

Abstract
The excited-state dynamics of the RNA homopolymer of cytosine and of the 18-mer (dC)18 were studied by steady-state and time-resolved absorption and emission spectroscopy. At pH 6.8, excitation of poly(rC) by a femtosecond UV pump pulse produces excited states that decay up to one order of magnitude more slowly than the excited states formed in the mononucleotide cytidine 5’-monophosphate under the same conditions. Even slower relaxation is observed for the hemiprotonated, self-associated form of poly(rC), which is stable at acidic pH. Transient absorption and time-resolved fluorescence signals for (dC)18 at pH 6.8 are similar to ones observed for poly(rC) near pH 4, indicating that hemiprotonated structures are found in DNA C tracts at neutral pH. In both systems, there is evidence for two kinds of emitting states with lifetimes of ~100 ps and slightly more than 1 ns. The former states are responsible for the bulk of emission from the hemiprotonated structures. Evidence suggests that slow electronic relaxation in these self-complexes is the result of vertical base stacking. The similar signals from RNA and DNA C tracts suggest a common base-stacked structure, which may be identical with that of i-motif DNA.
1. Introduction
Excited electronic states of nucleic acids have been studied intensely for decades because of the role they play in genetic damage to organisms exposed to UV light [1,2]. In recent years, ultrafast laser spectroscopy has provided many new insights into excited states in nucleic acids [3–10]. Experiments on single bases are easier to interpret than those on multiple base systems (“base multimers”), and are more amenable to quantum chemical modeling [11–15]. The combination of experiment and theory has provided considerable insight into ultrafast internal conversion, the major excited state decay pathway for single bases.
There is growing evidence that both the nature and dynamics of excited electronic states in base multimers differ radically from those in single bases [6,7,9,16]. For many years, emission signals with lifetimes of up to several nanoseconds have been reported for DNA multimers in room-temperature solution [17–19]. More recently, excited states with lifetimes of ≥100 ps have been observed in femtosecond transient absorption experiments on polynucleotides [20] and in AT-containing oligonucleotides [7]. We have suggested that the long-lived excited states, which represent a major decay pathway, are localized on two stacked bases and have a significant degree of charge transfer character [7]. Markovitsi et al. [21] and Buchvarov et al. [9] have proposed an alternative assignment of the long-lived states to delocalized Frenkel excitons. Other researchers have emphasized the possibility of hydrogen atom or proton transfer within base pairs as a potential relaxation pathway for excess electronic energy [22–24]. Further experimental work on base multimers is urgently needed to better understand the effects of electronic coupling between proximal bases.
In this report, we present results from femtosecond transient absorption and time-correlated single-photon counting experiments on oligo- and polynucleotides containing only cytosine (“C tracts”). A preliminary account of some results was given elsewhere [16]. Tracts of repeated cytosines spontaneously self-associate into higher-order structures as a function of pH. The basic unit of these structures is the base pair formed between cytosine and a second cytosine residue protonated at N3 (CH+·C, Scheme 1).
Scheme 1.
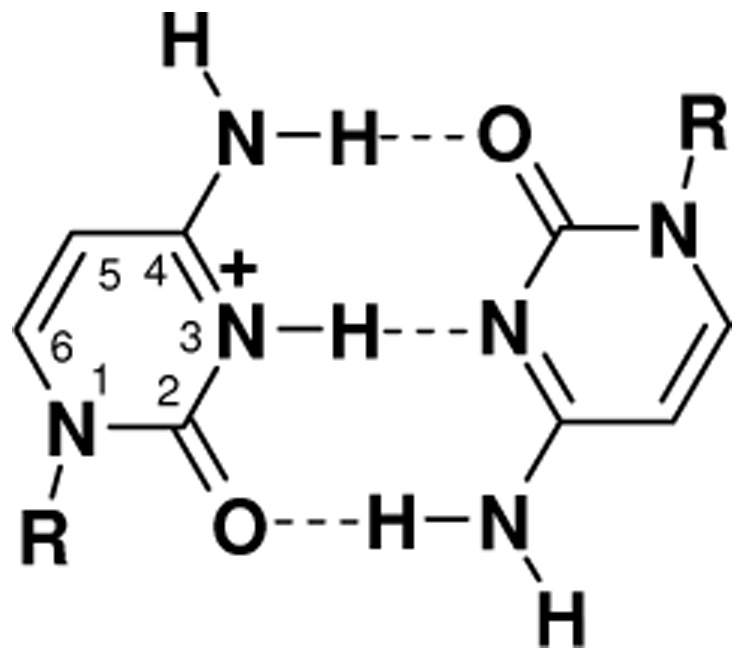
Hemiprotonated cytosine-cytosine base pair. R denotes ribose or 2’-deoxyribose.
Base stacking and base pairing are readily tuned in C tracts by altering pH. It was proposed more than forty years ago that poly(rC) can adopt a double-stranded conformation with parallel strands when the pH is between 4 and 6 [25,26]. More recently, structural NMR studies have shown that cytosine-rich DNAs form an unusual quadruplex structure (“i-motif DNA”) consisting of two intercalated duplexes made up of hemiprotonated base pairs [27,28]. This structure, which may be biologically significant [29–31], is formed when two parallel-stranded duplexes associate in head-to-tail fashion, forming intercalated hemiprotonated base pairs.
Current uncertainties about the structure of RNA and DNA C tracts form the backdrop for this study of their photophysical properties. It was shown earlier that the double-stranded form of poly(rC) is significantly more fluorescent than either the C monomer or the single-stranded form of poly(rC) [32,33], but the underlying reasons have never been elucidated. In this contribution, we use current models of C tract structures to discuss, at least preliminarily, how base pairing and base stacking affect the excited-state properties we observe in these multichromophoric molecules.
2. Experimental Methods
Cytidine 5’-monophosphate (CMP) and polycytidylic acid (poly(rC)) were purchased from Sigma Chemical Co. (St. Louis, MO, USA) and used as received. The oligodeoxynucleotide (dC)18 was purchased from Midland Certified Reagent Company (Midland, TX, USA) as a lyophilized powder.
Samples were prepared in phosphate buffer (pH = 6.8 ±0.1) with 0.1 M added NaCl. The buffer was prepared by dissolving 1.77 g of Na2HPO4 and 1.70 g of KH2PO4 in 500 mL of ultrapurified water from a commercial water purification system. Solutions were prepared at room temperature and were not thermally annealed. The pH was adjusted by drop-wise addition of dilute H3PO4. Steady-state measurements were carried out at room temperature (T = 22 ±1°C) using a Lambda 25 double-beam spectrophotometer from PerkinElmer (Wellesley, MA, USA). Circular dichroism (CD) spectra were recorded on an Aviv model 202 circular dichroism spectropolarimeter (Aviv Instruments, Proterion Corporation, Pistacaway, NJ).
Solution concentrations for transient absorption measurements were adjusted to achieve an absorbance between 1 and 2 OD in a 1 mm path length at the pump wavelength of 263 nm, corresponding to concentrations of 1 – 3 mM per nucleotide. Transient absorption signals were recorded by the pump-probe technique using amplified titanium-sapphire laser spectrometers that have been described elsewhere [3,7]. Briefly, pump pulses with a center wavelength of ~263 nm were obtained from the third harmonic laser output. Probe pulses at visible wavelengths were derived from a white light continuum generated in a 1 cm path length cell filled with water. UV probe pulses at 280 nm were obtained from an optical parametric amplifier. The instrument response function for the UV pump / UV probe measurements was ~300 fs. The optical delay line used for the longest delay measurements (up to 2.5 ns) was aligned carefully to avoid pump-probe walkoff. Signals measured months apart gave reasonably consistent long-time dynamics, and we estimate that the signal decays at times greater than 1 ns are accurate to within 10%.
Some transient absorption signals were analyzed by fitting to a sum of exponentials convoluted with a Gaussian function of full-width half maximum of 200 fs. This value was chosen to model the finite response time of the apparatus based on difference-frequency mixing signals between the third harmonic pump pulse and the fundamental in the probe arm. Transient absorption signals at visible probe wavelengths were corrected for the two-photon ionization of the solvent by the pump pulse as previously described [20]. Signals corrected in this manner are denoted by ΔAs in the figures.
Time-resolved fluorescence measurements were performed using the time-correlated single photon counting technique. The experimental set up is fully described elsewhere [34]. A portion of the output of a synchronously-pumped cavity-dumped dye laser was routed to a fast photodiode to establish the time position of each pulse. The remainder of the pulse train was frequency doubled in an appropriate nonlinear crystal to generate UV excitation pulses. Fluorescence was collected at 90° and directed through a polarizer oriented at 54.7° relative to the polarization of the laser excitation, a depolarizer, and a subtractive double monochromator. Samples were held in a 1 cm path length cuvette and stirred continuously during measurements. The instrument response time was approximately 40 ps.
3. Results
3.1. Steady-State Spectra
UV-vis absorption spectra of cytidine 5’-monophosphate (CMP) and its homopolymer poly(rC) were recorded at pH 6.8 (Fig. 1a) and pH 4 (Fig. 1b). At both pH values, the wavelength of maximum absorption (λmax) of the polymer is blue shifted by several nm compared to its mononucleotide. The poly(rC) absorption spectrum also has greater intensity on the red side of the long-wavelength absorption band. When the pH is reduced from 7 to 4, λmax of CMP increases from 271 to 279 nm, while λmax of poly(rC) increases from 268 to 275 nm. The absorption spectrum of (dC)18 at pH 6.8 closely resembles the absorption spectrum of poly(rC) at pH 4 (Fig. 1c). At alkaline pH values greater than 8, the absorption spectra of the DNA C tracts shift to shorter wavelengths and closely resemble the spectrum of poly(rC) at neutral conditions.
Figure 1.

Normalized absorption spectra of poly(rC) (circles) and CMP (squares) at (a) pH 6.8 and (b) pH 4. The wavelength of maximum absorption (λmax) is indicated by a vertical line. (c) Comparison of the pH 4 absorption spectrum of poly(rC) (circles) and (dC)18 at pH 6.8 (diamonds). The solid line is the emission spectrum of poly(rC) at pH 4 excited at 290 nm.
The acid form of poly(rC) is much more fluorescent than the homopolymer at pH 6.8, as first reported by Favre in 1972 [32]. The solid line in Fig. 1c is the emission spectrum of poly(rC) at pH 4 excited at 290 nm. The wavelength of maximum emission is 429 nm in reasonable agreement with ref. [32]. The CD spectrum of (dC)18 is shown in Fig. 2 at pH 6.8 and 8.5. At pH 6.8, a positive band with a maximum at 290 nm, and a weaker negative band with a maximum at 263 nm are observed. This spectrum is a signature for hemiprotonated base pairing, and our results confirm earlier reports that deoxycytidylates can form self-associated structures at neutral pH [35,36]. At pH 8.5, the CD spectrum changes dramatically, signaling a structural transition to the single-stranded form [36].
Figure 2.

Circular dichroism spectrum of (dC)18 at pH 6.8 (solid curve) and pH 8.5 (dashed curve).
3.2. Transient absorption measurements
Transient absorption signals recorded at a probe wavelength of 570 nm are shown in Fig. 3 for poly(rC) and CMP at pH 6.8. Similar decays were observed at other visible probe wavelengths. A nonlinear least-squares fit of a single exponential convoluted with the instrument response function to the CMP transient in Fig. 3 gave a time constant of 700 ± 80 fs (2σ). The poly(rC) signal at pH 6.8 could not be fit to a single exponential. A biexponential fit gave time constants of 2.5 ± 0.4 ps (71% amplitude) and 12 ± 4 ps (29%).
Figure 3.

Transient absorption signals at 570 nm following excitation at 263 nm from poly(rC) (○) and CMP (□) in pH 6.8 buffer solution. Solid curves are least-squares fits described in the text. The signals were corrected for ionization of neat water using the procedure described in ref. [20]
Transient absorption signals from poly(rC) at acidic pH decay orders of magnitude more slowly than at pH 6.8 as reported earlier [16]. Fig. 4 shows transient absorption signals probed at 280 nm and 570 nm at pH 4. These signals agree reasonably well with ones recorded earlier by our group at pH 5 [16]. As shown in Fig. 4, the same dynamics were observed for (dC)18 at pH 6.8 and for poly(rC) at pH 4 within experimental uncertainty. Bleach recovery signals at 280 nm (bottom panel of Fig. 4c) for both hemiprotonated systems largely mirror the dynamics seen at visible wavelengths, including 570 nm. A rapid decay component with a time constant of no more than a few ps is seen near time zero in all of the transients. This component is most pronounced in the bleach recovery signals, which drop by nearly 30% in the first 10 ps after excitation.
Figure 4.

Transient absorption from poly(rC) at pH 4 (circles) and (dC)18 at pH 6.8 (diamonds) at the indicated probe wavelengths after excitation at 263 nm. The solid line in the middle panel is a least squares-fit curve to guide the eye.
The longest delay time of 2.5 ns in Fig. 4 is four times longer than in the earlier measurements. Despite the longer time range, the signals still fail to recover to the baseline. Two exponentials could not adequately fit the data. When three or more exponentials were used, the time constants were poorly determined due to the nonzero signal level at the longest delay times. The signals for poly(rC) and (dC)18 in Fig. 4 reach 50% of the initial signal level approximately 600 ps after excitation. This decay time (t1/2) and the 1/e-decay time (t1/e) (~1.0 ns) are model independent and hence more robust estimates of the signal kinetics than fits to sums of exponentials.
Transient absorption signals for poly(rC) were recorded as a function of temperature at neutral and acidic pH. Fig. 5a shows results at 20 and 70°C for the single-stranded form of poly(rC) (neutral pH), and Fig. 5b shows results for the hemiprotonated structure (pH 4). Thermal denaturation experiments (data not shown) showed non-cooperative melting of poly(rC) at pH 6.8 and cooperative melting of poly(rC) at pH 4 with a melting temperature of approximately 65°C. At pH 4, the signal at 70°C decays faster than at 20°C and is well described by a single exponential with a time constant of 1.33 ± 0.15 ps. In view of the higher noise level at this temperature, we do not consider this decay to differ significantly from that of monomeric CMP at room temperature.
Figure 5.

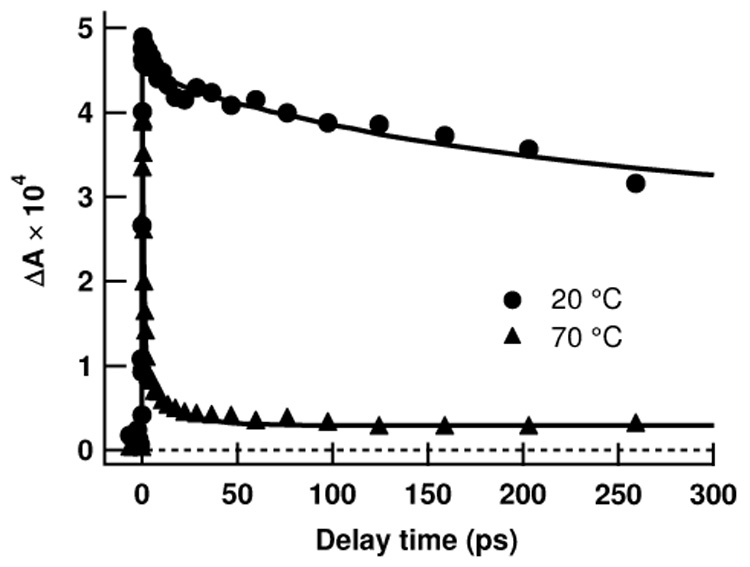
Transient absorption at 20° and 70°C from poly(rC) at (a) pH 6.8 and (b) pH 4. The pump wavelength was 263 nm and the probe wavelength was 570 nm.
Raising the temperature of the acidic form of poly(rC) to 70°C leads to a complete loss of the long-time signal (Fig. 5b). At this elevated temperature, about 80% of the signal decays with a CMP-like decay time of ~1 ps. An offset is seen at long times (also in Fig. 5a) because the 70°C signals were not corrected for the two photon ionization of water. It is also possible that some of the long-time signal seen in Fig. 5b is due to residual hemiprotonated structures.
Fig. 6 compares transient absorption signals measured at 570 nm from (dC)18 at pH 6.8 and pH 8.5. The pH value of 8.5 was chosen because it has been shown previously that poly(dC) is single-stranded at these conditions [36]. At neutral pH, transient absorption signals from (dC)18 resemble those of poly(rC) at pH 4 as shown already in Fig. 4. When the pH is raised to 8.5, the hemiprotonated structure disappears and the signal resembles that of single-stranded poly(rC) at neutral conditions.
Figure 6.

Transient absorption signals of (dC)18 at pH 6.8 and pH 8.5 pumped at 263 nm and probed at 570 nm.
3.3. Time-resolved emission measurements
Emission of poly(rC) at acidic pH and of (dC)18 at neutral pH was studied on the ps and ns time scales by the time-correlated single photon counting (TCSPC) technique. Reliable signals could not be obtained from single-stranded C tracts (i.e. poly(rC) at neutral pH and (dC)18 at alkaline pH) due to their ultraweak emission. In contrast, emission from the self-associated structures is easy to detect by TCSPC. Fig. 7 compares time-resolved emission signals of poly(rC) (pH 6.8) and (dC)18 (pH 4) at 440 nm excited at 300 nm. The emission in both cases is strongly biphasic with a rapid decay, followed by a slower decay of much lower amplitude. The emission signals from the respective C tracts are similar, but differences are observed. The initial fluorescence decay for (dC)18 occurs more rapidly than for poly(rC), but the trend is reversed at later times. As in the case of the transient absorption signals, it was not possible to obtain satisfactory fits to sums of a few exponentials. The t1/2 values for emission at 440 nm, a value near the wavelength of maximum emission (see Fig. 1c), were 103 ps and 68 ps for poly(rC) and (dC)18, respectively.
Figure 7.

Emission decays measured by TCSPC at 440 nm for poly(rC) at pH 5 (circles) and (dC)18 at pH 6.8 (squares). The instrument response function (FWHM of 46 ps) is shown by the solid black curve without markers. The dashed black curve is the 570 nm transient absorption signal of poly(rC) at pH 4. The inset shows all curves on a logarithmic axis.
4. Discussion
The transient absorption and time-resolved emission in Fig. 3 – Fig. 7 demonstrate that excited electronic states in RNA and DNA C tracts decay one to three orders of magnitude more slowly than the excited singlet state of monomeric cytidine, which has a lifetime of ~1 ps [37]. Two classes of long-lived excitations are evident in these experiments. In hemiprotonated C tracts (poly(rC) at acidic pH and (dC)18 at pH 6.8), excited states relax on a time scale of hundreds of picoseconds. For single-stranded forms, on the other hand, decay takes place on a time scale of ~10 ps at room temperature. These differences must originate in the spatial arrangement of individual cytosines in the single-stranded and hemiprotonated, self-associated forms. In the following sections, we discuss what is known about these structures and how they may affect electronic energy relaxation.
4.1. Single-stranded C tracts
The TA signals of poly(rC) at pH 6.8 decay an order of magnitude more slowly than those of the mononucleotide CMP. At 70°C, the signal for poly(rC) at pH 6.8 is dominated by a monomer-like lifetime within experimental uncertainty. At 70°C, the helix-coil equilibrium is shifted in favor of coiled regions and the total fraction of stacked bases decreases. The loss of the 10 ps signal component at this temperature therefore strongly indicates that vertical base stacking is responsible for the slower decay. The formation of longer-lived singlet excited states as a result of base stacking has been seen previously in poly(rA) and poly(dA) [20]. These observations strengthen the conclusion that ultrafast internal conversion is either impossible or inhibited in base stacks.
Recently, it was shown that electronically excited pyrimidine mononucleotides can relax to the ground state via a relatively long-lived ¹nπ* state in addition to decay via ultrafast internal conversion [38]. The ¹nπ* state does not absorb at visible probe wavelengths, and so cannot contribute to transient absorption signals at visible wavelengths. This state is also radiatively dark, and cannot be the source of the fluorescence seen in our experiments. The lifetime of the ¹nπ* state of CMP is 34 ± 3 ps in aqueous solution, but this value is a poor match for the decay times seen in poly(rC) at neutral and acidic pH, respectively. For CMP pronounced differences were observed in decay dynamics between visible and UV probe wavelengths. Here, no such differences are seen. The transient at 570 nm and the bleach recovery signal at 280 nm (Fig. 4) have approximate mirror symmetry, indicating that the species absorbing at 570 nm repopulates the electronic ground state. Our experiments also show quenching of the long-lived excited states at elevated temperatures (Fig. 5a), and this behavior is inconsistent with monomer-localized ¹nπ* states. All of these considerations indicate that the long-lived excited states observed in poly(rC) are not ¹nπ* states.
Experiments have established that poly(rC) is partially ordered and single-stranded at neutral pH, but the precise structure is unknown. Arnott et al. studied poly(rC) fibers using x-ray diffraction, and proposed that the polynucleotide adopts a right-handed, six-fold helix with a rise between bases of 0.31 nm [39]. Broido and Kearns, however, found aspects of the x-ray structure of Arnott et al. incompatible with their two-dimensional NMR results. They proposed instead that neutral poly(rC) forms an ordered left-handed single helix in aqueous solution at room temperature [40]. The latter structure has 8 nucleotides per turn and a rise of 0.29 nm. Bases in the Broido and Kearns structure are inclined by 45° from the helix axis, are located on the outside of the helix, and are hydrogen-bonded, but not stacked [40]. Some support for this unusual structure came from a recent study of vibrational Raman optical activity of poly(rC) [41]. However, there has never been any evidence for this structure in DNA C tracts.
We believe that the appearance of a similar decay component of approximately 10 ps in single-stranded poly(C) (Fig. 3) and (dC)18 (Fig. 6, pH 8.5) is best explained by conventional base stacking. In our current model, the fast decay time (~1 ps) in the transient absorption signals of the single-stranded C tracts is assigned to monomer-like excited states in residues that are not base stacked [7]. The longer decay component of ~10 ps is assigned to excitations in vertical stacks of two or more bases [7]. As shown in Fig. 5, thermal disordering of the single helix and the resulting loss of base stacking increase the extent of monomer-like decay.
4.2. Hemiprotonated self-associated structures
Poly(rC) forms a self-complex in which half of the residues are protonated at pH values slightly above cytidine’s pKa of 4.2 [42]. Although this hemiprotonated structure was originally proposed to be double-stranded [25,26], the precise structure is uncertain, as we discuss in more detail below. The acid form of poly(rC) and (dC)18 at neutral pH have very similar absorption spectra (Fig. 1c). Furthermore, the CD spectrum of (dC)18 (Fig. 2) indicates the presence of hemiprotonated base pairs (see Scheme 1). The fluorescence of the poly(rC) sample dropped precipitously when the pH was increased from 4 to 7. A similar sharp decrease in emission was observed when the pH of the (dC)18 sample was increased from 7 to 8.5. Collectively, these observations indicate that the RNA C tracts at slightly acidic pH and the DNA C tracts near neutral pH are both present as hemiprotonated self-associated structures and have similar photophysical properties.
The temperature-dependent measurements in Fig. 5 reveal that secondary structure induced by hemiprotonation and not just the presence of protonated cytidine residues is responsible for the long-lived excited states. Thermal denaturation of poly(rC) at pH 4 is accompanied by proton uptake, leading to an increased number of protonated cytosine residues [43]. Despite the increase in protonation that occurs with increasing temperature, the lifetime of the transient absorption signal at pH 4 decreases dramatically on going from room temperature to 70°C. This indicates that protonated cytosine residues are not inherently more fluorescent than the neutral molecules. Indeed, the lifetime of protonated cytidine is 630 ± 60 fs compared to ~1 ps for the neutral nucleoside [37]. In contrast to guanosine, where protonation dramatically increases the excited state lifetime [3], protonation of the cytidine monomer thus actually reduces the excited state lifetime in aqueous solution [37].
The possibility that base pairing facilitates electronic energy relaxation on an ultrafast time scale by proton or hydrogen atom transfer has been actively discussed recently [23,24,44]. By virtue of the slow energy relaxation seen in the hemiprotonated forms, the CH+·C base pair joins the growing list of base pairs that support excited states, which are longer-lived than those of monomeric bases—at least for base pairs present in base stacks. In addition to the CH+·C base pair, this list includes the base pairs found in the acid duplex of poly(A) [20] and the A·T base pairs in various AT-containing oligonucleotides [7].
A distinctive feature of the hemiprotonated C tract assemblies is their fluorescence. The increased fluorescence of the hemiprotonated form of the RNA homopolymer poly(rC) was discovered by Favre in 1972 [32]. Although it has been known for many years that poly(dC) can self-associate [35], our measurements are the first to our knowledge to demonstrate that deoxycytidylates such as (dC)18 have enhanced fluorescence as a result of the hemiprotonated structure. The fluorescence quantum yield of the acid form of poly(rC) at pH 4 was reported variously to be 4 × 10−3 [32] and 8 × 10−4 [33]. The larger of the two values is about 25-times greater than the fluorescence quantum yield reported for single-stranded poly(rC) (1.56×10−4).[45] This increase is considerably less than expected given the increase in lifetime of up to three orders of magnitude (~1 ns compared to ~1 ps). The bleach recovery signal at 280nm suggests that a substantial fraction of excited states in the hemiprotonated form of poly(rC) relax slowly. This in turn implies that these states have reduced radiative character compared to the short-lived ¹ππ* state of monomeric bases. An obvious possibility, which is supported by the strongly red-shifted emission spectrum of the hemiprotonated form (Fig. 1c), is a charge transfer state [46]. Similar conclusions were reached for single-stranded tracts of A bases [7].
Plessow et al. studied emission from (dC)n (n = 2 – 15) oligonucleotides (pH 7.1, 150 mM NaCl) excited by 80 ps, 283 nm laser pulses using a picosecond streak camera coupled to an imaging spectrometer [19]. They reported a dramatic enhancement of fluorescence from (dC)15 compared to (dC)2 and CMP [19]. The emission decays from their study decayed biexponentially with a long time constant of 1 – 2 ns that was independent of length, and a short time constant of < 100 ps that depended on the length of the oligonucleotide and on the emission wavelength [19]. Plessow et al. assumed that (dC)15 is single-stranded under their conditions. They assigned the long lifetime component to excimers formed in base stacks, and the short-lived component to monomer-like fluorescence. They wrote in their study that a ~100 ps life time is rather long for monomer-like fluorescence, but suggested that this could be due to restricted solvent exposure [19]. This explanation is contradicted, however, by recent results showing fluorescence lifetimes of ≤1 ps for monomeric bases in a variety of solvents [47–49].
Based on our findings there is little doubt that the emission observed by Plessow et al. [19] should be reassigned to the hemiprotonated self-associated structure present at neutral pH. Several observations support this conclusion. First, the emission spectrum of ref. [19] agrees well with our spectrum in Fig. 1c, and does not match the spectrum of single-stranded poly(rC) recorded at pH 7 [45]. Second, the enhanced absorption reported by Plessow et al. for (dC)15 between 280 and 320 nm compared to dCMP and (dC)2 is consistent with the enhancement of the red tail of the spectrum that accompanies hemiprotonation (see Fig. 1). Finally, the nearly total quenching of long-lived excited states on raising the pH of (dC)18 from 7 to 8.5 is further evidence that a self-complex is responsible for the emission.
The TCSPC emission signals in Fig. 7 are strongly biphasic. Integration of the TCSPC emission decay curves shows that about half of the total emission detected between 0 and 2.5 ns occurs in the first 200 ps. In contrast, Plessow et al. observed that approximately two thirds of the emission of (dC)15 occurs on a nanosecond time scale [19]. The lower time resolution in the Plessow et al. study can explain the attenuation of the subnanosecond component in their measurements. According to our findings, the relative amplitude of the ns component to the shorter-lived component is greater in the transient absorption signals than in the emission decays (Fig. 7). This difference rules out complex kinetics by a single population as an explanation for the biphasic decays. Instead, the two decay times are best explained by two kinds of excited states. The faster component, which has a t1/2 time of less than 100 ps, is much too slow to originate from monomer emission. Both states may be CT states based on their low radiative transition rates as mentioned earlier. These different states could result from two different base stacking environments in the polymer, but it is not possible to further define the two emitting states at present.
4.3. Structural implications
Understanding excited states of polycytidylates is hampered by limited knowledge of the structure or structures present in buffer solution. It has been assumed in the past that poly(rC) and poly(dC) form hemiprotonated duplexes [36], but there is no decisive structural proof. About fifteen years ago, NMR [27,50] and later x-ray crystal structures [51,52] showed that dC-rich oligonucleotides adopt a quadruplex form (“i-motif DNA”) composed of two intercalated duplexes, each of which consists of CH+·C base pairs. Crystals of d(CT)3 grown at neutral pH adopt the quadruplex structure [52], showing that i-motif structures do not require acidic conditions for deoxycytidylates.
It is worth considering whether the similar excited-state dynamics of RNA and DNA C tracts and their similar CD spectra [36] are a consequence of common structures. Despite considerable efforts, the necessary conditions for i-motif formation are poorly understood. In particular, the question of whether the i-motif can form in C tracts of oligoribonucleotides is not completely settled. Initial evidence suggested that RNAs cannot form C quadruplex structures [53], but Snoussi et al. observed features in NMR spectra of C-rich RNA sequences at pH 4.3 that match ones seen previously in i-motif DNA [54]. For long RNA C tracts like poly(rC), the duplex is thought to be more stable than alternative quadruplex structures [55]. Experiments by Petrovic et al. favor this viewpoint for acidic poly(rC), but cannot rule out a quadruplex structure [56].
A further complication is the possibility that multiple, self-associated structures exist in equilibrium. Raman measurements on (dC)10 were interpreted in terms of a complex equilibrium between duplex, tetraplex, and even octaplex structures [57]. The two-dimensional NMR spectra of Snoussi et al. suggest that (rC)5 adopts multiple intercalation topologies perhaps including an i-motif core terminated by double-stranded hemiprotonated duplexes or unpaired dangling strands [54]. Additional experiments are planned to investigate whether structural heterogeneity contributes to the complex photophysical dynamics of hemiprotonated C tracts.
Photophysical observations such as those reported here can ultimately shed light on structural issues, although this goal is not attainable at present. Our results show that the acid and neutral hemiprotonated forms of RNA and DNA C tracts, respectively, are very similar in their excited-state properties. It is tempting to assign the long-lived and fluorescent excited states to excitations in CH+·C base pairs since this motif is present in both the duplex and quadruplex hemiprotonated structures. However, this is undercut by the finding that the triplex poly(CH+)·poly(I)·poly(C) is fluorescent [58]. In this structure, CH+ is paired to inosine and no CH+·C base pairs are present. This is strong evidence that vertical base stacks, and not horizontal base pairs, are responsible for the fluorescent properties as suggested previously [32,46].
If base stacking governs excited-state dynamics, then differences might be expected for duplex and quadruplex C tracts in light of their different base stacking geometries. Fig. 8a shows the overlap of two hemiprotonated base pairs taken from the crystal structure of a deoxycytidine dimer with a modified backbone [59]. In this structure, which the authors proposed as a model for a parallel-stranded duplex, successive base pairs are separated by 3.44 Å and a twist angle of 34°. As shown in Fig. 8a, there is partial overlap of pyrimidine rings in this structure. In contrast, stacked base pairs in the quadruplex or i-motif structure have zero overlap of pyrimidine rings (Fig. 8b). Instead, the amino groups of one CH+·C base pair are stacked in nearly antiparallel fashion above the amino groups of the next one. Carbonyl groups are also approximately antiparallel (Fig. 8b). This antiparallel arrangement of bond dipoles may contribute to the overall electrostatic stabilization. Stacking of exocyclic groups allows the hemiprotonated base pairs to approach to a distance of 3.1 Å [52], much closer than the ~3.4 Å typically found in double-stranded nucleic acids.
Figure 8.

(a) CH+·C base pair stacking in the Egli et al. (ref. [59]) crystal structure of a deoxycytidylyl-(3′–5′)-deoxycytidine analog containing intranucleosidyl C(3′)–C(5′) ethylene bridges, and (b) in the quadruplex (“i-motif”) crystal structure of Chen et al. (ref. [51]). Color scheme: hydrogen (white), nitrogen (blue), and oxygen (red). The three hydrogen bonds of the base pair on top (bottom) are shown in black (orange). The proton between the N3 atoms of each base pair has been omitted. These images were produced using the UCSF Chimera package (ref. [67]) from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
Both duplex and quadruplex structures position the electron-rich amino group very near the electron-deficient pyrimidine ring of the base below it (Fig. 8). This is a favorable geometry for a charge transfer interaction. Charge separation is also likely to be enhanced by the shared proton in the CH+·C base pair. However, the very different stacking geometries and electrostatic local environment for duplex vs. quadruplex C tracts (Fig. 8) would almost certainly affect the lifetime of a charge transfer state due to differences in electronic coupling and reorganization energy. Even allowing for structural reorganization after UV excitation, it seems highly unlikely that the base stacks in these different self-associated structures could ever achieve the same local geometry.
The very similar excited-state lifetimes seen in poly(rC) at pH 4 and (dC)18 at pH 6.8 thus suggest to us that these systems are present in the same structure—a structure which is probably a quadruplex given the preference of deoxycytidylates for this form. This conclusion is clearly at odds with the dominant view in the literature that long RNA C tracts do not form quadruplex structures. Support for this unorthodox viewpoint comes from the absorption spectrum of hemiprotonated poly(rC) (Fig. 1b), which shows a larger enhancement on the red edge compared to single-stranded poly(rC) (Fig. 1a). This difference is suggestive of a stronger excitonic interaction due to closer approach between bases in a quadruplex structure. Importantly, the absorption spectrum of protonated cytidine shows no such exciton splitting, but is simply red-shifted relative to the neutral form [60].
A further possibility is that RNA and DNA C tracts have identical excited-state dynamics despite very different base-stacking geometries. However, this appears unlikely given the sensitivity of excited-state dynamics to secondary structure seen here and elsewhere [7–9,20]. Calculations aimed at understanding how excited states of nucleobase multimers depend on local structure are just beginning to appear [61–64], and these will help to resolve these issues. A few studies have even examined cytosine-cytosine base stacks [65,66], but have not yet used geometries from structural biology. Clearly, progress in understanding the electronic structure of these multichromophoric systems will be facilitated by calculations that incorporate realistic structural models.
5. Conclusions
The time-resolved transient absorption and emission results presented here demonstrate that excited states of cytidine and 2’-deoxycytidine multimers decay orders of magnitude more slowly than excited states in the corresponding monomers. These systems establish that variation in nucleic acid secondary structure profoundly affects photophysical properties. As a consequence of this sensitivity, improved structural characterization of model nucleic acid systems will be indispensable for fully understanding electronic structure.
In the case of the single-stranded C tracts, base stacking slows relaxation by an order of magnitude. Even longer-lived excited states are observed in the self-complexes of RNA and DNA C tracts. Results suggest that the long-lived and relatively more fluorescent excited states are not characteristic of a single hemiprotonated CC base pair, but arise from base stacking interactions between two such base pairs. Time-resolved emission experiments show that emission occurs predominantly on a subnanosecond time scale. Two kinds of excited states are suggested to be present, but elucidation of these states requires further study.
We have shown that deoxycytidylates at physiological conditions of pH and ionic strength have similar excited-state dynamics as the acid form of poly(rC). This increases the possibility that dC-rich sequences can adopt hemiprotonated structures in biological systems. It also suggests that transient spectroscopy may be a useful diagnostic for self-associated structures, including the i-motif. Improved understanding of how structure affects excited states could allow ultrafast spectroscopic methods to develop into tools for studying nucleic acid structures with high time resolution.
Acknowledgements
This research was made possible by a grant from the National Institutes of Health (R01 GM64563). Some measurements were performed in Ohio State’s Center for Chemical and Biophysical Dynamics, using equipment funded by the National Science Foundation, and the Ohio Board of Regents. MHL acknowledges an undergraduate research scholarship from the Colleges of Arts and Sciences Honors Committee of The Ohio State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Callis PR. Ann. Rev. Phys. Chem. 1983;34:329. [Google Scholar]
- 2.Crespo-Hernández CE, Cohen B, Hare PM, Kohler B. Chem. Rev. 2004;104:1977. doi: 10.1021/cr0206770. [DOI] [PubMed] [Google Scholar]
- 3.Pecourt J-ML, Peon J, Kohler B. J. Am. Chem. Soc. 2001;123:10370. doi: 10.1021/ja0161453. [DOI] [PubMed] [Google Scholar]
- 4.Peon J, Zewail AH. Chem. Phys. Lett. 2001;348:255. [Google Scholar]
- 5.Onidas D, Markovitsi D, Marguet S, Sharonov A, Gustavsson T. J. Phys. Chem. B. 2002;106:11367. [Google Scholar]
- 6.Markovitsi D, Sharonov A, Onidas D, Gustavsson T. ChemPhysChem. 2003;4:303. doi: 10.1002/cphc.200390050. [DOI] [PubMed] [Google Scholar]
- 7.Crespo-Hernández CE, Cohen B, Kohler B. Nature. 2005;436:1141. doi: 10.1038/nature03933. [DOI] [PubMed] [Google Scholar]
- 8.Kwok W-M, Ma C, Phillips DL. J. Am. Chem. Soc. 2006;128:11894. doi: 10.1021/ja0622002. [DOI] [PubMed] [Google Scholar]
- 9.Buchvarov I, Wang Q, Raytchev M, Trifonov A, Fiebig T. Proc. Natl. Acad. Sci. USA. 2007;104:4794. doi: 10.1073/pnas.0606757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schreier WJ, Schrader TE, Koller FO, Gilch P, Crespo-Hernández CE, Swaminathan VN, Carell T, Zinth W, Kohler B. Science. 2007;315:625. doi: 10.1126/science.1135428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sobolewski AL, Domcke W. Eur. Phys. J. D. 2002;20:369. [Google Scholar]
- 12.Ismail N, Blancafort L, Olivucci M, Kohler B, Robb MA. J. Am. Chem. Soc. 2002;124:6818. doi: 10.1021/ja0258273. [DOI] [PubMed] [Google Scholar]
- 13.Matsika S. J. Phys. Chem. A. 2004;108:7584. [Google Scholar]
- 14.Blancafort L. J. Am. Chem. Soc. 2006;128:210. doi: 10.1021/ja054998f. [DOI] [PubMed] [Google Scholar]
- 15.Serrano-Andrés L, Merchán M, Borin AC. Proc. Natl. Acad. Sci. USA. 2006;103:8691. doi: 10.1073/pnas.0602991103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen B, Crespo-Hernández CE, Hare PM, Kohler B. In: Femtochemistry and Femtobiology : Ultrafast Events in Molecular Science. Martin MM, Hynes JT, editors. Amsterdam: Elsevier Science; 2004. p. 463. [Google Scholar]
- 17.Ballini JP, Vigny P, Daniels M. Biophys. Chem. 1983;18:61. doi: 10.1016/0301-4622(83)80027-1. [DOI] [PubMed] [Google Scholar]
- 18.Ballini JP, Daniels M, Vigny P. Biophys. Chem. 1991;39:253. doi: 10.1016/0301-4622(91)80003-a. [DOI] [PubMed] [Google Scholar]
- 19.Plessow R, Brockhinke A, Eimer W, Kohse-Höinghaus K. J. Phys. Chem. B. 2000;104:3695. [Google Scholar]
- 20.Crespo-Hernández CE, Kohler B. J. Phys. Chem. B. 2004;108:11182. [Google Scholar]
- 21.Markovitsi D, Talbot F, Gustavsson T, Onidas D, Lazzarotto E, Marguet S. Nature. 2006;441:E7. doi: 10.1038/nature04903. [DOI] [PubMed] [Google Scholar]
- 22.Schultz T, Samoylova E, Radloff W, Hertel IV, Sobolewski AL, Domcke W. Science. 2004;306:1765. doi: 10.1126/science.1104038. [DOI] [PubMed] [Google Scholar]
- 23.Abo-Riziq A, Grace L, Nir E, Kabelac M, Hobza P, de Vries MS. Proc. Natl. Acad. Sci. USA. 2005;102:20. doi: 10.1073/pnas.0408574102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwalb NK, Temps F. J. Am. Chem. Soc. 2007;129:9272. doi: 10.1021/ja073448+. [DOI] [PubMed] [Google Scholar]
- 25.Langridge R, Rich A. Nature. 1963;198:725. doi: 10.1038/198725a0. [DOI] [PubMed] [Google Scholar]
- 26.Akinrimisi EO, Sander C. P.O.P. Ts'o, Biochemistry. 1963;2:340. doi: 10.1021/bi00902a028. [DOI] [PubMed] [Google Scholar]
- 27.Gehring K, Leroy JL, Guéron M. Nature. 1993;363:561. doi: 10.1038/363561a0. [DOI] [PubMed] [Google Scholar]
- 28.Guéron M, Leroy J-L. Current Opinion in Structural Biology. 2000;10:326. doi: 10.1016/s0959-440x(00)00091-9. [DOI] [PubMed] [Google Scholar]
- 29.Mills M, Lacroix L, Arimondo PB, Leroy J-L, Francois J-C, Klump H, Mergny J-L. Current Medicinal Chemistry: Anti-Cancer Agents. 2002;2:627. doi: 10.2174/1568011023353877. [DOI] [PubMed] [Google Scholar]
- 30.Volker J, Klump HH, Breslauer KJ. Biopolymers. 2007;86:136. doi: 10.1002/bip.20712. [DOI] [PubMed] [Google Scholar]
- 31.Kaushik M, Suehl N, Marky LA. Biophys. Chem. 2007;126:154. doi: 10.1016/j.bpc.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 32.Favre A. FEBS Lett. 1972;22:280. doi: 10.1016/0014-5793(72)80250-3. [DOI] [PubMed] [Google Scholar]
- 33.Vigny P, Favre A. Photochem. Photobiol. 1974;20:345. doi: 10.1111/j.1751-1097.1974.tb06586.x. [DOI] [PubMed] [Google Scholar]
- 34.Buterbaugh JS, Toscano JP, Weaver WL, Gord JR, Hadad CM, Gustafson TL, Platz MS. J. Am. Chem. Soc. 1997;119:3580. [Google Scholar]
- 35.Inman RB. J Mol. Biol. 1964;9:624. doi: 10.1016/s0022-2836(64)80171-6. [DOI] [PubMed] [Google Scholar]
- 36.Antao VP, Gray DM. J. Biomolecular Struct. & Dynamics. 1993;10:819. doi: 10.1080/07391102.1993.10508677. [DOI] [PubMed] [Google Scholar]
- 37.Malone RJ, Miller AM, Kohler B. Photochem. Photobiol. 2003;77:158. doi: 10.1562/0031-8655(2003)077<0158:sesloc>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 38.Hare PM, Crespo-Hernández CE, Kohler B. Proc. Natl. Acad. Sci. USA. 2007;104:435. doi: 10.1073/pnas.0608055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnott S, Chandrasekaran R, Leslie AGW. J. Mol. Biol. 1976;106:735. doi: 10.1016/0022-2836(76)90262-x. [DOI] [PubMed] [Google Scholar]
- 40.Broido MS, Kearns DR. J. Am. Chem. Soc. 1982;104:5207. [Google Scholar]
- 41.Bell AF, Hecht L, Barron LD. J. Am. Chem. Soc. 1997;119:6006. [Google Scholar]
- 42.Christensen JJ, Rytting JH, Izatt RM. J. Phys. Chem. 1967;71:2700. doi: 10.1021/j100867a047. [DOI] [PubMed] [Google Scholar]
- 43.Guschlbauer W. Proc. Natl. Acad. Sci. USA. 1967;57:1441. doi: 10.1073/pnas.57.5.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sobolewski AL, Domcke W, Hattig C. Proc. Natl. Acad. Sci. USA. 2005;102:17903. doi: 10.1073/pnas.0504087102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morgan JP, Daniels M. Photochem. Photobiol. 1980;31:207. doi: 10.1111/j.1751-1097.1984.tb08854.x. [DOI] [PubMed] [Google Scholar]
- 46.Montenay-Garestier T, Hélène C. Biochemistry. 1970;9:2865. doi: 10.1021/bi00816a017. [DOI] [PubMed] [Google Scholar]
- 47.Sharonov A, Gustavsson T, Carré V, Renault E, Markovitsi D. Chem. Phys. Lett. 2003;380:173. [Google Scholar]
- 48.Gustavsson T, Banyasz A, Lazzarotto E, Markovitsi D, Scalmani G, Frisch MJ, Barone V, Improta R. J. Am. Chem. Soc. 2006;128:607. doi: 10.1021/ja056181s. [DOI] [PubMed] [Google Scholar]
- 49.Hare PM, Crespo-Hernández CE, Kohler B. J. Phys. Chem. B. 2006;110:18641. doi: 10.1021/jp064714t. [DOI] [PubMed] [Google Scholar]
- 50.Leroy JL, Gehring K, Kettani A, Guéron M. Biochemistry. 1993;32:6019. doi: 10.1021/bi00074a013. [DOI] [PubMed] [Google Scholar]
- 51.Chen L, Cai L, Zhang X, Rich A. Biochemistry. 1994;33:13540. doi: 10.1021/bi00250a005. [DOI] [PubMed] [Google Scholar]
- 52.Kang C, Berger I, Lockshin C, Ratliff R, Moyzis R, Rich A. Proc. Natl. Acad. Sci. USA. 1994;91:11636. doi: 10.1073/pnas.91.24.11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lacroix L, Mergny JL, Leroy JL, Hélène C. Biochemistry. 1996;35:8715. doi: 10.1021/bi960107s. [DOI] [PubMed] [Google Scholar]
- 54.Snoussi K, Nonin-Lecomte S, Leroy JL. J. Mol. Biol. 2001;309:139. doi: 10.1006/jmbi.2001.4618. [DOI] [PubMed] [Google Scholar]
- 55.Collin D, Gehring K. J. Am. Chem. Soc. 1998;120:4069. [Google Scholar]
- 56.Petrovic AG, Polavarapu PL. J. Phys. Chem. B. 2006;110:22826. doi: 10.1021/jp063221l. [DOI] [PubMed] [Google Scholar]
- 57.Rush T, III, Yong H, Peticolas WL. Biopolymers. 1997;41:121. [Google Scholar]
- 58.Thiele D, Guschlbauer W, Favre A. Biochim. Biophys. Acta, Nucleic Acids and Protein Synthesis. 1972;272:22. [PubMed] [Google Scholar]
- 59.Egli M, Lubini P, Bolli M, Dobler M, Leumann C. J. Am. Chem. Soc. 1993;115:5855. [Google Scholar]
- 60.Voet D, Gratzer WB, Cox RA, Doty P. Biopolymers. 1963;1:193. [Google Scholar]
- 61.Bouvier B, Dognon J-P, Lavery R, Markovitsi D, Millié P, Onidas D, Zakrzewska K. J. Phys. Chem. B. 2003;107:13512. [Google Scholar]
- 62.Emanuele E, Zakrzewska K, Markovitsi D, Lavery R, Millié P. J. Phys. Chem. B. 2005;109:16109. doi: 10.1021/jp051833k. [DOI] [PubMed] [Google Scholar]
- 63.Bittner ER. J. Chem. Phys. 2006;125:094909/1. doi: 10.1063/1.2335452. [DOI] [PubMed] [Google Scholar]
- 64.Santoro F, Barone V, Improta R. Proc. Natl. Acad. Sci. USA. 2007;104:9931. doi: 10.1073/pnas.0703298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Danilov VI, Slyusarchuk ON, Alderfer JL, Stewart JJP, Callis PR. Photochem. Photobiol. 1994;59:125. [Google Scholar]
- 66.Olaso-González G, Roca-Sanjuán D, Serrano-Andrés L, Merchán M. J. Chem. Phys. 2006;125:231102. doi: 10.1063/1.2408411. [DOI] [PubMed] [Google Scholar]
- 67.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J. Comput. Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]