 25 kDa using linkers with 1–5 Gly residues. The release characteristics of the conjugates in pH 4.0 and 7.4 buffers, blood, liver lysosomes, and various lysosomal proteinases were determined using a size-exclusion and/or a newly-developed reversed-phase HPLC method capable of simultaneous quantitation of MP, MPS, and all five possible MPS-peptidyl intermediates. We synthesized conjugates with ≥ 90% purity and 6.9–9.5% (w/w) degree of MP substitution. The conjugates were stable at pH 4.0, but released MP and intact MPS-peptidyl intermediates in the pH 7.4 buffer and rat blood, with faster degradation rates for longer linkers. Rat lysosomal fractions degraded the conjugates to MP and all the possible intermediates also at a rate directly proportional to the length of the peptide. Whereas the degradation of the conjugates by cysteine peptidases (papain or cathepsin B) was relatively substantial, no degradation was observed in the presence of aspartic (cathepsin D) or serine (trypsin) proteinases, which do not cleave peptide bonds with Gly. These newly-developed dextran conjugates of MP show promise for controlled delivery of MP in lysosomes.
Keywords: controlled delivery, controlled release, targeted drug delivery, prodrugs, synthesis, HPLC, stability, linker, spacer
INTRODUCTION
Methylprednisolone (MP) is a glucocorticoid with well-defined anti-inflammatory and immunosuppressive effects.1 In liver transplantation, MP is used as a first line therapy in the treatment of acute rejection.2,3 For this purpose, large intravenous doses of the drug (~ 1 g/day) are administered as pulse therapy, usually over 3–5 days. However, this regimen has been associated with significant life-threatening toxicities.2,4 Therefore, a targeted delivery of MP to the liver in liver-transplanted patients may afford administration of smaller doses, resulting in prolonged local effects in the liver, with less toxicity to other organs.
A variety of approaches, such as the use of macromolecular carriers, have been explored to selectively deliver drugs to their target site.5,6 Among various macromolecules, dextrans are one of the most extensively investigated carriers because they are water-soluble and biodegradable natural polysaccharides, which have been used clinically as plasma volume expanders for several decades.7,8 Additionally, dextrans are available commercially as different molecular weights and have numerous hydroxyl groups that can be easily conjugated to the parent drug. Therefore, they have been investigated as macromolecular carriers for the delivery of a number of therapeutic agents, such as anticancer and steroidal and non-steroidal drugs.8
Recently, we conjugated MP to dextran with a Mw of 70 kDa, using succinic acid as a linker, and investigated its hydrolysis kinetics,9 pharmacokinetics,10 and pharmacodynamics11,12 in rats. It was demonstrated that the conjugate would selectively accumulate and gradually release MP in the liver and spleen, resulting in a more intense and sustained immunosuppressive activity in these organs, compared with administration of an equal dose of the parent drug. Additionally, studies in a rat liver transplantation model indicated that the conjugate is superior to MP for prevention of rejection of allograft in this model.13 However, despite very high accumulation of the conjugate in the liver and spleen, the release of MP from the conjugate was slow and incomplete.11,12 The conjugate:MP concentration ratios in the liver and spleen tissues were relatively large and increased with time, resulting in the disappearance of immunosuppressive activity at later time points despite the presence of large concentrations of the conjugate in these tissues.11,12 Therefore, the present study was designed to develop and test the second generation dextran prodrugs of MP containing various peptides as linkers between dextran and MP succinate (MPS). Previous studies have shown that peptide linkers of various length and amino acid type and sequence may be degraded at different rates by lysosomal enzymes.14–16 Therefore, the hypothesis of this investigation was that the rate of regeneration of MP from its dextran prodrug can be controlled using different peptide linkers. Hydrolysis of MPS-peptide-dextran prodrugs would potentially lead to the formation of MP, MPS, and MPS-peptidyl intermediates. Therefore, for stability, purity, and release investigations, it is desirable to quantitate all possible components, including intermediates, in the sample.
In this article, synthesis and characterization of MPS-peptide-dextran prodrugs (DMP) with five different peptides are described. Additionally, a new HPLC method for simultaneous quantitation of MP, MPS, and five MPS-peptidyl intermediates is developed and validated. The assay is used for the determination of the in vitro release profiles of all the possible intermediates from prodrugs in the presence of various peptidases, rat liver lysosomes, or buffers with different pH values. The stability of the conjugates in rat blood was also quantitated using size-exclusion chromatography.
EXPERIMENTAL SECTION
Chemicals
Dextran with an average Mw of 23,500 was obtained from Dextran Products Ltd. (Scarborouh, Ontario, Canada). 6α-Methylprednisolone (MP) was purchased from Steraloids (Newport, RI, USA). Fmoc-Gly-Wang resin, coupling reagents, and Fmoc-amino acid building blocks, Fmoc-Gly-OH and Fmoc-methyl Gly (mGly)-OH, were purchased from Novabiochem (San Diego, CA). Papain from papaya latex (33 U/mg), cathepsin B from bovine spleen (24 U/mg), cathepsin D from bovine spleen (5 U/mg), trypsin from bovine pancreas (10 KU/mg), rat liver lysosome isolation kit, and acid phosphatase assay kit were purchased from Sigma (St. Louis, MO). For chromatography HPLC grade acetonitrile (EMD) was obtained from VWR Scientific (Minneapolis, MN, USA). All other reagents were analytical grade and obtained through commercial sources.
Animals
Adult, male Sprague-Dawley rats were used as blood donors and for the preparation of liver lysosomal fractions described later. All the procedures involving animals in this study were consistent with the “Principles of Laboratory Animal Care” (NIH publication Vol. 25, No. 28, revised 1996) and approved by the Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee.
Synthesis and Characterization of Dextran Prodrugs of Methylprednisolone (DMP)
The chemical structures of final products were characterized by nuclear magnetic resonance spectrometry (1H NMR, 13C NMR) determined on a Bruker NMR spectrometer (400 MHz). 13C NMR spectra are fully decoupled. Chemical shifts are reported in parts per millions (ppm). The chemical structures of final products were confirmed by a high-resolution PE Biosystems Mariner API time-of-flight electrospray mass spectrometer.
Synthesis of MP Succinate (MPS)
4-Dimethylaminopyridine (DMAP, 100 mg, 0.82 mmol) and succinic anhydride (290 mg, 2.90 mmol) were added to a solution of MP (1.08 g, 1.45 mmol) in dry pyridine (15.0 mL). The reaction mixture was stirred at room temperature overnight. After completion of the reaction, the solvent was evaporated under reduced pressure and the crude compound was purified by column chromatography over silica gel using dichloromethane/methanol as the eluents to yield MPS (1.35 g, 98.5%).
1H NMR (400 MHz, DMSO-d6, δ ppm) 12.25 (s, 1H,), 7.32 (d, J = 10.2 Hz, 1H), 6.18 (d, J = 10.2 Hz, 1H), 5.82 (s, 1H), 5.40 (s, 1H), 5.07 (d, J = 17.6 Hz, 1H), 4.76 (d, J = 17.6 Hz, 1H), 4.28 (s, 1H), 2.63–2.60 (m, 3H), 2.51–2.48 (m, 3H), 2.10–2.01 (m, 2H), 1.87 (d, J = 10.4 Hz, 1H), 1.66–1.57 (m, 3H), 1.45–1.41 (m, 1H) 1.38 (s, 3H), 1.35–1.28 (m, 1H), 1.04 (d, J = 6.1 Hz, 3H), 0.86–0.83 (m, 1H), 0.78 (s, 3H), 0.75–0.66 (m, 1H). 13C NMR (100 MHz, DMSO-d6, δ ppm) 206.00, 185.97, 174.25, 174.10, 172.49, 158.08, 127.53, 119.60, 89.04, 69.11, 68.49, 56.72, 51.79, 47.88, 44.84, 43.72, 33.94, 33.28, 31.60, 29.47, 29.27, 24.30, 22.17, 18.46, 17.38; HR-MS (ESI-TOF) (m/z): C26H34O8 calcd, 474.2254; found 497.3423 [M + Na + H]+.
Synthesis of MPS-Peptide Conjugates
Synthesized MPS-peptides contained mGly (MPS-mG-OH), mGly-Gly (MPS-mGG-OH), mGly-Gly-Gly (MPS-mGGG-OH), mGly-Gly-Gly-Gly (MPS-mGGGG-OH), or mGly-Gly-Gly-Gly-Gly (MPS-mGGGGG-OH). Additionally, two MPS-peptides without mGly (MPS-GG-OH and MPS-GGGG-OH) were synthesized for comparative purposes. As a representative example, the synthesis of MPS-mGGGGG-OH is given here (Scheme 1). The peptide was assembled on Fmoc-Gly-Wang resin (600 mg, 0.66 mmol/g) by Fmoc solid phase peptide synthesis strategy on a PS3 automated peptide synthesizer (Rainin Instrument Co., Oakland, CA) at room temperature using Fmoc protected amino acids [Fmoc-Gly-OH (C+1), Fmoc-Gly-OH (C+2), Fmoc-Gly-OH (C+3), Fmoc-mGly-OH (C+4)] (1.58 mmol) and MPS (1.58 mmol). 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (1.58 mmol) and N-methylmorpholine (NMM) (1.58 mmol) in N,N-dimethylformamide (DMF) were used as coupling and activating reagents, respectively. Fmoc deprotection at each step was carried out using piperidine in DMF (20%). MPS-mGGGGG-OH was cleaved from the resin by a mixture of TFA/anisole/water (95:2.5:2.5), precipitated by the addition of cold diethyl ether (Et2O). The crude peptide conjugates were purified by HPLC (Shimadzu LC-8A preparative liquid chromatograph; Shimadzu fraction collector 10A) on a Phenomenex® Prodigy 10μm ODS reversed-phase column. All MPS-peptide conjugates were separated by eluting the crude peptide at 4.0 mL/min using a gradient of 0–100% acetonitrile (0.1% TFA) and water (0.1% TFA) over 60 min and were lyophilized.
Scheme 1.

Synthesis of MPS-mGGGGG-OH and MPS-mGGGGG-dextran. Reagents: (i) Piperidine (20%), DMF, (ii) Fmoc-Gly-OH, HBTU, DMF, (iii) Fmoc-mGly-OH, HBTU, DMF, (iv) MPS, HBTU.
MPS-mGGGGG-OH: 1H NMR (400 MHz, DMSO-d6, δ ppm) 8.21–8.12 (m, 4H), 7.32 (d, J = 10.1 Hz, 1H), 6.18 (d, J = 10.1, 1H), 5.81 (s, 1H), 5.05 (dd, J = 17.6 Hz, 3.8 Hz, 1H), 4.76 (d, J = 17.6 Hz, 2H), 4.28 (s, 1H), 4.05 (s, 1H), 3.95 (s, 1H), 3.83–3.68 (m, 8H), 2.89 (s, 1H), 2.79 (s, 1H), 2.73 (s, 1H), 2.64–2.45 (m, 3H), 2.10–2.01 (m, 2H), 1.88 (d, J = 10.7 Hz, 1H), 1.66–1.61 (m, 3H), 1.45–1.30 (m, 5H), 1.04 (d, J = 6.2, 3H), 0.86–0.66 (m, 5H); 13C NMR (100 MHz, DMSO-d6, δ ppm) 205.28, 185.15, 173.48, 172.03, 171.33, 171.13, 169.24, 169.10, 168.86, 168.55, 157.27, 126.67, 118.75, 88.58, 68.28, 67.52, 55.91, 50.96, 50.35, 47.04, 44.01, 42.90, 42.02, 41.73, 40.57, 40.11, 35.98, 34.34, 33.07, 32.44, 30.77, 28.56, 27.54, 23.45, 21.32, 17.60,16.51; HR-MS (ESI-TOF) (m/z): C37H51N5O13 calcd, 773.3483; found 773.1624 [M]+, 1543.3510 [2M – 3H]+.
1H NMR, 13C NMR, and HR-MS (ESI-TOF) (m/z) for other MPS-peptide conjugates are provided in Supporting Information.
Synthesis of MPS-Peptide-Dextran (DMP) Conjugates
The following conjugates were synthesized: MPS-mG-Dex, MPS-mGG-Dex, MPS-mGGG-Dex, MPS-mGGGG-Dex, and MPS-mGGGGG-Dex. Additionally, in preliminary studies, MPS-GG-Dex and MPS-GGGG-Dex were synthesized. As a representative example, the synthesis of MPS-mGGGGG-Dex is given here (Scheme 1). To the stirring solution of MPS-mGGGGG-OH (350 mg), dextran (200 mg), and 4-dimethylaminopyridine (DMAP) (30 mg. 0.25 mmol) in dry DMSO (3.0 mL) in a dry round bottom flask under nitrogen atmosphere was added N,N-diisopropylethylamine (DIPEA, 200 μL, 1.21 mmol) followed by N,N′-diisopropylcarbodiimide (DIC, 60 μL, 0.39 mmol). The reaction mixture was stirred at 40°C for 48 h and poured into a cold ethanol (30 mL). The precipitate was centrifuged and washed twice with cold ethanol:diethyl ether (50:50, v/v), and finally with cold ethanol:acetonitrile (70:30, v/v). The solid was centrifuged and dried under vacuum to give MPS-mGGGGG-Dex conjugate.
Further Characterization of the Conjugates
Purities of the powders were determined using the size-exclusion chromatographic (SEC) method described below. The degree of substitution of MP in various conjugates was determined by hydrolysis of the conjugate under basic conditions. To 1.0 mg of the conjugate were added 1 mL of 0.1 N NaOH and 0.6 mL of methanol. After leaving the mixture at room temperature for 5 min, 100 μL of the sample was micropipetted into a microcentrifuge tube containing 100 μL of 0.1 M HCl. An aliquot (50 μL) was then injected into a reversed-phase HPLC method described below.
Degradation/Hydrolysis of Conjugates
Chemical Hydrolysis in Buffers
Hydrolysis of various DMP conjugates at a concentration equivalent to 100 μg/mL MP was studied at 37°C in pH 7.4 (100 mM phosphate buffer) and 4.0 (50 mM acetate buffer), simulating the physiological and lysosomal pH, respectively. Samples (100 μL, n = 3) were taken at different times after incubations (0–12 h) into siliconized microcentrifuge tubes, and processed as described below before analysis by both SEC and reversed-phase assays for quantitation of the intact conjugates and released intermediates, respectively.
Blood Hydrolysis
Blood was obtained from six anesthetized (ketamine:xylazine; 80:8 mg/kg, im), untreated rats by cardiac puncture. Approximately 4 IU of heparin was added to each mL of blood to prevent coagulation. Immediately after the collection of blood, DMP conjugates (in isotonic phosphate buffer at pH 7.4) were added to produce a blood concentration of 10 μg/mL (MP equivalent). After mixing blood with the conjugates, samples were incubated at 37°C. Preliminary studies indicated that incubation of rat blood at 37°C for longer than 2 h might be associated with a progressive decrease in pH, which could affect the degree of hydrolysis. Therefore, we limited our blood studies to 6 h and kept the pH within 7.35–7.45, by addition of small total volumes of 5–10 μl of isotonic sodium bicarbonate solution (1.5%, w/v) to the incubation media (~4 mL). Samples were then taken at 0, 1, 3, and 6 h and immediately centrifuged to separate plasma. Plasma samples were then processed as described below before analysis of the intact conjugates by the SEC method.
Hydrolysis in the Presence of Peptidases
Hydrolysis of DMP conjugates in the presence of various types of peptidases was studied at peptidase concentrations of approximately 8 μM. The tested peptidases included cysteine (papain and cathepsin B), aspartic (cathepsin D), and serine (trypsin) proteinases. The latter two peptidases were included as negative controls because, based on their substrate specificity,17,18 they are not expected to cleave peptide bonds with Gly. DMP conjugates at a concentration equivalent to 100 μg/mL MP were incubated in 50 mM acetate buffer (pH 4), 5 mM reduced glutathione, and respective peptidases at 37°C. In the case of trypsin, CaCl2 at a final concentration of 10 mM was also added. Samples were then taken at 0, 3, 6, 12, 24, and 48 h and treated as described below before injection into the reversed-phase HPLC.
Liver Lysosome Hydrolysis
Crude lysosomal fractions were prepared from the liver of untreated rats according to the procedure described in the lysosome isolation kit. Briefly, rats (n = 3) were anesthetized by an im injection of ketamine:xylazine (80:8 mg/kg), and after cannulation of the portal vein, the livers were perfused with ice-cold PBS and removed. The livers were then homogenized in 4 volumes of the extraction buffer, followed by differential centrifugation for isolating the lysosomal fraction. The protein concentrations in lysosomal preparations were determined by Bio-Rad protein assay (Bio-Rad, Herecules, CA, USA). The activity of acid phosphatase, a lysosomal marker, in the preparation was tested using a commercial kit (Sigma). The specific enzyme activity in the lysosomal fraction was >9-fold that in the liver homogenate.
For lysosomal hydrolysis studies, DMP conjugates (100 μg/mL, MP equivalent) were incubated at 37°C in 50 mM acetate buffer (pH 4.0) in the presence of 5 mM reduced glutathione and 5 mg/mL lysosomal protein. Samples (100 μL) were then taken at 0, 3, 6, 12, 24, and 48 h and treated as described below before reversed-phase HPLC analysis.
Sample Preparation
Except for blood, 100 μL of methanol and 20 μL of 10% (v/v) acetic acid were added to all the samples (100 μL) immediately after their collection to stop further hydrolysis. Preliminary studies indicated that the treatment of samples with acetic acid and methanol renders them stable for at least 24 h at room temperature. Addition of methanol and acetic acid also caused precipitation of proteins in the peptidase and lysosomal studies. After vortex mixing (5 sec) and centrifugation (10,000 rpm for 5 min), the resultant supernatants were transferred to autosampler inserts and a 50 or 150 μL aliquot was injected into the SEC or reversed-phase HPLC methods, respectively, described below. All the samples were analyzed within 10 h after collection.
Plasma samples were only analyzed by SEC method because endogenous peaks in plasma interfered with the reversed-phase assay of the intermediates. Immediately after collection, 20 μL of 10% acetic acid was added to 100 μL of plasma, and the samples were stored at −80°C until analysis within 24 h. To precipitate proteins before HPLC analysis, 80 μL of cold methanol and 20 μL of 20% (v/v) perchloric acid (70%) were added to the samples. After a brief vortex mixing and centrifugation, 150 μL of the supernatant was transferred to a new microcentrifuge and 100 μL of HPLC water and 75 μL of 0.5 M phosphate buffer (pH 7.0) were added, and a 100 μL aliquot was subjected to the SEC method described below.
Analytical Methods
The concentrations of MP, MPS, and MPS-peptidyl intermediates, including MPS-mG-OH, MPS-mGG-OH, MPS-mGGG-OH, MPS-mGGGG-OH, and MPS-mGGGGG-OH, in the samples were determined by a reversed-phase HPLC method developed and validated in our laboratory. The samples were analyzed at ambient temperature using a 25 cm × 4.6 mm C18 (5 μm) column (Partisil ODS-3, Whatman, Florham Park, NJ), preceded by a guard column packed with spherical C18 silica gel (20–45 μm). The isocratic mobile phase consisted of 0.1 M phosphate buffer (pH 4.6):acetonitrile (73:27), which was pumped at a flow rate of 1 mL/min.
The validity of the assay was investigated by determination of the accuracy and precision of the assay based on the reported guidelines.19 The inter-run validity was determined by analyzing five replicates of quality control samples at each concentration of 0.5, 5, and 100 μg/mL against the calibration standards in the range of 0.5–100 μg/mL on different days. Calibration standards were prepared in lysosomal matrix after deactivation of the enzymes at 100°C for 60 minutes. The accuracy and precision values were then calculated by percent errors and CVs, respectively. To determine the recovery of the analytes from lysosomes after protein precipitation, lysosomal samples (n = 3) containing 5 or 100 μg/mL of all the five MPS-peptidyl linkers, MP, and MPS were analyzed using the above assay. The peak areas obtained from these samples were then compared with those containing equivalent concentrations of the analytes in HPLC water.
Concentrations of the intact prodrugs were analyzed using a slightly modified size-exclusion chromatographic assay reported before for the assay of MP-succinate-dextran conjugates.20 Briefly, conjugates were separated from impurities or other interfering peaks using a 30 cm × 7.8 mm analytical, gel chromatography column (PolySep-GFC 3000; Phenomenex, Torrance, CA) at ambient temperature. The mobile phase consisted of KH2PO4 (10 mM) and acetonitrile (65:35) and was pumped at a flow rate of 1.0 mL/min.
The HPLC instrument (Waters, Milford, MA, USA) consisted of a 515 pump, a 717 autosampler, and a 997 photodiode array detector, operated in the range of 245–255 nm. The chromatographic data was managed using Empower software (Waters).
Data Analysis
The time-dependent decline in the conjugate concentration after incubation in various media was fitted using a first-order kinetic model, and the degradation half life was estimated from the slope of the fitted lines. The statistical differences among various conjugates were analyzed using ANOVA with post-hoc Scheffe’s F test. All tests were performed at a significance level (α) of 0.05. Data are presented as mean ± SD.
RESULTS
Synthesis and Characterization
MP was conjugated to dextran using peptide linkers in three major steps: (i) synthesis of 5′-O-succinate ester of the drug (MPS), (ii) the reaction of MPS with resin-bound peptides and cleavage, and (iii) the reaction of MPS-peptide conjugates with dextran (Scheme 1). MP was converted to MPS in the presence of pyridine and succinic anhydride. MPS was conjugated to dextran through the peptide linkers. In general, all peptides were assembled on Fmoc-Gly-Wang resin by the solid-phase synthesis strategy employing Fmoc-based chemistry and Fmoc-L-amino acid building blocks (i.e., Fmoc-Gly-OH, Fmoc-mGly-OH). After the Fmoc deprotection with piperidine, the free N-terminal of the peptides was conjugated with MPS to afford polymer-bound MPS-peptide conjugates. MPS-mG-OH was synthesized by anchoring Fmoc-mG-OH on Wang resin, followed by MPS. Acidic cleavage of the conjugates from Wang resin, followed by reaction with dextran in the presence of DIC and DIEA afforded MPS-mG-dextran, MPS-mGG-dextran, MPS-mGGG-dextran, MPS-mGGGG-dextran, MPS-mGGGGG-dextran, MPS-GG-dextran, and MPS-GGGG-dextran (Scheme 1). The reaction between dextran and MPS-peptide can potentially occur via any of its three hydroxyl groups present in each glucose molecule (Scheme 1). However, the exact site of substitution was not determined in this study.
The degree of substitution and purity of different conjugates used in this study are reported in Table 1. The purity, determined by SEC of the intact conjugate, was ≥90% for all the conjugates. The degrees of MP substitution were also very close for all the conjugates and ranged from 6.9% to 9.5% (w/w). For consistency, batches of conjugates that had degrees of substitution higher than 10% or lower than 6.5% were not used in this study.
Table 1.
Average ± SD values (%) of degree of substitution (DS) and purity of the MPS-peptide-dextran conjugates (n = 3).
| Conjugate | DS (%) | Purity (%) |
|---|---|---|
| MPS-mG-Dex | 9.4 ± 0.2 | 91 |
| MPS-mGG-Dex | 7.0 ± 0.2 | 91 |
| MPS-mGGG-Dex | 9.5 ± 0.9 | 95 |
| MPS-mGGGG-Dex | 7.4 ± 0.1 | 90 |
| MPS-mGGGGG-Dex | 6.9 ± 0.1 | 93 |
HPLC Analysis of Intermediates
Chromatograms of a blank lysosomal sample, a standard lysosomal sample containing 5 μg/mL of MP, MPS, MPS-mG-OH, MPS-mGG-OH, MPS-mGGG-OH, MPS-mGGGG-OH, and MPS-mGGGGG-OH, and a lysosomal sample taken 48 h after incubation (37°C) with MPS-mGGGGG-Dex (100 μg/mL of MP equivalent) are depicted in Figure 1. Under the stated chromatographic conditions all the 7 analytes of interest were separated from each other with retention times of 14, 15, 16, 18, 20, 25, and 47 min for MPS-mGGGGG-OH, MPS-mGGGG-OH, MPS-mGGG-OH, MPS-mGG-OH, MPS-mG-OH, MP, and MPS, respectively (Fig. 1).
Figure 1.
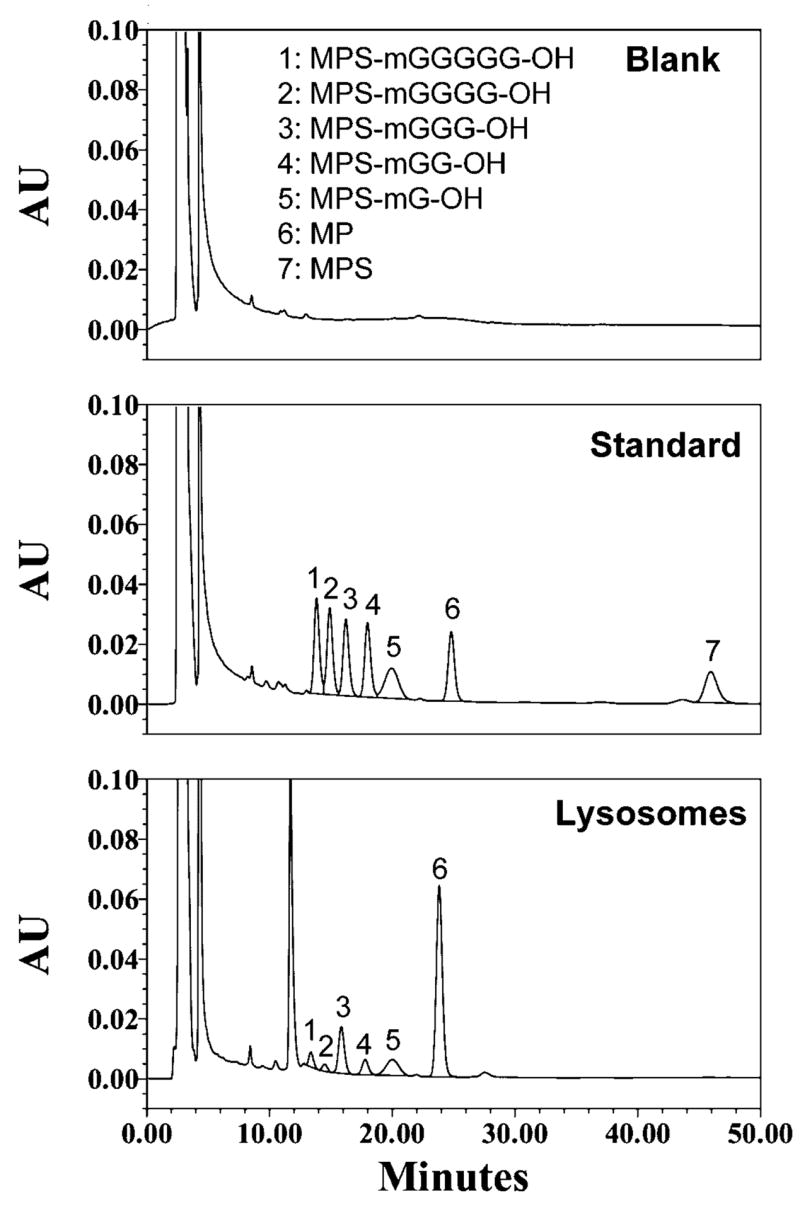
Chromatograms of a blank deactivated lysosomal sample (top), a standard deactivated lysosomal sample containing 5 μg/mL of MP, MPS, MPS-mG-OH, MPS-mGG-OH, MPS-mGGG-OH, MPS-mGGGG-OH, and MPS-mGGGGG-OH (middle), and a sample taken 48 h after incubation (37°C) of 100 μg/mL (MP equivalent) MPS-mGGGGG-Dex with active lysosomes (bottom).
Five calibration curves were used to determine the inter-run validation of the assay, the results of which are presented in Table 2. The responses of the detector to analytes were linear (r2≥0.99) over the studied range of 0.5 – 100 μg/mL for all the components. The accuracy of the assay was demonstrated by error values of <14 % for all the components at all the concentrations, including the lower limit of quantitation of 0.5 μg/mL. Additionally, the precision of the assay was demonstrated by the CV values of ≤12 %.
Table 2.
Inter-run accuracy (%Error) and precision (%CV) of the reversed-phase assay (n =5)
| Analyte | Added Conc. (μg/mL) | Calculated Conc. (μg/mL) | % Error | % CV |
|---|---|---|---|---|
| MP | 0.5 | 0.50 | 0.5 | 6.3 |
| MPS | 0.5 | 0.49 | −2.9 | 3.1 |
| MPS-mG-OH | 0.5 | 0.53 | 6.9 | 5.5 |
| MPS-mGG-OH | 0.5 | 0.48 | −3.6 | 3.5 |
| MPS-mGGG-OH | 0.5 | 0.48 | −4.7 | 8.7 |
| MPS-mGGGG-OH | 0.5 | 0.44 | −12.8 | 12.0 |
| MPS-mGGGGG-OH | 0.5 | 0.43 | −13.2 | 9.8 |
| MP | 5 | 5.06 | 1.1 | 1.2 |
| MPS | 5 | 4.82 | −3.6 | 2.4 |
| MPS-mG-OH | 5 | 4.79 | −4.2 | 1.6 |
| MPS-mGG-OH | 5 | 4.81 | −3.8 | 1.0 |
| MPS-mGGG-OH | 5 | 4.82 | −3.6 | 2.2 |
| MPS-mGGGG-OH | 5 | 4.82 | −3.6 | 0.7 |
| MPS-mGGGGG-OH | 5 | 4.87 | −2.6 | 1.3 |
| MP | 100 | 108 | 7.6 | 1.6 |
| MPS | 100 | 107 | 7.2 | 1.7 |
| MPS-mG-OH | 100 | 108 | 7.7 | 3.2 |
| MPS-mGG-OH | 100 | 104 | 3.9 | 3.1 |
| MPS-mGGG-OH | 100 | 103 | 3.2 | 3.2 |
| MPS-mGGGG-OH | 100 | 102 | 2.0 | 2.8 |
| MPS-mGGGGG-OH | 100 | 101 | 0.6 | 3.5 |
The recoveries of all the components from the lysosomal samples were almost complete with efficiencies ranging from 94 to 104% and 96 to 102% at the lowest (0.5 μg/mL) and highest (100 μg/mL) concentrations, respectively.
Hydrolysis of DMP Conjugates in Buffer
The concentrations of DMP conjugates as a function of incubation (37°C) time in pH 7.4 buffer, measured using the SEC assay, are depicted in Fig. 2, and the corresponding degradation half lives are reported in Table 3. The degradation of DMP conjugates in pH 7.4 buffer was dependent on the length of the linker (p < 0.05, ANOVA); an increase in the length of linker from one (MPS-mG-Dex) to five (MPS-mGGGGG-Dex) amino acids decreased the degradation half life from 10.7 ± 0.1 h to 4.27 ± 0.02 h (Fig. 2 and Table 3). However, except for MPS-mG-Dex, which showed a degradation half life twice as long, the half lives were close for all the other linkers (Table 3). Consistent with the half life values, the concentrations of the DMP conjugates remaining after 12 h of incubation at pH 7.4 and 37°C ranged from 15% (MPS-mGGGGG-Dex) to 46% (MPS-mG-Dex) (Fig. 2).
Figure 2.
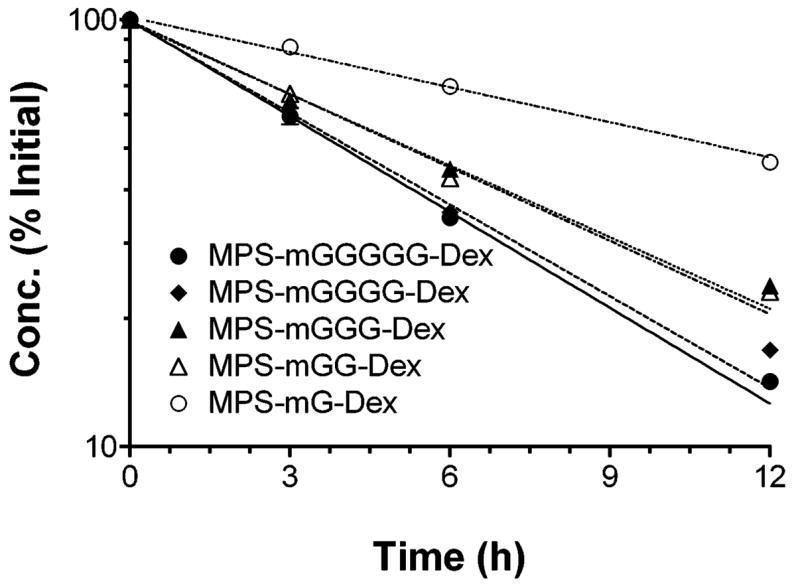
Concentration-time courses of DMP conjugates with five different linkers after incubation (37°C) in pH 7.4 buffer. Symbols and bars represent mean and standard deviations, respectively. Lines represent first-order fits to the data (n = 3).
Table 3.
Mean ± SD values of degradation half lives (h) of DMP conjugates in pH 7.4 buffer and rat blood (n = 3).
| pH 7.4 | Blood | |
|---|---|---|
| MPS-mG-Dex | 10.7 ± 0.1a,b,c,d,e | 24.5 ± 3.0e,g,h,i |
| MPS-mGG-Dex | 5.67 ± 0.03a | 8.36 ± 0.45g |
| MPS-mGGG-Dex | 5.87 ± 0.03b,f | 9.80 ± 0.25f,h,j |
| MPS-mGGGG-Dex | 4.69 ± 0.03c | 6.89 ± 0.25i |
| MPS-mGGGGG-Dex | 4.27 ± 0.02d | 4.67 ± 0.21g,j |
Means with similar letters are significantly different each other: ANOVA with post-hoc Scheffe test..
Time courses of the release of MP and its peptidyl intermediates at pH 7.4, measured using the reversed-phase assay, are demonstrated in Figure 3. For all the conjugates, the released moieties were only MP and the MPS attached to the intact peptidyl spacer, indicating chemical hydrolysis of ester bonds only (Scheme 1). The total concentration of MP released, relative to the initial conjugate concentration, during 12 h of incubation at pH 7.4 was substantial, ranging from 38% for MPS-mG-Dex to 66% for MPS-mGGGG-Dex (Fig. 3). Although the concentrations of total MP (MP plus MPS-peptidyl derivatives) varied with the length of the linker, those of the free MP seemed not much different among different conjugates (Fig. 3).
Figure 3.
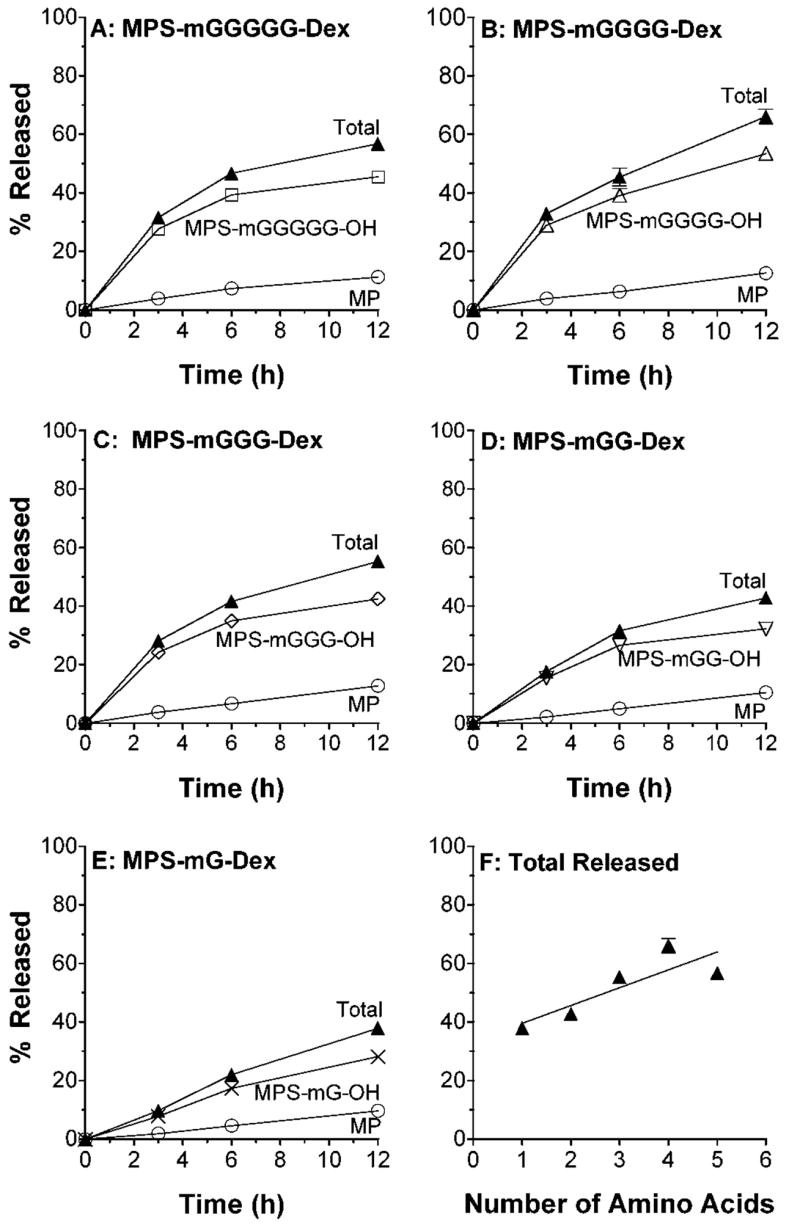
A–E: In vitro release profiles of MP and MPS-peptidyl intermediates after incubation (37°C) of DMP conjugates with five different linkers in pH 7.4 buffer. F: Total MP equivalent released at 12 h versus the number of amino acids in the linker. Symbols and bars represent mean and standard deviations, respectively (n = 3).
In contrast to the relatively significant degradation of DMP conjugates at pH 7.4 (Fig. 3), no discernable decline in the concentrations of the conjugates was observed in the SEC assay after incubation at pH 4.0 (data not shown). Additionally, only minor amounts (≤3%) of the released products of DMP were seen at pH 4.0 using the reversed-phase assay. However, in agreement with the release patterns at pH 7.4 (Fig. 3), only MP and MPS attached to the intact linker were observed for all the conjugates.
Hydrolysis of DMP in Blood
The results of degradation of DMP conjugates in blood, based on measurements of the parent conjugates, are presented in Fig. 4 and Table 3. The degradation of the conjugates in blood was dependent on the length of the peptide linker, with MPS-mG-Dex and MPS-mGGGGG-Dex exhibiting the slowest and fastest degradation, respectively (Fig. 4 and Table 3). However, the differences in the degradation half lives among the conjugates with 2–4 amino acids were not significant (Fig. 4 and Table 3). The conjugate concentrations remaining at 6 h and corresponding half lives ranged from 38 to 81% (Fig. 4) and 4.67 to 24.5 h (Table 3), respectively. The degradation half lives of MPS-mG-Dex and MPS-mGGG-Dex in blood were significantly longer than their corresponding values at pH 7.4 buffer (Table 3).
Figure 4.

Concentration-time courses of DMP conjugates with five different linkers after incubation (37°C) in rat blood. Symbols and bars represent mean and standard deviations, respectively. Lines represent first-order fits to the data (n = 3).
Hydrolysis of DMP by Peptidases
Papain
Papain-mediated release of MP and MPS-peptidyl intermediates is shown in Fig. 5. Except for MPS-mG-OH, all the other possible MPS-peptidyl intermediates were observed in relatively high concentrations for all the conjugates (Fig. 5). The total MP released from the conjugates during 48 h of incubation with papain ranged from 31% for MPS-mG-Dex to 71% for MPS-mGGGGG-Dex (Figure 5). Overall, the % total MP release appeared to be dependent on the length of the peptide linker, whereas the free MP release appeared to be similar for all the conjugates (Fig. 5).
Figure 5.
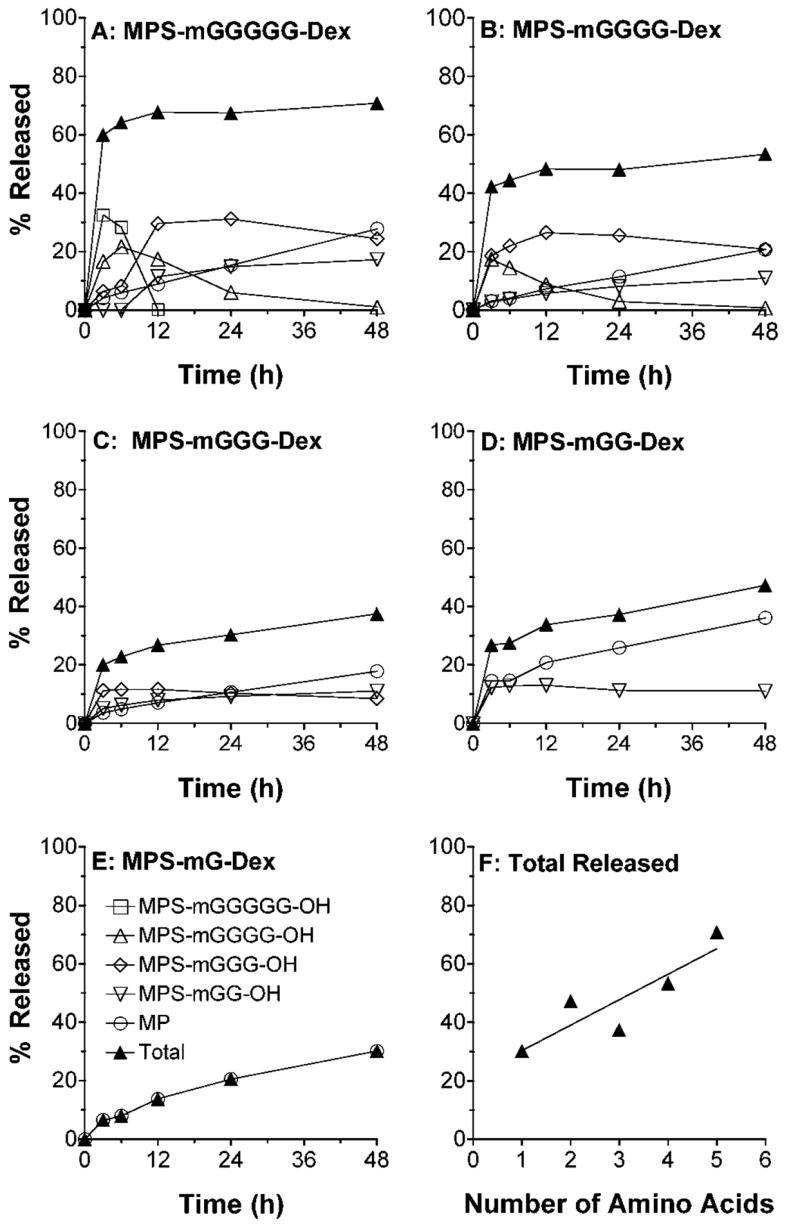
A–E: In vitro release profiles of MP and MPS-peptidyl intermediates after incubation (37°C) of DMP conjugates with five different linkers in the presence of papain. F: Total MP equivalent released at 48 h versus the number of amino acids in the linker. Symbols and bars represent mean and standard deviations, respectively (n = 3).
Cathepsin-B
Cathepsin-B-mediated degradation of the dextran conjugates of MP is demonstrated in Fig. 6. With the exception of appearance of cathepsin-B-mediated MPS-mG-OH intermediate for most conjugates, the qualitative pattern of conjugate hydrolysis by cathepsin-B (Fig. 6) was similar to that observed for papain (Fig. 5). However, the % total MP release (5–17%, Fig. 6f) by cathepsin B was much lower than that seen by papain incubation (Fig. 5f). A decrease in the length of the linker caused a progressive decrease in the cathepsin-B-mediated MP and MPS-peptidyl intermediate release (Fig. 6f).
Figure 6.
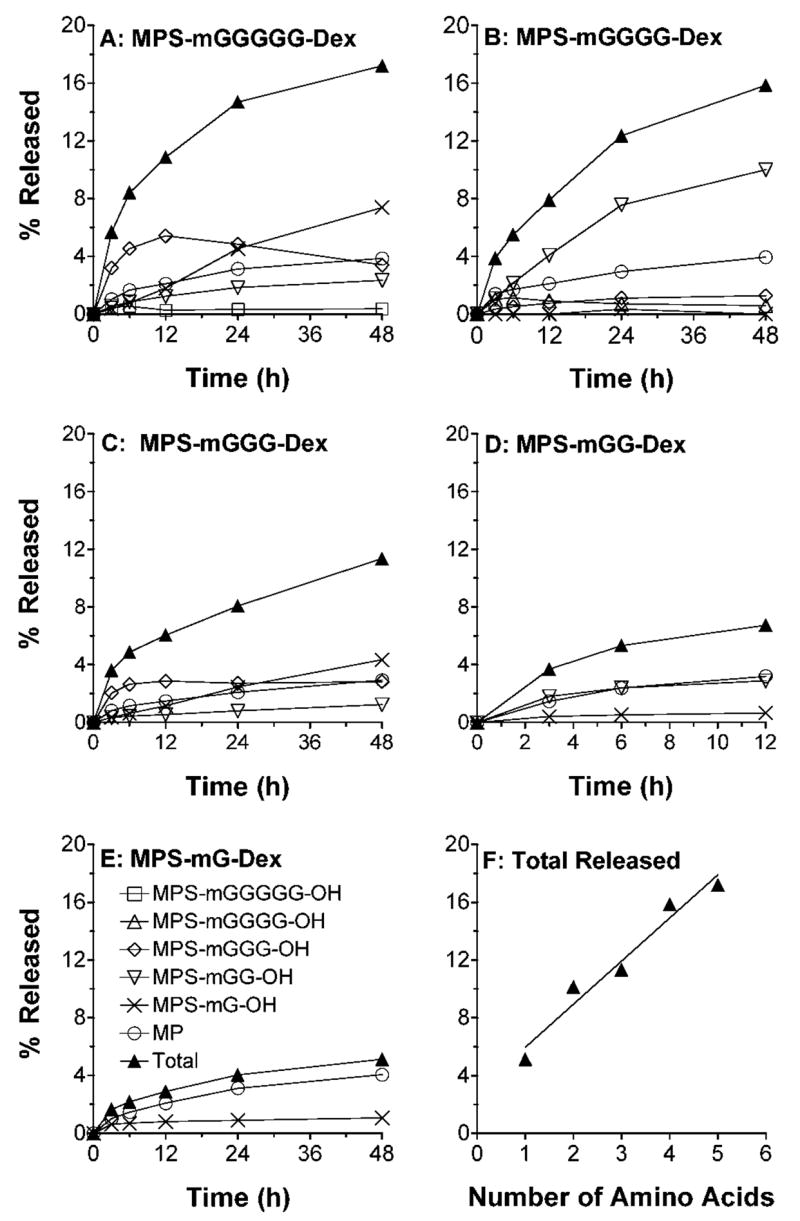
A–E: In vitro release profiles of MP and MPS-peptidyl intermediates after incubation (37°C) of DMP conjugates with five different linkers in the presence of cathepsin-B. F: Total MP equivalent released at 48 h versus the number of amino acids in the linker. Symbols and bars represent mean and standard deviations, respectively (n = 3).
Cathepsin-D and Trypsin
To understand the enzyme specificity for the degradation of the conjugate, we included cathepsin D, as an aspartyl proteinase, and trypsin, as a serine proteinase, in our studies as negative controls. As expected, the rate of release of free MP and MPS-peptidyl intermediates was very slow and negligible with both enzymes (data not shown).
Hydrolysis of DMP in Liver Lysosomes
The hydrolysis patterns of conjugates in the presence of lysosomal fraction are demonstrated in Fig. 7. Lysosomal degradation caused release of free MP and all the possible MPS-peptidyl intermediates, including relatively high concentrations of MPS-mG-OH. However, for most conjugates, MP was by far the most abundant released moiety (Fig. 7). The total amount of MP and MPS-peptidyl intermediates released in the presence of lysosomes ranged from 3% (MPS-mG-Dex) to 11% (MPS-mGGGGG-Dex) (Fig. 7f). An increase in the peptide link caused an increase in the release of MP and/or its peptidyl intermediates, although hydrolysis of MPS-mGG-Dex deviated from this pattern (Fig. 7f).
Figure 7.
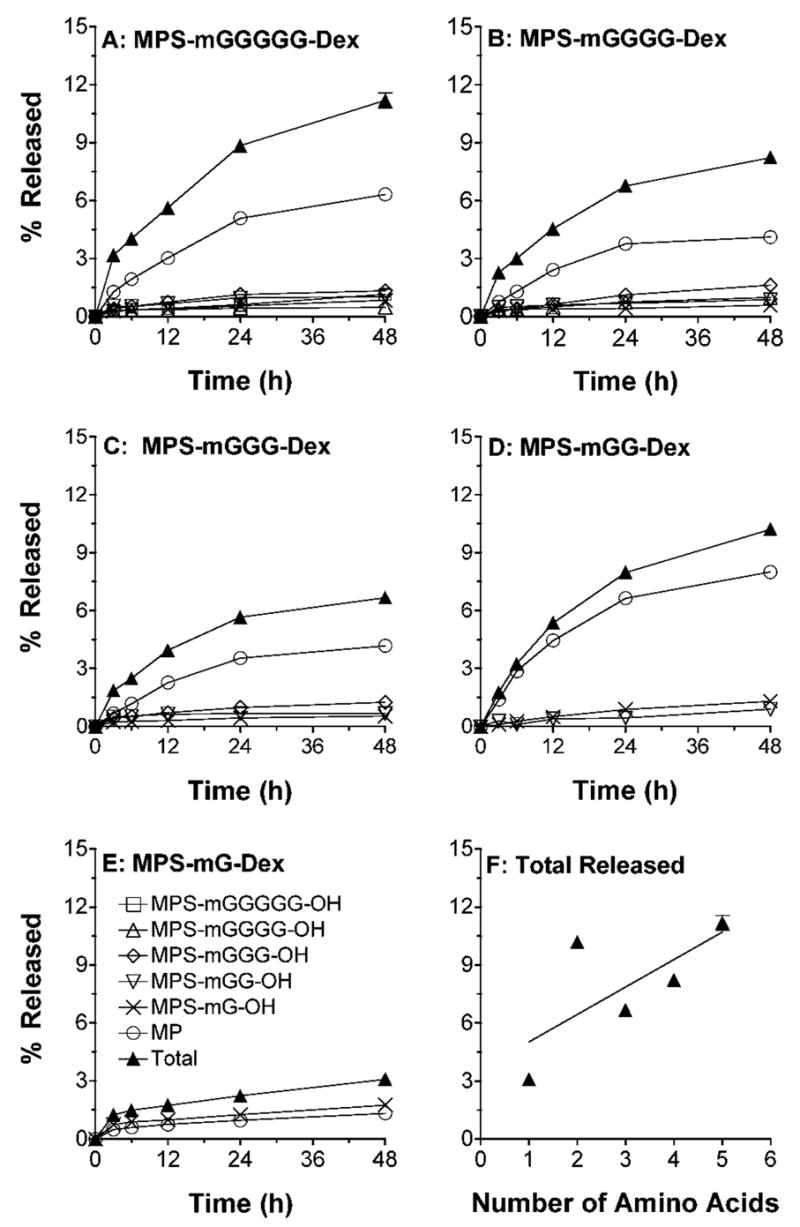
A–E: In vitro release profiles of MP and MPS-peptidyl intermediates after incubation (37°C) of DMP conjugates with five different linkers in the presence of rat lysosomal fraction. F: Total MP equivalent released at 48 h versus the number of amino acids in the linker. Symbols and bars represent mean and standard deviations, respectively (n = 3).
DISCUSSION
Dextran macromolecular prodrugs are usually synthesized by covalent attachment of drugs, directly or through a spacer arm, to the dextran polymer.7,8 The drug is then released in vivo by hydrolytic and/or enzymatic cleavage of the linking bond. Therefore, the rate of the drug release is dependent on the nature of the linking bond and the type and length of the spacer group. Our previous studies on a 70 kDa dextran prodrug of MP with a succinate linker, although promising, demonstrated some shortcomings. First, the conjugate showed a slow and incomplete release of MP both in vitro9 and in vivo,11,12 reducing the potential duration and intensity of action of the conjugate. Additionally, in vivo pharmacokinetic studies indicated some degree of nonlinearity in the elimination of the conjugate, most likely due to the large Mw of the dextran carrier.21 This study was therefore undertaken to synthesize new dextran conjugates of MP with necessary modifications that would be devoid of the above shortcomings.
Our previous works on dextran carriers have shown that although liver accumulation of dextran 70 kDa is modestly higher than that of dextran 20 kDa, the lower Mw dextran is expected to follow linear pharmacokinetics because of more involvement of a linear renal clearance in its elimination.22,23 Additionally, peptide linkers are expected to be superior to the succinate linker because the rate of release of drugs from macromolecular prodrugs with such linkers may be controlled by the length, type, and sequence of amino acids.15,16 Therefore, modifications incorporated in the synthesis of the new conjugates included the use of dextran ~25 kDa, instead of 70 kDa, as a carrier and peptides of various lengths, instead of succinic acid, as linkers between MP and dextran.
Our initial attempts at synthesis of MPS-peptide-dextran conjugates using Gly-Gly or Gly-Gly-Gly-Gly as linkers resulted in conjugates with very low (<1%) degrees of MP substitution, which were very unstable in aqueous solutions (data not shown). Changes in the synthetic procedures such as increasing the length of reactions and/or changing the reagents or activation methods did not improve these shortcomings. The rapid hydrolysis of these conjugates is consistent with a recent report24 in which authors demonstrated cyclization of a prednisolone succinate β-cyclodextrin amide conjugate, resulting in a rapid release of prednisolone. Although the release of prednisolone from prednisolone-21-hemisuccinate was very slow with a half life of 69 h at pH 7.0 and 37°C, the hydrolysis of prednisolone 21-hemisuccinate-β-cyclodextrin amide conjugate, even at a lower temperature of 25°C, was very fast with a half life of 6.5 min in the same buffer. The authors attributed this behavior to an intramolecular nucleophilic catalysis of the amide group, which occurs in the alkaline region. The same intramolecular rearrangement could theoretically be responsible for the fast release of MP from our initial conjugates with Gly-Gly or Gly-Gly-Gly-Gly linkers, resulting in a low degree of substitution. If true, incorporation of N-methyl glycine, instead of glycine, at the N-terminal of peptide linker is expected to prevent this undesired cyclization. Indeed, when we used this latter approach (Scheme 1), we were able to obtain conjugates with relatively high degree of MP substitution (Table 3) with improved stability in aqueous solutions (Fig. 2).
Simultaneous quantitation of hydrolyzed intermediates of MP, including five MPS-peptidyl intermediates, and MP and MPS required for stability and release studies was challenging. Previously, we reported a method for the simultaneous measurement of MP and MPS,25 which could not be adapted to separate all seven analytes of interest in the current study. Because of the succinyl moiety, the retention times of MPS and all the MPS-peptidyl intermediates are sensitive to both the organic modifier and pH of the mobile phase, whereas the retention time of MP is pH-independent. We took advantage of these characteristics to resolve all seven analytes of interest within a run time of <50 min (Fig. 1).
The hydrolysis of DMP conjugates in pH 7.4 buffer at 37°C to release only MP and intact MPS-peptide linker (Fig. 3) indicates chemical hydrolysis of the two ester bonds in the structure of the conjugates (Scheme 1). Whereas hydrolysis of the ester bond between dextran hydroxyl groups and the C-terminal of the linker amino acid would result in the release of intact MPS-peptide linker, hydrolysis of the ester bond between MP and succinic acid would yield free MP. Free MP may also be released indirectly from the regenerated MPS-peptide linker. The linker length-dependent degradation half life of conjugates at pH 7.4 (Fig. 2, Table 3) suggests that the ester bonds in the conjugates with longer linkers are more prone to hydrolysis, probably due to the electronic effect of the linker. Longer linkers have more electron withdrawing groups because of the presence of multiple amide bonds. Therefore, it is possible that the ester moieties of the conjugates with longer linkers become more polarized with the presence of partially positive and negative charges on the carbon and oxygen, respectively, rendering them more susceptible to nucleophilic attack by water.
It is commonly assumed that the degradation of prodrugs may be faster in blood, compared with pH 7.4 buffers, due to the presence of enzymes. However, in our case, we did not make such an assumption because of two reasons. First, proteases responsible for degradation of similar peptide linkers of other prodrugs are shown to be present in tissue lysosomes with an optimal pH of ~4.16 Furthermore, previous works on dextran-methylprednisolone succinate conjugates9,10 showed that esterases in blood could not release MP from its dextran conjugate. Therefore, we anticipated similar degradation kinetics in the pH 7.4 buffer and blood for our newly-synthesized conjugates. Although the degradation half life values of the conjugates with mGG, mGGGG, and mGGGGG were similar in the pH 7.4 buffer and blood, those of the conjugates with mG and mGGG linkers showed longer half lives in blood (Table 3). The mechanisms responsible for the apparent increased stability of MPS-mG-Dex and MPS-mGGG-Dex in blood are not known, although minor differences in the actual pH of the two media during the experiments may have been a contributing factor. Nevertheless, similar to the degradation data in the pH 7.4 buffer, the degradation of the conjugates in blood was affected by the length of the linker (Fig. 4). Overall, hydrolysis kinetics of most of the studied DMP conjugates in blood (Table 3) are slow enough to allow substantial hepatic accumulation of the intact conjugates after their in vivo administration.
It is generally believed that dextran conjugates enter cells via endocytosis, ultimately arriving in the lysosomal compartment of the cell.26,27 Therefore, it is necessary to study the fate of dextran conjugates in the presence of lysosomal enzymes. In vitro incubation of the conjugates with a crude lysosomal fraction obtained from rats resulted in degradation of the conjugate, releasing all the possible intermediates (Fig. 7). The dependency of the release rate on the length of the peptide, observed in our studies (Fig. 7f), is in agreement with previous studies14–16,28 using peptides as linkers for conjugation of macromolecular carriers to drugs, such as anticancer agents. For example, Harada et al.16 attached T-0128, a camptothecin analogue, to carboxymethyl-dextran using peptides with various peptide linkers. In agreement with the results presented here (Fig. 7), these authors showed that both in vitro release in the presence of lysosomal fractions and in vivo release in tumors and livers of tumor-bearing rats increased with lengthening the Gly chain. Therefore, using the conjugates developed in our studies containing different lengths of Gly-chain linker, the rate of release of MP in vivo may be controlled and an optimum linker selected in future in vivo studies.
To determine the mechanism(s) of hydrolysis of peptidic bonds in lysosomes, further studies were carried out using individual peptidases. In addition to lysosomal proteinases (cathepsin B, cathepsin D, and trypsin), we also included papain in our studies because a number of lysosomal cathepsins in humans and animals are cysteine peptidases that belong to the papain family.29,30 Relatively significant hydrolysis of the peptide linkers by papain (Fig. 5) and cathepsin B (Fig. 6) and lack of hydrolysis by cathepsin D or trypsin (data not shown) strongly suggest enzymatic hydrolysis of the linkers by cysteine (papain and cathepsin B) proteinases. This is in agreement with the report of Harada et al.,16 who demonstrated a significant role for cathepsin B in the release of a camptothecin analogue from a conjugate with Gly-Gly-Gly linker.
Our previous study31 clearly showed that MP attached to dextran macromolecule via a succinate linker is not effective by itself and has to release MP in order to be effective. In our current studies with new conjugates containing peptide linkers, hydrolysis of the conjugates in different media resulted in the generation of both free MP and its peptidyl derivatives (Figs. 3, 5, 6, and 7). Whether peptidyl derivatives are effective by themselves or need to release MP to be effective has not been studied. However, considering the interaction of most ligands with their receptors, it is unlikely that the intact MPS-peptidyl derivatives can react with the cytosolic glucocorticoid receptors. Nevertheless, additional investigations are needed to study the effectiveness, if any, of MPS-peptidyl derivatives.
In conclusion, dextran prodrugs of methylprednisolone with peptide linkers containing one to five Gly moieties were synthesized. A reversed-phase HPLC method was developed and validated to quantitate all seven possible hydrolysis products of the conjugates. The in vitro hydrolytic patterns of DMP conjugates to MP and MPS-peptidyl intermediates were determined in buffers, blood, liver lysosomes, and in the presence of various peptidases. The conjugates were relatively stable in rat blood. However, they generated all the possible intermediates in the presence of liver lysosomes, with the rate of release increasing with an increase in the length of the peptide linker. Among studied lysosomal enzymes, cysteine peptidases, such as cathepsin B, appear to play a major role in the hydrolysis of these conjugates in the lysosomes. The results of this study indicate that the dextran-peptide-methylprednisolone conjugates may be of interest for therapeutic lysomotropic delivery of MP. Future studies are underway to investigate the in vivo behavior of these conjugates.
Supplementary Material
Supporting Information Available. 1H NMR, 13C NMR, and HR-MS (ESI-TOF) (m/z) for MPS-mG-OH, MPS-mGG-OH, MPS-mGGG-OH, MPS-mGGGG-OH, MPS-GG-OH, and MPS-GGGG-OH. This material is available free of charge via the Internet at http://www.interscience.wiley.com.
Acknowledgments
This study was supported by a grant from the National Institute of General Medical Sciences of NIH (R01 GM069869). The authors would like to thank Chaitanya Chimalakonda and Nguyen-Hai Nam for conducting some preliminary experiments.
References
- 1.Czock D, Keller F, Rasche FM, Haussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44(1):61–98. doi: 10.2165/00003088-200544010-00003. [DOI] [PubMed] [Google Scholar]
- 2.Volpin R, Angeli P, Galioto A, Fasolato S, Neri D, Barbazza F, Merenda R, Del Piccolo F, Strazzabosco M, Casagrande F, Feltracco P, Sticca A, Merkel C, Gerunda G, Gatta A. Comparison between two high-dose methylprednisolone schedules in the treatment of acute hepatic cellular rejection in liver transplant recipients: a controlled clinical trial. Liver Transpl. 2002;8(6):527–534. doi: 10.1053/jlts.2002.33456. [DOI] [PubMed] [Google Scholar]
- 3.Encke J, Uhl W, Stremmel W, Sauer P. Immunosuppression and modulation in liver transplantation. Nephrol Dial Transplant. 2004;19(Suppl 4):IV22–IV25. doi: 10.1093/ndt/gfh1037. [DOI] [PubMed] [Google Scholar]
- 4.Guillen EL, Ruiz AM, Bugallo JB. Hypotension, bradycardia, and asystole after high-dose intravenous methylprednisolone in a monitored patient. Am J Kidney Dis. 1998;32(2):E4. doi: 10.1053/ajkd.1998.v32.pm10074612. [DOI] [PubMed] [Google Scholar]
- 5.Mehvar R. Recent trends in the use of polysaccharides for improved delivery of therapeutic agents: pharmacokinetic and pharmacodynamic perspectives. Curr Pharm Biotechnol. 2003;4(5):283–302. doi: 10.2174/1389201033489685. [DOI] [PubMed] [Google Scholar]
- 6.Takakura Y, Hashida M. Macromolecular carrier systems for targeted drug delivery: pharmacokinetic considerations on biodistribution. Pharm Res. 1996;13(6):820–831. doi: 10.1023/a:1016084508097. [DOI] [PubMed] [Google Scholar]
- 7.Larsen C. Dextran prodrugs-structure and stability in relation to therapeutic activity. Adv Drug Deliv Rev. 1989;3:103–154. [Google Scholar]
- 8.Mehvar R. Dextrans for targeted and sustained delivery of therapeutic and imaging agents. J Control Release. 2000;69(1):1–25. doi: 10.1016/s0168-3659(00)00302-3. [DOI] [PubMed] [Google Scholar]
- 9.Mehvar R, Dann RO, Hoganson DA. Kinetics of hydrolysis of dextran-methylprednisolone succinate, a macromolecular prodrug of methylprednisolone, in rat blood and liver lysosomes. J Control Release. 2000;68:53–61. doi: 10.1016/s0168-3659(00)00234-0. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X, Mehvar R. Dextran-methylprednisolone succinate as a prodrug of methylprednisolone: plasma and tissue disposition. J Pharm Sci. 2001;90(12):2078–2087. doi: 10.1002/jps.1158. [DOI] [PubMed] [Google Scholar]
- 11.Mehvar R, Hoganson DA. Dextran-methylprednisolone succinate as a prodrug of methylprednisolone: immunosuppressive effects after in vivo administration to rats. Pharm Res. 2000;17(11):1402–1407. doi: 10.1023/a:1007555107691. [DOI] [PubMed] [Google Scholar]
- 12.Chimalakonda AP, Mehvar R. Dextran-methylprednisolone succinate as a prodrug of methylprednisolone: local immunosuppressive effects in liver after systemic administration to rats. Pharm Res. 2003;20(2):198–204. doi: 10.1023/a:1022358702643. [DOI] [PubMed] [Google Scholar]
- 13.Chimalakonda AP, Montgomery DL, Weidanz JA, Shaik IH, Nguyen JH, Lemasters JJ, Kobayashi E, Mehvar R. Attenuation of acute rejection in a rat liver transplantation model by a liver-targeted dextran prodrug of methylprednisolone. Transplantation. 2006;81(5):678–685. doi: 10.1097/01.tp.0000177654.48112.b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subr V, Strohalm J, Ulbrich K, Duncan R, Hume IC. Polymers containing enzymatically degradable bonds, XII. Effect of spacer structure on the rate of release of daunomycin and adriamaycin from poly [N-(2-hydroxypropyl)-methylacrylamide] copolymer drug carriers in vitro and antitumor activity measured in vivo. J Control Release. 1992;18:123–132. [Google Scholar]
- 15.Soyez H, Schacht E, Vanderkerken S. The crucial role of spacer groups in macromolecular prodrug design. Advan Drug Delivery Rev. 1996;21(2):81–106. [Google Scholar]
- 16.Harada M, Sakakibara H, Yano T, Suzuki T, Okuno S. Determinants for the drug release from T-0128, camptothecin analogue-carboxymethyl dextran conjugate. J Control Release. 2000;69(3):399–412. doi: 10.1016/s0168-3659(00)00321-7. [DOI] [PubMed] [Google Scholar]
- 17.Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol Cell Proteomics. 2004;3(6):608–614. doi: 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Scarborough PE, Guruprasad K, Topham C, Richo GR, Conner GE, Blundell TL, Dunn BM. Exploration of subsite binding specificity of human cathepsin D through kinetics and rule-based molecular modeling. Protein Sci. 1993;2(2):264–276. doi: 10.1002/pro.5560020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah VP, Midha KK, Dighe S, McGilveray IJ, Skelly JP, Yacobi A, Layoff T, Viswanathan CT, Cook CE, McDowall RD, Pittman KA, Spector S. Analytical methods validation: bioavailability, bioequivalence, and pharmacokinetics. J Pharm Sci. 1992;81(3):309–312. doi: 10.1007/BF03189968. [DOI] [PubMed] [Google Scholar]
- 20.Mehvar R. High-performance size-exclusion chromatographic analysis of dextran-methylprednisolone hemisuccinate in rat plasma. J Chromatogr B. 2000;744(2):293–298. doi: 10.1016/s0378-4347(00)00256-5. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Mehvar R. Dextran-methylprednisolone succinate as a prodrug of methylprednisolone: dose-dependent pharmacokinetics in rats. Int J Pharm. 2001;229(1–2):173–182. doi: 10.1016/s0378-5173(01)00854-7. [DOI] [PubMed] [Google Scholar]
- 22.Mehvar R, Robinson MA, Reynolds JM. Molecular weight dependent tissue accumulation of dextrans: in vivo studies in rats. J Pharm Sci. 1994;83(10):1495–1499. doi: 10.1002/jps.2600831024. [DOI] [PubMed] [Google Scholar]
- 23.Mehvar R, Robinson MA, Reynolds JM. Dose dependency of the kinetics of dextrans in rats: effects of molecular weight. J Pharm Sci. 1995;84(7):815–818. doi: 10.1002/jps.2600840706. [DOI] [PubMed] [Google Scholar]
- 24.Yano H, Hirayama F, Arima H, Uekama K. Hydrolysis behavior of prednisolone 21-hemisuccinate/beta-cyclodextrin amide conjugate: involvement of intramolecular catalysis of amide group in drug release. Chem Pharm Bull. 2000;48(8):1125–1128. doi: 10.1248/cpb.48.1125. [DOI] [PubMed] [Google Scholar]
- 25.Mehvar R, Dann RO, Hoganson DA. Simultaneous analysis of methylprednisolone, methylprednisolone succinate, and endogenous corticosterone in rat plasma. J Pharmaceut Biomed Anal. 2000;22(6):1015–1022. doi: 10.1016/s0731-7085(00)00253-3. [DOI] [PubMed] [Google Scholar]
- 26.Lake JR, Licko V, Van Dyke RW, Scharschmidt BF. Biliary secretion of fluid-phase markers by the isolated perfused rat liver. Role of transcellular vesicular transport. J Clin Invest. 1985;76(2):676–684. doi: 10.1172/JCI112021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stock RJ, Cilento EV, McCuskey RS. A quantitative study of fluorescein isothiocyanate-dextran transport in the microcirculation of the isolated perfused rat liver. Hepatology. 1989;9(1):75–82. doi: 10.1002/hep.1840090112. [DOI] [PubMed] [Google Scholar]
- 28.Kopecek J. Controlled biodegradability of polymers--a key to drug delivery systems. Biomaterials. 1984;5(1):19–25. doi: 10.1016/0142-9612(84)90062-0. [DOI] [PubMed] [Google Scholar]
- 29.Nagler DK, Tam W, Storer AC, Krupa JC, Mort JS, Menard R. Interdependency of sequence and positional specificities for cysteine proteases of the papain family. Biochemistry. 1999;38(15):4868–4874. doi: 10.1021/bi982632s. [DOI] [PubMed] [Google Scholar]
- 30.Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta. 2000;1477(1–2):98–111. doi: 10.1016/s0167-4838(99)00263-0. [DOI] [PubMed] [Google Scholar]
- 31.Rensberger KL, Hoganson DA, Mehvar R. Dextran-methylprednisolone succinate as a prodrug of methylprednisolone: in vitro immunosuppressive effects on rat blood and spleen lymphocytes. Int J Pharm. 2000;207(1–2):71–76. doi: 10.1016/s0378-5173(00)00544-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available. 1H NMR, 13C NMR, and HR-MS (ESI-TOF) (m/z) for MPS-mG-OH, MPS-mGG-OH, MPS-mGGG-OH, MPS-mGGGG-OH, MPS-GG-OH, and MPS-GGGG-OH. This material is available free of charge via the Internet at http://www.interscience.wiley.com.