

Abstract
Signal transduction from the insulin receptor to downstream effectors is attenuated by phosphorylation at a number of Ser/Thr residues of insulin receptor substrate-1 (IRS-1) resulting in resistance to insulin action, the hallmark of type II diabetes. Ser/Thr residues can also be reversibly glycosylated by O-linked β-N-acetylglucosamine (O-GlcNAc) monosaccharide, a dynamic posttranslational modification that offers an alternative means of protein regulation to phosphorylation. To identify sites of O-GlcNAc modification in IRS-1, recombinant rat IRS-1 isolated from HEK293 cells was analyzed by two complementary mass spectrometric methods. Using data-dependent neutral loss MS3 mass spectrometry, MS/MS data were scanned for peptides that exhibited a neutral loss corresponding to the mass of N-acetylglucosamine upon dissociation in an ion trap. This methodology provided sequence coverage of 84% of the protein, permitted identification of a novel site of phosphorylation at Thr-1045, and facilitated the detection of an O-GlcNAc-modified peptide of IRS-1 at residues 1027−1073. The level of O-GlcNAc modification of this peptide increased when cells were grown under conditions of high glucose with or without chronic insulin stimulation or in the presence of an inhibitor of the O-GlcNAcase enzyme. To map the exact site of O-GlcNAc modification, IRS-1 peptides were chemically derivatized with dithiothreitol following β-elimination and Michael addition prior to LC-MS/MS. This approach revealed Ser-1036 as the site of O-GlcNAc modification. Site-directed mutagenesis and Western blotting with an anti-O-GlcNAc antibody suggested that Ser-1036 is the major site of O-GlcNAc modification of IRS-1. Identification of this site will facilitate exploring the biological significance of the O-GlcNAc modification.
The O-linked glycosylation of serine and threonine residues by a single monosaccharide, β-N-acetylglucosamine (GlcNAc),1 is a dynamic and reversible posttranslational modification occurring on cytoplasmic and nuclear proteins (1). The posttranslational O-GlcNAc modification (O-GlcNAcylation) of transcription factors and signaling molecules has been proposed to influence transcriptional regulation (2) and insulin signaling in a manner analogous to phosphorylation (3). The enzymes catalyzing the incorporation and removal of GlcNAc from proteins, O-GlcNAc-transferase and O-GlcNAcase, are highly conserved in eukaryotes and ubiquitously distributed in tissues (4). Transgenic embryos deficient in O-GlcNAc-transferase are not viable, suggesting the crucial nature of this enzymatic activity (5). The sugar donor for O-GlcNAc modification, UDP-GlcNAc, is generated by the metabolism of glucose through the hexosamine biosynthetic pathway. Under normal conditions 2−3% of the glucose entering the cell is committed to this pathway (6). However, in states of nutrient excess, such as type II diabetes, UDP-GlcNAc levels and the extent of protein O-GlcNAc modification rise (7). Increased protein O-GlcNAcylation has also been observed in models of amyotrophic lateral sclerosis (8), cardiac dysfunction (9), and Alzheimer disease (10).
Insulin receptor substrate-1 (IRS-1), a 131-kDa protein central to the insulin signaling pathway, has been shown by immunological methods to be O-GlcNAc-modified under conditions that promote insulin resistance in cultured adipocytes (11) and endothelial cells (12) and in skeletal muscle in vivo (13). Phosphorylation at specific Tyr residues of IRS-1 by the insulin receptor mediates binding of IRS-1 to downstream effectors, such as phosphatidylinositol 3-kinase, Grb2, and SHP-2 (14). These interactions are further modulated by Ser/Thr kinases. For instance, Ser/Thr phosphorylation at residues 302, 307, 318, 612, 632, 789, or 1101 reduce insulin signaling (for a review, see Ref. 15). This interplay of posttranslational modifications of IRS-1 is responsible, in part, for the diverse effects elicited by insulin. To further investigate the effect of O-GlcNAc modification on insulin signaling at the level of IRS-1, we have used complementary mass spectrometric methods to identify the sites of O-GlcNAc modification of IRS-1.
Although many proteins possess the O-GlcNAc modification, the effect of this posttranslational modification on protein function has only been determined for a handful of proteins. This is due to the relatively low abundance of O-GlcNAc-modified peptides and to the labile nature of this modification that render the identification of the sites of modification by tandem mass spectrometry challenging. Direct mass spectrometric analyses of purified proteins have taken advantage of the labile nature of this modification by scanning for peptides that readily lose the monosaccharide during fragmentation (16–18). Upon CID of an O-GlcNAc-modified peptide in the mass spectrometer, typically the O-linked N-acetylglucosamine moiety is the first to fragment from the peptide leaving an intact Ser (or Thr) residue. This provides a means of using the mass spectrometer to selectively detect O-GlcNAc-modified peptides, which demonstrate a neutral loss of 203.2 Da from the peptide by data-dependent neutral loss MS3 or which fragment to form an N-acetylglucosamine oxonium ion (m/z 204.1) by precursor ion scanning. The fragmentation of peptides by the recently developed electron transfer dissociation method offers the advantage that ions retain the GlcNAc permitting mapping the site of modification (19). The utility of these methods depends on the abundance of the modified peptide within a mixture of peptides because both methods rely on the O-GlcNAc-modified peptide being selected by the instrument for fragmentation by tandem mass spectrometry. Strategies utilized to enrich O-GlcNAc-modified peptides or proteins for mass spectrometric analysis including wheat germ agglutinin lectin affinity chromatography, immunoprecipitation, and the enzymatic incorporation of galactose by galactosyltransferase for ricin affinity chromatography are limited by nonselectivity and inefficiency (20–22). Recent developments in enzymatic labeling approaches represent promising advances in the identification of O-GlcNAc-modified proteins from complex protein mixtures (23–25). However, these methods may not provide information on the site of modification and require reagents that are not presently commercially available. Alternatively a chemical derivatization strategy involving β-elimination of the GlcNAc moiety and replacement with a thiol-reactive handle by Michael addition (BEMAD) facilitates enrichment and generates a stable derivative amenable to mapping the sites of modification (21). However, careful controls are necessary to discriminate among Ser/Thr residues that are phosphorylated, O-sulfonated (26), O-GlcNAc-modified, or O-glycosylated by other monosaccharides or polysaccharide chains (27).
The present study examined the use of data-dependent neutral loss MS3 mass spectrometry and chemical derivatization prior to mass spectrometry to identify novel sites of O-GlcNAc modification of IRS-1. These methods were first optimized using mixtures of protein standards spiked with a synthetic O-GlcNAc-modified peptide. Data-dependent neutral loss MS3 provided a simple straightforward approach for the detection of an O-GlcNAc-modified peptide of IRS-1. Chemical derivatization by the BEMAD approach revealed the exact site of modification and offers an alternative methodology for the detection of sites of phosphorylation. These complementary methods permitted identification of novel sites of O-GlcNAc modification and phosphorylation within the C terminus of IRS-1. Site-directed mutagenesis suggests that the site of O-GlcNAc modification identified, Ser-1036, is the major site of this modification in IRS-1.
EXPERIMENTAL PROCEDURES
Protein Standards
A synthetic O-GlcNAc-modified peptide, PSVPVS(O-GlcNAc)GSAPGR, derived from UL32 protein of human cytomegalovirus was a gift from Dr. G. W. Hart, Johns Hopkins University. Standard protein mixtures that included phosphorylated and N-linked glycoproteins contained BSA, β-casein, RNase B, lysozyme, and mono- and tetraphosphorylated peptides of casein (Sigma). Protein mixtures were spiked with the O-GlcNAcylated peptide at varying molar ratios including 1:1, 1:10, and 1:50 (peptide:protein). For each LC-MS/MS analysis using the neutral loss methodology, 2 pmol of synthetic peptide in varying ratios were analyzed, whereas 5 times this amount was analyzed by LC-MS/MS following the BEMAD chemical derivatization approach.
Generation of IRS-1 cDNA
The cDNA encoding rat IRS-1 (a gift from Dr. Morris White (28)) was cut out of the pCMV vector with SacI and HindIII and subcloned into pBluescript SK–. To aid in cloning IRS-1 into the EcoRV and HindIII sites of the mammalian expression vector pTriEX4 (Novagen), an EcoRV site was incorporated at nucleotide 19 by site-directed mutagenesis (QuikChange XL, Stratagene) with the following primer 5′-CCCTCCGGATATCGATGGCTTCTCAGACG-3′. A single nucleotide was inserted to align the reading frames of the His and S-tags with the coding sequence of IRS-1 using the primer 5′-CCTCAACGATATCGATCGGCTTCTCAGACG-3′. IRS-1, amino acid residues 6−1235, was expressed as an N-terminally tagged His and S-tag fusion protein in HEK293 cells. Site-directed mutagenesis was performed to substitute Ser-1035, Ser-1036, and Thr-1037 with alanines (IRS-1 AAA) using the primer 5′-GGAGCTGCCCCCCCTCCCGCGGCGGCGGCCTCTGC-3′.
Cell Culture and Expression of IRS-1
HEK293 cells were maintained in improved minimal essential medium (BioSource) containing 10% heat-inactivated fetal calf serum with 1% antibiotic/antimycotic (Invitrogen). Cells were transiently transfected with IRS-1 cDNA using Effectine reagent according to the manufacturer's recommendations (Qiagen). Eighteen hours prior to cell lysis, transfected cells were treated with 5 mm glucose ± the O-GlcNAcase inhibitor O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate (PUGNAc; 50 μm) (Toronto Chemicals) (29) or 25 mm glucose (high glucose) ± 100 nm insulin. Cells were lysed in lysis buffer (300 mm NaCl, 50 mm sodium phosphate, 10 mm imidazole, 10 mm β-mercaptoethanol, and 1% Nonidet P-40) supplemented with 50 μm PUGNAc, 10 mm sodium fluoride, 500 μm sodium vanadate, 1 mm pyrophosphate, and EDTA-free protease inhibitor mixture (Roche Applied Science) 48 h after transfection. The His-tagged protein was enriched by incubation of the lysate with nickel-nitrilotriacetic acid His bind resin (Novagen) for 1 h at room temperature followed by extensive washing with lysis buffer containing 20 mm imidazole. IRS-1 was eluted in lysis buffer containing 250 mm imidazole and purified by SDS-PAGE on 4−10% XT Criterion gels (Bio-Rad). The IRS-1 protein band was visualized using Ezinc protein stain (Pierce) and cut out of the gel for further manipulation.
Protein Reduction or Oxidation and Proteolysis
IRS-1 to be analyzed by data-dependent neutral loss MS3 was reduced with DTT and alkylated with iodoacetamide. In-gel digestion was performed with trypsin (Promega), Lys-C (Roche Applied Science), Glu-C (Roche Applied Science), or chymotrypsin (Roche Applied Science) for 18 h at 37 °C.
Mixtures of protein standards spiked with the synthetic O-GlcNAc-modified peptide were dried, oxidized with performic acid (PFA) vapor in a vacuum chamber containing 1 ml of PFA for 1 h at room temperature as described by McLachlin and Chait (30), and digested with trypsin (Promega) overnight at 37 °C. Phosphates were removed with 20 units of calf alkaline phosphatase (Promega) in the presence of 1 mm MgCl2 at 37 °C for 4 h. Peptides were desalted by C18 microspin columns (Nest Group) and dried under vacuum. Peptides were oxidized again by incubation in 50 μl of 5% (v/v) 30% hydrogen peroxide, 45% (v/v) 88% formic acid for 1 h at room temperature and dried under vacuum.
Gel pieces containing IRS-1 were destained, washed with 100 mm ammonium bicarbonate, and oxidized for 1 h on ice with a PFA solution containing 5 μl of 30% H2O2 and 95 μl of 95% formic acid that had been incubated for 1 h at room temperature and chilled prior to use. Gel pieces were washed twice for 10 min with 100 mm ammonium bicarbonate, dehydrated with acetonitrile, rehydrated with ammonium bicarbonate, dehydrated with acetonitrile, and dried completely before proteolytic digestion. Extracted peptides were treated with 2 units of shrimp alkaline phosphatase (Promega) by incubation in reaction buffer supplied by the manufacturer at 37 °C for 18 h or with 10 units of β-N-acetylhexosaminidase (New England Biolabs) in reaction buffer supplied by the manufacturer at 37 °C for 18 h. Mixtures were acidified, desalted by C18 microspin columns, dried under vacuum, and oxidized by PFA vapor (as described above). Peptides were analyzed by MALDI-TOF MS and LC-MS/MS or subjected to β-elimination chemistry as described below.
Chemical Derivatization of O-GlcNAc-modified Peptides with DTT
Peptides that had been oxidized and digested were subjected to BEMAD for the removal of the N-acetylglucosamine from Ser/Thr residues and replacement with DTT (21). O-GlcNAc Ser and O-GlcNAc Thr undergo β-elimination to dehydroalanine and dehydroamino-2-butyric acid, respectively, followed by derivatization with DTT. The reaction was initiated with the addition of 50 μl of 0.2% (w/v) NaOH, 20 mm dithiothreitol (Sigma), and 2% (v/v) triethylamine to the dried peptide sample and incubated at 56 °C for 2 h at pH 12.5. Excess DTT was removed by C18 microspin columns, and the peptides were dried under vacuum. The mixture was brought up in degassed PBS with 1 mm EDTA (PBS/EDTA), and the DTT-derivatized peptides were purified by incubation for 2 h with 25 μl of a 50% slurry of thiopropyl-Sepharose (Amersham Biosciences) at room temperature. The sulfhydryl-reactive peptides retained on the column were washed extensively with PBS/EDTA, and the DTT-derivatized peptides were eluted by incubation for 1 h at room temperature with 50 μl of 20 mm DTT in PBS/EDTA. Peptides were desalted, dried, and resuspended for LC-MS/MS in the presence of 10 mm DTT.
Reversed Phase HPLC and Mass Spectrometry
Mixtures of standard protein digests were separated and analyzed with a Series 1100 HPLC system (Hewlett Packard) in-line with an LCQ ion trap mass spectrometer (ThermoFinnigan). Peptides were separated on a 300-μm × 10-cm C18 column (Vydac, Grace) with a gradient of 2−60% B in 120 min, 60−98% B in 40 min, and 98% B for 10 min with 0.1% acetic acid and 0.005% heptafluorobutyric acid in water as solvent A or in 60% acetonitrile as solvent B at a flow rate of 8 μl/min. For recovery experiments each sample was subjected to at least three separate LC-MS/MS analyses using the data-dependent top three experiment or the neutral loss methodology described below.
IRS-1 peptides were separated on a 75-μm × 15-cm C18 column (Vydac, Grace) with a gradient of 5−60% B in 110 min, 60−95% B in 25 min, and 98% B for 15 min (B = 0.02% heptafluorobutyric acid in acetonitrile) at 180 nl/min with an Ultimate 2D HPLC system (LC Packings) and mass-analyzed with an LTQ ion trap mass spectrometer (ThermoFinnigan). For the analysis of DTT-derivatized peptides of IRS-1, the mass spectrometer was operated in data-dependent mode with one survey MS scan followed by five MS/MS scans on the five most intense ions. MS/MS data were collected on precursor ions ±1.5 Da that were above a threshold of 100 using 35% collision energy and 30-ms ion activation. Dynamic exclusion was enabled with a repeat count of two and a repeat duration of 30 s. Peptides were also analyzed by MALDI MS (Voyager DE-STR) using α-cyano-4-hydroxycinnamic acid matrix in 70% acetonitrile and 0.1% TFA. The instrument was calibrated using external calibration.
Data-dependent Neutral Loss
To identify O-GlcNAc-modified peptides using neutral loss technology, the mass spectrometer was programmed to collect MS/MS/MS (MS3) data on precursor ions that exhibited a loss upon MS/MS corresponding to N-acetylglucosamine (203.2, 101.6, 67.7, or 51 ± 0.5 Da from different charge state peptides). The experiment consisted of a survey MS scan followed by three MS/MS scans. When a neutral loss ion was detected in the MS/MS scan and it was among the top five most intense ions, a MS3 scan event was triggered. The mass range selected for the survey scan was 600−1800 m/z. The MS/MS parameters used are described above.
Data Analysis
Tandem mass spectra were evaluated by SEQUEST (31) supplied as part of BioWorks 3.1 SR1 (ThermoFinnigan). Searches were performed against the rat IRS-1 protein sequence (Swiss-Prot accession number P35570) with the enzyme specified and allowing for one missed cleavage with the exception of chymotrypsin digests where no enzyme was specified. For DTT-derivatized peptides, the SEQUEST parameters included differential modification of Ser and Thr residues (+136.2 Da) and static modifications accounting for oxidation at Cys (+48 Da), Met (+32 Da), and Trp (+16, 32, or 48 Da). The MS/MS spectra of peaks observed following BEMAD derivatization and thiol chromatography that did not correspond to predicted DTT-derivatized peptides of IRS-1 were searched for incomplete proteolysis, trypsin autolysis, incomplete oxidation of Cys-containing peptides, and products of acid hydrolysis. For data-dependent neutral loss experiments the parameters included carboxyamidomethylated Cys (+57 Da) specified as a static modification, differential phosphorylation of Ser/Thr (+80 Da), and differential O-GlcNAc modification of Ser/Thr (+203.2 Da). SEQUEST results were filtered by Xcorr scores of 1.0, 2.0, and 2.5 for 1+,2+, and 3+ charge state peptides, respectively. Tandem mass spectra of O-GlcNAc-modified, phosphorylated, or DTT-derivatized peptides identified by SEQUEST were examined manually, and the assignments of the sites of modification were based on comparisons between the tandem mass spectra and predicted fragments generated by Sherpa 4.0 (32) or the MS-Product component of ProteinProspector (prospector.ucsf.edu).
Estimation of O-GlcNAc Modification Stoichiometry
Changes in the level of O-GlcNAc modification of the tryptic peptide 1027−1073 under various treatment conditions were assessed by calculating the area under the peak of the extracted ion chromatograms corresponding to the 2+ and 3+ charge states of the unmodified peptide and the O-GlcNAc-modified peptide (33). The peak areas were manually selected from unsmoothed extracted ion chromatograms using the XCalibur Qual Browser software (ThermoFinnigan). The values reported represent the ion intensity of the O-GlcNAc-modified peptide as a percentage of the total ion intensity for the modified and unmodified peptide. The average percent modification ± S.E. was determined from multiple analyses where n equals the number of LC-MS/MS analyses. Because the effect of O-GlcNAc modification on the ionization efficiency of this peptide is not known this semiquantitative analysis may not reflect absolute quantification but is useful for comparison purposes.
Immunoblotting
Wild type and mutant IRS-1 proteins, enriched by nickel affinity chromatography, were boiled under reducing conditions in XT MOPS sample buffer (Bio-Rad) and resolved by SDS-PAGE. Proteins were transferred to nitrocellulose and immunoblotted with a monoclonal anti-O-GlcNAc 110.6 antibody (34) according to the manufacturer's protocol (Pierce). Blots were stripped and reprobed with the S-protein conjugated to HRP (Novagen).
RESULTS
Detection of an O-GlcNAc-modified Peptide by Data-dependent Neutral Loss MS3
To test and optimize the neutral loss MS3 methodology for the identification of O-GlcNAc-modified peptides, preliminary studies were conducted using a synthetic O-GlcNAc-modified peptide, PSVPVS(O-GlcNAc)GSAPGR. Mixtures of protein standards were spiked with the O-GlcNAcylated peptide at varying molar ratios ranging from 1:1 to 1:50. Trypsin-digested samples were analyzed by LC-MS/MS using data-dependent neutral loss MS3 mass spectrometry. CID of the synthetic O-GlcNAc-modified peptide at m/z 657.5 (calculated molecular mass, 1313.7 Da; observed molecular mass, 1313.0 Da) resulted in characteristic loss of N-acetylglucosamine (203.2 Da) yielding the doubly charged deglycosylated peptide at m/z 555.9 (calculated molecular mass, 1109.2 Da; observed molecular mass, 1109.8 Da) as the predominant ion in the tandem mass spectrum. The N-acetylglucosamine oxonium ion at 203.8 Da was also observed; the CID spectrum of this peptide has been shown previously (16). In the LC-MS/MS analysis of a 1:1 molar ratio of this peptide to each protein in the mixture, a scenario that mimics 100% modification of one peptide from a tryptic digestion of a ∼100-kDa protein, the O-GlcNAc-modified peptide was detected by the neutral loss method. Neutral loss of 101.6 Da from the doubly charged ion at m/z 657.5−555.9 triggered the acquisition of a MS3 spectrum providing further sequence confirmation and facilitating detection of the O-GlcNAc-modified peptide. However, in the presence of 10-fold molar excess of protein standards to the peptide, the O-GlcNAc peptide co-eluted with more abundant peptides, was not selected for MS/MS, and therefore was not detected by the neutral loss methodology. These studies confirmed that, although the neutral loss technology was amenable to detection and identification of O-GlcNAc-modified peptides, it did not provide information of the site of modification and had a limited dynamic range of detection.
Recovery and Analysis of an O-GlcNAc-modified Peptide following Chemical Derivatization
To address the limited dynamic range of detection of an O-GlcNAc-modified peptide in a mixture and the inability to discern the exact site of modification, a chemical derivatization strategy in which the O-GlcNAc moiety was replaced with DTT (BEMAD) was tested (21). The protein mixtures, spiked with the synthetic peptide, were oxidized with PFA vapor and digested with trypsin. Because the alkaline conditions of β-elimination resulted in partial derivatization of phosphopeptide standards, peptides were treated with alkaline phosphatase to increase the specificity of the reaction for O-GlcNAcylated peptides. Based on the previous observations and suggestions of McLachlin and Chait (30) with respect to the presence of contaminant peaks following β-elimination/derivatization and thiol chromatography, a second step of performic acid oxidation following enzymatic digestion was performed. This resulted in less background contamination with thiol-reactive products of trypsin autolysis. Upon β-elimination and derivatization, the mass of the O-GlcNAc-modified peptide shifted from 1313.7 to 1247.4 Da due to the loss of N-acetylglucosamine (203.2 Da) and the addition of DTT (136.2 Da) as described previously (21). LC-MS/MS analysis of the eluate from the thiol column revealed chromatographically resolved peaks detected at m/z 623.6 that had identical fragmentation patterns, consistent with the expected presence of isomeric forms of the derivatized peptide. A series of y ions, including the underivatized y6 ion and the DTT-derivatized y7–y11 ions, permitted mapping the site of derivatization. Using this approach, the derivatized peptide was recovered and sequenced from a mixture of protein standards at a molar ratio of 1:50 peptide to each protein in the mixture, analogous to an abundance of 2% O-GlcNAc modification at one site of a 100-kDa protein. This was the highest ratio tested. Enrichment and sequence analysis by tandem mass spectrometry enabled confirmation of the site of derivatization, PSVPVS(DTT)GSAPGR.
O-GlcNAc Modification of IRS-1 Identified by Neutral Loss MS3 Mass Spectrometry
Analysis of peptide digests resulting from proteolysis of IRS-1 with trypsin, Lys-C, chymotrypsin, or Glu-C by neutral loss MS3 mass spectrometry revealed a C-terminal peptide as a major site of O-GlcNAc modification. During the analysis of tryptic peptides, a MS3 scan event was triggered when dissociation of a triply charged ion at m/z 1510.3 generated a fragment ion at m/z 1442.6. The signal at m/z 1510.3 corresponds to IRS-1 residues 1027−1073 modified with O-GlcNAc, and the fragment ion at m/z 1442.6 corresponds to this peptide following dissociation of the monosaccharide. The elution profiles, mass spectrum, and MS/MS spectra confirming the identity of this peptide as residues 1027−1073 with and without O-GlcNAc modification are shown in Fig. 1. The observed molecular masses of the unmodified and O-GlcNAc-modified peptides are 4324.8 and 4527.6 Da, respectively (calculated molecular masses, 4323.7 and 4526.9 Da, respectively). The increase in mass of the O-GlcNAc-modified peptide, 202.8 Da, corresponds to the calculated average mass (203.2 Da) of N-acetylhexosamine. Analysis of Lys-C-digested IRS-1 by neutral loss MS3 scanning aided in the detection of the O-GlcNAc-modified residues 1022−1085 (calculated molecular mass, 6370.0 Da; observed molecular mass, 6371.2 Da). The neutral loss of 51 Da from the quadruply charged O-GlcNAc-modified peptide at m/z 1593.8 to the deglycosylated peptide at m/z 1542.8 triggered the acquisition of a MS3 spectrum (data not shown). Unlike trypsin and Lys-C digestion, proteolytic cleavage of IRS-1 with Glu-C resulted in an O-GlcNAc-modified peptide spanning residues 1021−1051 that partially retained N-acetylglucosamine upon CID. This permitted the correct assignment of the O-GlcNAc-modified peptide by SEQUEST; however, due to the complexity of the spectrum a low XCorr value of 1.8 was obtained and typically would not be observed when the results were filtered by the XCorr score. Fragmentation of the doubly charged O-GlcNAc-modified peptide 1021−1051 at m/z 1034.4 generated the neutral loss ion (m/z 966.1) as the second most abundant ion in the MS/MS spectrum triggering the acquisition of MS3. The fragmentation pattern was consistent with the presence of O-GlcNAc modification within residues 1032−1042 (PPPSSTASASA) (Supplemental Fig. 1). Analysis of chymotrypsin-digested IRS-1 peptide 1029−1055 by neutral loss MS3 methodology further confirmed O-GlcNAc modification within this C-terminal region of IRS-1. Under each digestion condition, the neutral loss methodology consistently detected IRS-1 peptides containing residues 1032−1042. See Table I for the calculated and observed masses of the O-GlcNAc-modified peptides detected under each digestion condition.
Fig. 1. LC-MS/MS of trypsin-digested IRS-1 peptide 1027−1073 with and without O-GlcNAc modification.
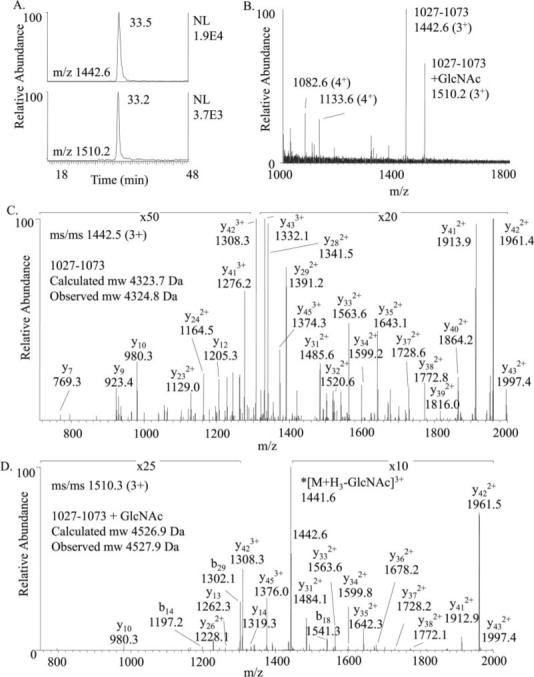
A, extracted ion chromatograms showing the elution profiles of residues 1027−1073 with (m/z 1510.2) and without (m/z 1442.6) O-GlcNAc modification. B, mass spectrum acquired at 33−33.2 min. C, MS/MS of precursor at m/z 1442.5 confirming the identity of the peptide 1027−1073 (TTGAAPPPSSTASASASVTPQGAAEQAAHSSLLGGPQGPGGMSAFTR). D, MS/MS of O-GlcNAcylated precursor at m/z 1510.3 confirmed the sequence of 1027−1073 and shows loss of the monosaccharide upon CID. Generation of the neutral loss ion at 1442.6 triggered MS3 and facilitated detection of this peptide. The expected mass shift due to O-GlcNAc modification is 203.2 Da. Fragment ions in both MS/MS spectra are labeled according the predicted b and y ions of the unmodified peptide.
Table I.
Observed O-GlcNAc-modified and phosphorylated peptides of IRS-1 residues 1021−1085 identified by data-dependent neutral loss MS3 under various proteolytic digestion conditions Partial or complete loss of the N-acetylglucosamine upon CID permitted detection of these O-GlcNAc-modified peptides by data-dependent neutral loss. The expected increases in mass due to O-GlcNAc modification (GlcNAc) and phosphorylation (P) are 203.2 and 80 Da. The sequence of each peptide was confirmed by MS/MS, and the retention times are given in parentheses. ND, not detected.
| Residues | [M + H]+ |
Observed [M + GlcNAc]+ | Observed [M + P]+ | Observed [M + GlcNAc + P]+ | ||
|---|---|---|---|---|---|---|
| CalcAve | Observed | |||||
| Trypsin | 1027−1073 | 4324.7 | 4325.0 (33.5 min) | 4527.5 (33.2 min) | ND | ND |
| Glu-C | 1021−1051 | 2896.2 | 2895.8 (30.7 min) | 3098.8 (30.1 min) | ND | ND |
| Lys-C | 1022−1085 | 6167.8 | 6167.8 (44.2 min) | 6372.6 (44.2 min) | 6247.8 (43.6 min) | 6457.0 (43.6 min) |
| Chymotrypsin | 1029−1055 | 2420.5 | 2420.6 (24.6 min) | 2623.4 23.9 min) | ND | ND |
| Chymotrypsin | 1039−1055 | 1583.6 | ND | ND | 1665.2 (19.3 min) | ND |
Changes in the Level of IRS-1 O-GlcNAc Modification with High Glucose and Insulin
The extent of O-GlcNAc modification of residues 1027−1073 increased when IRS-1-transfected cells were grown in the presence of high glucose, chronic insulin, or the O-GlcNAcase inhibitor PUGNAc. Semiquantitative analysis, based on the peak areas of extracted ion chromatograms of the 2+ and 3+ charge states of the peptide 1027−1073 with and without O-GlcNAc modification, indicated that under basal conditions (5 mm glucose) 6.0 ± 0.8% (n = 3) of the peptide was O-GlcNAc-modified. Following an 18-h incubation in 25 mm glucose in the presence or absence of 100 nm insulin, the level of O-GlcNAc modification increased to 12.8 ± 0.8% (n = 2) or 9.6 ± 0.9% (n = 3), respectively. Treatment of the cells with PUGNAc increased the extent of modification to 13.7 ± 0.6% (n = 7). These trends were consistent with the levels of total protein O-GlcNAc modification of intact recombinant IRS-1 as determined by immunoblot analysis with the anti-O-GlcNAc antibody (data not shown). The level of modification induced by treatment with PUGNAc consistently permitted detection by neutral loss MS3 methodology, whereas under basal conditions the O-GlcNAc-modified peptide was present but was selected for MS/MS and detected by the neutral loss MS3 methodology only once of six analyses.
Sites of Phosphorylation within Residues 1021−1085
Because O-GlcNAcylation has been proposed to serve as a reciprocal means of protein regulation to phosphorylation at identical or adjacent residues, the phosphorylation state of residues 1021−1085 was assessed. The neutral loss methodology aided in the detection of the phosphorylated and O-GlcNAcylated peptide 1022−1085 generated by Lys-C digestion. Fragmentation of the precursor ion at m/z 1615.0 to the deglycosylated fragment ion at m/z 1562.0 triggered the acquisition of a MS3 scan. Although Ser/Thr phosphopeptides may undergo neutral loss of phosphoric acid (98 Da) during CID, the loss of the monosaccharide generated the predominant fragment ion in the spectrum. Based on the MS/MS spectrum of precursor ion at m/z 1615.0 (4+), the site of phosphorylation was narrowed down to residues Ser-1072, Ser-1077, or Ser-1083 (data not shown). MS/MS of the phosphorylated (nonglycosylated) form of peptide 1022−1085 at m/z 1562.7 (4+) confirmed phosphorylation at Ser-1077 (calculated molecular mass, 6246.7 Da; observed molecular mass, 6244.8 Da), a recently reported site of phosphorylation (35). LC-MS/MS analysis of chymotrypsin-digested IRS-1 residues 1039−1055 revealed a novel site of phosphorylation at Thr-1045 (Fig. 2). The calculated and observed molecular masses of phosphorylated 1039−1055 are 1662.6 and 1664.2 Da, respectively. The major fragment ion in the CID spectrum, at m/z 783.4, corresponds to the loss of phosphoric acid from the precursor ion consistent with the assignment of this modification as phosphorylation rather than sulfonation (26). The signal intensities of these phosphopeptides were not sufficient for semiquantitative analysis of the relative level of phosphorylation under the various treatment conditions, e.g. treatment with PUGNAc or high glucose plus insulin.
Fig. 2. LC-MS/MS of phosphopeptide 1039−1055 (SASASVpTPQGAAEQAAH where pT is phosphothreonine).
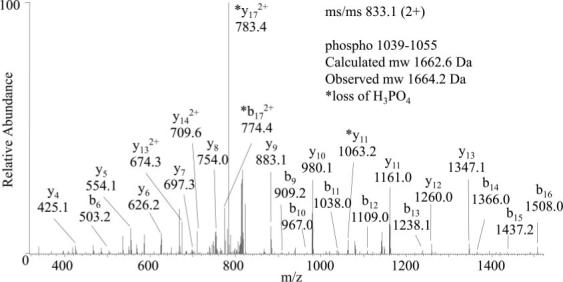
MS/MS of the doubly charged precursor ion at m/z 833.1 revealed Thr-1045 as the site of phosphorylation. The asterisks indicate ions that exhibited neutral loss of phosphoric acid (98 Da).
No Other Sites of O-GlcNAc Modification Detected by the Neutral Loss Methodology
During each LC-MS/MS analysis the neutral loss MS3 scan was triggered between five and 17 times permitting the acquisition of tandem mass spectra of other peptides of IRS-1. This yielded 78.2, 21, 24, or 42% sequence coverage following trypsin, Lys-C, Glu-C, or chymotrypsin digestion, respectively. In total 84% of the IRS-1 amino acid sequence was identified by tandem mass spec-trometry. The only true positive hits detected by the neutral loss methodology corresponded to those peptides containing residue 1036 that were shifted in mass by 203.2 Da and underwent a neutral loss of 203.2 Da consistent with O-GlcNAc modification. One scenario of an unmodified peptide that consistently triggered the MS3 was that of a peptide that began with the sequence Ser-Ile-Pro where preferential fragmentation N-terminal to proline generated a y ion 200 Da less than the precursor ion as the most intense ion in the spectrum. A similar situation was observed due to the generation of a y ion that lost 202 Da upon dissociation of two N-terminal Thr residues. Other false positive hits were disregarded based on discrepancies between the charge state indicated by the neutral loss and the charge state of a predicted IRS-1 peptide.
Mapping the Site of O-GlcNAc Modification by Chemical Derivatization of IRS-1
To expand the dynamic range of detection of O-GlcNAc-modified peptides and to map the exact site of modification within residues 1032−1042, IRS-1 peptides were subjected to β-elimination and derivatization with DTT. Gel-purified IRS-1 isolated from PUGNAc-treated cells was oxidized by performic acid, proteolytically digested, and treated with alkaline phosphatase or hexosaminidase. DTT-derivatized peptides purified by thiol chromatography were analyzed by MALDI MS and LC-MS/MS. The MALDI mass spectra of phosphatase-treated oxidized, tryptic peptides of IRS-1 before and after derivatization and thiol chromatography are shown in Fig. 3. Before derivatization, the oxidized forms of the tryptic peptide 1027−1073 (calculated molecular mass, 4355.7 Da; observed molecular mass, 4353.4 Da) and O-GlcNAcylated 1027−1073 (calculated molecular mass, 4558.9; observed molecular mass, 4555.6 Da) were observed. After derivatization, the signal at m/z 4556.6 was absent, and a new signal at m/z 4493.0 was detected that corresponds to DTT-derivatized 1027−1073 (calculated molecular mass, 4492.9 Da). The 136.2-Da shift corresponds to the expected shift in mass (136.2 Da) due to derivatization of serine or threonine with DTT. This DTT-derivatized peptide was enriched by the thiol chromatography (Fig. 3). LC-MS/MS of the DTT-derivatized peptide 1027−1073 at m/z 1498.0 (3+) confirmed the assignment of the peptide and revealed the site of modification at Ser-1036 (Fig. 4). A series of doubly charged y ions and the presence of the doubly charged y38 ion at m/z 1856.2 are consistent with the presence of DTT at residue 1036. This assignment was further confirmed by MS/MS of the DTT-modified peptides 1023−1055 (calculated molecular mass, 3211.5 Da; observed molecular mass, 3210.0 Da) and 1029−1055 (calculated molecular mass, 2555.8 Da; observed molecular mass, 2554.2 Da) generated after chymotrypsin digestion. Modification at residue 1036 of the peptide 1029−1055 (GAAPPPSSTASASASVTPQGAAEQAAH) was confirmed by tandem mass spectrometry based on the presence of the b7 ion without DTT at m/z 578.3, the b8 ion with DTT at m/z 801.4, the y19 ion without DTT at m/z 1754.6, and the y20 ion with DTT at m/z 1977.8 (data not shown).
Fig. 3. MALDI-TOF MS of IRS-1 tryptic peptides before and after β-elimination/derivatization with DTT and after thiol chromatography.
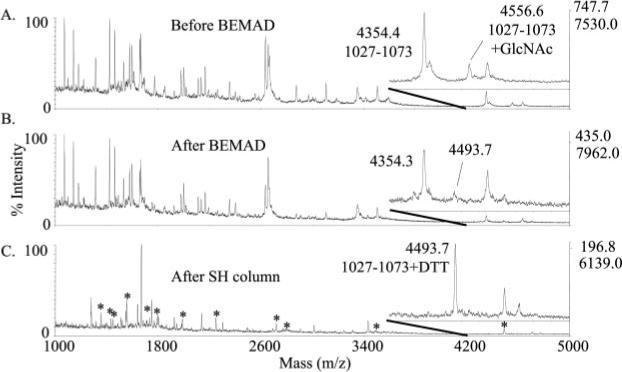
The insets highlight mass range 4200−5000. A, trypsin digest of oxidized, alkaline phosphatase-treated IRS-1. The average calculated [M + H]+ of the oxidized peptide 1027−1073 with and without O-GlcNAc modification is 4559.8 and 4356.7, respectively. B, IRS-1 peptides following β-elimination and Michael addition with DTT. The average calculated [M + H]+ of oxidized, DTT-derivatized 1027−1073 is 4492.0. C, sulfhydryl-reactive peptides purified by thiol chromatography. Masses that correspond to predicted DTT-derivatized peptides of IRS-1 are indicated by asterisks and are listed in Table II. The expected increases in mass due to modification by N-acetylglucosamine or derivatization with DTT are 203.2 and 136.2 Da, respectively.
Fig. 4. MS/MS of the DTT-derivatized peptide 1027−1073 precursor ion at m/z 1498.0 (3+).
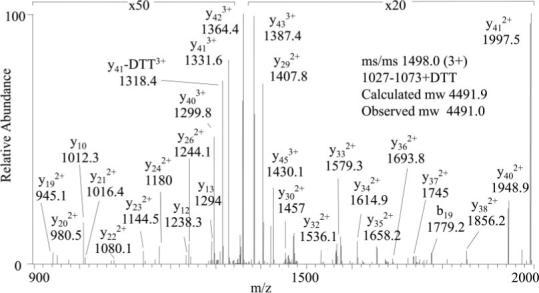
The fragmentation pattern is consistent with DTT modification at Ser-1036 (TTGAAPPPSSTASASASVTPQGAAEQAAHSSLLGGPQGPGGMSAFTR).
There were other DTT-derivatized peptides of IRS-1 observed by MALDI MS and confirmed by MS/MS (Fig. 3, indicated by asterisks). However, comparison of MALDI MS spectra following pretreatment of IRS-1 with alkaline phosphatase or hexosaminidase did not readily discriminate whether these peptides were phosphorylated or O-GlcNAc-modified. This could be due to O-GlcNAc modification and phosphorylation occurring on the same peptide as was seen with peptide 1027−1073 or to inefficient digestion with alkaline phosphatase. Most of the other DTT-derivatized peptides of IRS-1 observed by MALDI MS (indicated in Fig. 3) contain known sites of phosphorylation (residues 307, 343, 570, 612, 632, 1077 and 1099 or 1100 and 1215 or 1216) (Table II). Phosphorylation at all of these sites was observed in LC-MS/MS analyses of IRS-1 digests prior to DTT derivatization suggesting incomplete removal of phosphates by alkaline phosphatase. Three DTT-derivatized IRS-1 peptides identified by LC-MS/MS were at sites that have not been shown previously to be posttranslationally modified, residues 469, 637, and 972 (Table II). Retrospective searches of MS/MS data and LC-MS/MS analyses with mass lists including the predicted masses of the corresponding putative O-GlcNAc-modified peptides did not yield any further evidence that these DTT-derivatized peptides had been O-GlcNAc-modified previously. Thus, the nature of the modification of these three peptides prior to DTT derivatization is unresolved.
Table II.
The calculated and observed [M + H]+ of DTT-derivatized tryptic peptides of oxidized IRS-1 detected by MALDI MS (Fig. 3, lower panel) LC-MS/MS confirmed the sequence of these peptides and revealed the site of DTT derivatization (indicated by DTT followed by the residue number). LC-MS/MS analyses prior to β-elimination and DTT derivatization revealed these sites were phosphorylated before β-elimination (indicated by p followed by the residue number). References are given in parentheses. The proven site of O-GlcNAc modification is shown in bold. (R) or (K) indicates the N-terminal site of cleavage by trypsin.
| DTT-modified peptides of IRS-1 identified by MALDI MS | [M + DTT]+ |
Modification confirmed by MS/MS | |
|---|---|---|---|
| Calculated | Observed | ||
| 300−320, (R)TESITATSPASMVGGKPGSFR | 2250.5 | 2251.5 | p307a (38) |
| 323−348, (R)ASSDGEGTMSRPASVDGSPVSPSTNR | 2718.9 | 2720.6 | p343a (35) |
| 467−482, (K)GASTLTAPNGHYILSR | 1795.0 | 1796.6 | DTT469 |
| 569−590, (R)HSAFVPTHSYPEEGLEMHHLER | 2772.4 | 2772.7 | DTT570a (39) |
| 592−622, (R)GGHHRPDSSNLHTDDGYMPMSPGVAPVPSNR | 3487.8 | 3489.1 | p612a (40) |
| 624−634, (K)GNGDYMPMSPK | 1399.4 | 1397.6 | p632a (40) |
| 635−647, (K)SVSAPQQIINPIR | 1559.8 | 1557.5 | DTT637 |
| 963−978, (R)VGPAPPGAASICRPTR | 1735.0 | 1734.6 | DTT972 |
| 1027−1073, (R)TTGAAPPPSSTASASASVTPQGAAEQAAHSSLLGGPQGPGGMSAFTR | 4493.9 | 4493.7 | DTT1036 |
| 1074−1085, (R)VNLSPNHNQSAK | 1445.6 | 1450.5 | p1077a (35) |
| 1098−1108, (R)HSSETFSAPTR | 1356.5 | 1356.4 | DTT1099/1100a (41) |
| 1214−1229, (R)RSSEDLSTYASINFQK | 1983.2 | 1984.3 | DTT1215/1216a (35) |
Previously identified sites of phosphorylation.
Loss of O-GlcNAc Modification of IRS-1 following Site-directed Mutagenesis at Ser-1035, Ser-1036, and Thr-1037
To address the possibility that the major site of IRS-1 O-GlcNAc modification was within residues Ser-1035, Ser-1036, and Thr-1037, these residues were simultaneously substituted with alanine, and the mutated IRS-1 protein was analyzed by immunoblot analysis. The wild type and mutated IRS-1 proteins were isolated from transfected HEK293 cells that had been treated with PUGNAc to increase the level of O-GlcNAc modification. Immunoblot analysis with a monoclonal anti-O-GlcNAc antibody, which specifically recognizes O-linked β-N-acetylglucosamine monosaccharide, demonstrated a marked decrease in O-GlcNAc modification of the IRS-1 mutant (IRS-1 AAA) as compared with the wild type protein (Fig. 5). The total amount of protein loaded was assessed using S-protein-HRP, which recognizes the N-terminal S-tag fused to IRS-1. These data suggest that this region of IRS-1 contains the major site of O-GlcNAc modification and provide further evidence for the identification of the monosaccharide as N-acetylglucosamine.
Fig. 5. O-GlcNAc modification of recombinant wild type and mutant IRS-1 (IRS-1 AAA).

Wild type (wt) or mutant (AAA) IRS-1 proteins were expressed in HEK293 cells treated for 18 h with 50 μm PUGNAc to increase the level of protein O-GlcNAc modification. Nickel affinity-purified protein was separated by SDS-PAGE and transferred to nitrocellulose. The presence of O-GlcNAc modification was assessed by probing with an anti-O-GlcNAc antibody. Total IRS-1 protein loaded was assessed by probing the stripped blot with the S-protein-HRP conjugate. WB, Western blot.
DISCUSSION
Based on the Yin-yang2 algorithm for the prediction of sites of protein O-GlcNAc modification, among the 181 serine and 63 threonine residues in IRS-1, 65 Ser/Thr residues were predicted to be potential sites of O-GlcNAc modification with residues 1035, 1036, and 1037 given a high probability of modification. Thirty-five of the predicted O-GlcNAc sites are also known or predicted sites of phosphorylation. With the preponderance of possible sites of modification, two complementary mass spectrometric methods for the detection of O-GlcNAc modification were tested, and multiple proteases were used to maximize sequence coverage of IRS-1. To the best of our knowledge this is the first demonstration of the use of data-dependent neutral loss MS3 technology using an ion trap mass spectrometer for the detection of a novel site of O-GlcNAc modification within a protein. The neutral loss technology was advantageous in that it could be implemented during a typical LC-MS/MS acquisition. Therefore, in addition to selecting O-GlcNAc-modified peptides that lose 203.2 Da upon fragmentation for further sequencing and identification, CID spectra were collected on other peptides in the mixture permitting identification of other sites of posttranslational modification. The facile loss of the N-acetylglucosamine and the detection of primarily y ions of the trypsin-digested O-GlcNAcylated peptide prevented identification of the exact site of modification, whereas the Glu-C-digested peptide partially retained the monosaccharide. This permitted detection of the O-GlcNAcylated peptide by the neutral loss methodology and revealed that the site of modification was among residues 1032−1042. The BEMAD approach, although requiring more starting material and careful controls, permitted assignment of the exact site of O-GlcNAc modification.
Mass spectrometric and immunoblot analyses of wild type and mutant IRS-1 provided evidence for a novel site of O-GlcNAc modification at Ser-1036. Under each proteolytic digestion condition, the peptides containing residue 1036 were shifted in molecular mass by 203.2 Da and underwent a neutral loss of 203.2 Da, consistent with the presence of N-acetylglucosamine. Upon incubation of IRS-1 with β-N-acetylhexosaminidase the level of O-GlcNAc modification of residues 1027−1073 was reduced from 13.7 to 3%. Because neither the mass shift and neutral loss of 203.2 Da nor the enzymatic removal with N-acetylhexosaminidase discriminates between a GlcNAc or N-acetylgalactosamine, the anti-O-GlcNAc antibody, which specifically recognizes β-O-glycosidic linkage of N-acetylglucosamine to serine and threonine (34), confirmed the identity of the monosaccharide. The loss of immunoreactivity of the anti-O-GlcNAc antibody toward mutated IRS-1 confirmed the presence of the O-GlcNAc modification within residues 1035, 1036, and 1037. β-Elimination and derivatization with DTT prior to mass spectrometric analysis revealed the site of modification as Ser-1036. This assignment was based on tandem mass spectra of the DTT-derivatized peptides acquired after proteolysis with trypsin or chymotrypsin and treatment with alkaline phosphatase.
Based on the consistent detection of peptides spanning residue Ser-1036 as the only peptides that demonstrated a neutral loss corresponding to N-acetylglucosamine and the immunoblotting results of the IRS-1 mutant, Ser-1036 appears to be the major site of O-GlcNAc modification. This serine residue is conserved in mouse, rat, and human IRS-1. As with other proteins, O-GlcNAcylation may offer an alternative means of protein regulation to phosphorylation. Although Ser-1036 is a predicted site of phosphorylation by Cdc2 kinase (NetPhosK1.0) and Ser-1035 is a predicted site for phosphorylation by glycogen synthase 3 kinase (Motif Scan), phosphorylation at these sites was not detected under the treatment conditions used. However, phosphorylation of the O-GlcNAc-modified peptide was observed at other sites. A novel site of phosphorylation was observed at residue Thr-1045, and a previously reported site of phosphorylation was confirmed at Ser-1077. The functional significance of phosphorylation at these sites is not known. Interestingly the BEMAD approach also revealed sites of IRS-1 phosphorylation that are known to attenuate insulin signaling. Further application of the BEMAD chemistry utilizing isotope-coded DTT has been demonstrated for quantitative analysis of O-GlcNAc modification and phosphorylation (36). This would permit a quantitative assessment of site-specific changes in the extent of O-GlcNAc modification and phosphorylation of IRS-1.
The level of O-GlcNAc modification of the tryptic peptide 1027−1073 increased under conditions that model the diabetic state. The highest level of modification was observed following treatment of the cells with the O-GlcNAcase inhibitor PUGNAc or with high glucose and chronic insulin stimulation. In insulin-sensitive cell types, increased O-GlcNAc modification of IRS-1 observed under these conditions correlates with the development of insulin resistance to glucose transport. However, the direct effect of O-GlcNAc modification on the interaction of IRS-1 with binding partners is not known. The N-terminal phosphotyrosine binding domain of IRS-1 interacts with the insulin receptor kinase, whereas the phosphotyrosines within the C terminus of IRS-1 recruit downstream effectors including phosphatidylinositol 3-kinase, SHP-2, GRB2, NCK, CRK, and SHB (14). These proteins are involved in mediating and regulating the metabolic responses to insulin and activating the mitogen-activated protein kinase pathway (37). The site of O-GlcNAc modification occurs between the GRB2 binding site at phosphotyrosine at 895 and the two phosphotyrosine motifs at residues 1172 and 1222 that interact with SHP-2. Within this region of IRS-1 there are five other potential sites of tyrosine phosphorylation at residues 907, 939, 987, 999, and 1010, some of which cluster into known binding motifs (14). Identification of the major site of O-GlcNAc modification of IRS-1 will facilitate the investigation of the functional effects of this modification on signaling through IRS-1.
Acknowledgments
The data were acquired by L. E. B. on instrumentation maintained by the Biomolecular Mass Spectrometry Facility at the Medical University of South Carolina. We thank Dr. G. W. Hart and Dr. M. F. White for generously providing the O-GlcNAc standard peptide and the IRS-1-containing plasmid, respectively. We also thank Dr. L. Wells and Dr. K. Vosseller for helpful advice in developing the BEMAD procedure. We thank Katy A. Robinson for expert assistance in tissue culture.
Footnotes
The abbreviations used are: GlcNAc, β-N-acetylglucosamine; IRS-1, insulin receptor substrate-1; BEMAD, β-elimination followed by Michael addition with DTT; PFA, performic acid; PUGNAc, O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate; HRP, horseradish peroxidase; HEK, human kidney embryonic.
This work was supported in part by National Institutes of Health Grant DK02001 (to M. G. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
R. Gupta, J. Hansen, and S. Brunak, manuscript in preparation.
Supplementary Material
REFERENCES
- 1.Hart GW. Dynamic O-linked glycosylation of nuclear and cytoskeletal proteins. Annu. Rev. Biochem. 1997;66:315–335. doi: 10.1146/annurev.biochem.66.1.315. [DOI] [PubMed] [Google Scholar]
- 2.Comer FI, Hart GW. Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry. 2001;40:7845–7852. doi: 10.1021/bi0027480. [DOI] [PubMed] [Google Scholar]
- 3.Slawson C, Hart GW. Dynamic interplay between O-GlcNAc and O-phosphate: the sweet side of protein regulation. Curr. Opin. Struct. Biol. 2003;13:631–636. doi: 10.1016/j.sbi.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Iyer SP, Hart GW. Dynamic nuclear and cytoplasmic glycosylation: enzymes of O-GlcNAc cycling. Biochemistry. 2003;42:2493–2499. doi: 10.1021/bi020685a. [DOI] [PubMed] [Google Scholar]
- 5.Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991;266:4706–4712. [PubMed] [Google Scholar]
- 7.McClain DA. Hexosamines as mediators of nutrient sensing and regulation in diabetes. J. Diabetes Complicat. 2002;16:72–80. doi: 10.1016/s1056-8727(01)00188-x. [DOI] [PubMed] [Google Scholar]
- 8.Ludemann N, Clement A, Hans VH, Leschik J, Behl C, Brandt R. O-Glycosylation of the tail domain of neurofilament protein M in human neurons and in spinal cord tissue of a rat model of amyotrophic lateral sclerosis (ALS). J. Biol. Chem. 2005;280:31648–31658. doi: 10.1074/jbc.M504395200. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH. Adenovirus-mediated overexpression of O-Glc-NAcase improves contractile function in the diabetic heart. Circ. Res. 2005;96:1006–1013. doi: 10.1161/01.RES.0000165478.06813.58. [DOI] [PubMed] [Google Scholar]
- 10.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc. Natl. Acad. Sci. U. S. A. 2002;99:5313–5318. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G, Lauro R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. 2002;106:466–472. doi: 10.1161/01.cir.0000023043.02648.51. [DOI] [PubMed] [Google Scholar]
- 13.Patti ME, Virkamaki A, Landaker EJ, Kahn CR, Yki-Jarvinen H. Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance of early postreceptor insulin signaling events in skeletal muscle. Diabetes. 1999;48:1562–1571. doi: 10.2337/diabetes.48.8.1562. [DOI] [PubMed] [Google Scholar]
- 14.White MF. IRS proteins and the common path to diabetes. Am. J. Physiol. 2002;283:E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 15.Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie (Paris) 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Haynes PA, Aebersold R. Simultaneous detection and identification of O-GlcNAc-modified glycoproteins using liquid chromatography-tandem mass spectrometry. Anal. Chem. 2000;72:5402–5410. doi: 10.1021/ac000512w. [DOI] [PubMed] [Google Scholar]
- 17.Chalkley RJ, Burlingame AL. Identification of novel sites of O-N-acetylglucosamine modification of serum response factor using quadrupole time-of-flight mass spectrometry. Mol. Cell. Proteomics. 2003;2:182–190. doi: 10.1074/mcp.M300027-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Greis KD, Hayes BK, Comer FI, Kirk M, Barnes S, Lowary TL, Hart GW. Selective detection and site-analysis of O-Glc-NAc-modified glycopeptides by β-elimination and tandem electrospray mass spectrometry. Anal. Biochem. 1996;234:38–49. doi: 10.1006/abio.1996.0047. [DOI] [PubMed] [Google Scholar]
- 19.Schroeder J, Webb DJ, Shabanowitz J, Horwitz AF, Hunt DF. Methods for the detection of paxillin post-translational modifications and interacting proteins by mass spectrometry. J. Proteome Res. 2005;4:1832–1841. doi: 10.1021/pr0502020. [DOI] [PubMed] [Google Scholar]
- 20.Gao Y, Miyazaki J, Hart GW. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch. Biochem. Biophys. 2003;415:155–163. doi: 10.1016/s0003-9861(03)00234-0. [DOI] [PubMed] [Google Scholar]
- 21.Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW. Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol. Cell. Proteomics. 2002;1:791–804. doi: 10.1074/mcp.m200048-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.Roquemore EP, Chou TY, Hart GW. Detection of O-linked N-acetylglucosamine (O-GlcNAc) on cytoplasmic and nuclear proteins. Methods Enzymol. 1994;230:443–460. doi: 10.1016/0076-6879(94)30028-3. [DOI] [PubMed] [Google Scholar]
- 23.Sprung R, Nandi A, Chen Y, Kim SC, Barma D, Falck JR, Zhao Y. Tagging-via-substrate strategy for probing O-GlcNAc modified proteins. J. Proteome Res. 2005;4:950–957. doi: 10.1021/pr050033j. [DOI] [PubMed] [Google Scholar]
- 24.Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vocadlo DJ, Hang HC, Kim EJ, Hanover JA, Bertozzi CR. A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9116–9121. doi: 10.1073/pnas.1632821100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medzihradszky KF, Darula Z, Perlson E, Fainzilber M, Chalkley RJ, Ball H, Greenbaum D, Bogyo M, Tyson DR, Bradshaw RA, Burlingame AL. O-Sulfonation of serine and threonine: mass spectrometric detection and characterization of a new posttranslational modification in diverse proteins throughout the eukaryotes. Mol. Cell. Proteomics. 2004;3:429–440. doi: 10.1074/mcp.M300140-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Mirgorodskaya E, Hassan H, Clausen H, Roepstorff P. Mass spectrometric determination of O-glycosylation sites using β-elimination and partial acid hydrolysis. Anal. Chem. 2001;73:1263–1269. doi: 10.1021/ac001288d. [DOI] [PubMed] [Google Scholar]
- 28.Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, Cahill DA, Goldstein BJ, White MF. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352:73–77. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- 29.Haltiwanger RS, Grove K, Philipsberg GA. Modulation of O-linked N-acetylglucosamine levels on nuclear and cytoplasmic proteins in vivo using the peptide O-GlcNAc-β-N-acetylglucosaminidase inhibitor O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate. J. Biol. Chem. 1998;273:3611–3617. doi: 10.1074/jbc.273.6.3611. [DOI] [PubMed] [Google Scholar]
- 30.McLachlin DT, Chait BT. Improved β-elimination-based affinity purification strategy for enrichment of phosphopeptides. Anal. Chem. 2003;75:6826–6836. doi: 10.1021/ac034989u. [DOI] [PubMed] [Google Scholar]
- 31.Eng J, McCormack AL, Yates JR. An approach to correlated tandem mass spectral data of peptides with a sequence in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 32.Taylor JA, Walsh KA, Johnson RS. Sherpa: a Macintosh-based expert system for the interpretation of electrospray ionization LC/MS and MS/MS data from protein digests. Rapid Commun. Mass Spectrom. 1996;10:679–687. doi: 10.1002/(SICI)1097-0231(199604)10:6<679::AID-RCM528>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 33.Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14−3−3 binding. J. Biol. Chem. 2004;279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 34.Comer FI, Vosseller K, Wells L, Accavitti MA, Hart GW. Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anal. Biochem. 2001;293:169–177. doi: 10.1006/abio.2001.5132. [DOI] [PubMed] [Google Scholar]
- 35.Luo M, Reyna S, Wang L, Yi Z, Carroll C, Dong LQ, Langlais P, Weintraub ST, Mandarino LJ. Identification of IRS-1 serine/threonine phosphorylation sites using mass spectrometry analysis: regulatory role of serine 1223. Endocrinology. 2005;146:4410–4416. doi: 10.1210/en.2005-0260. [DOI] [PubMed] [Google Scholar]
- 36.Vosseller K, Hansen KC, Chalkley RJ, Trinidad JC, Wells L, Hart GW, Burlingame AL. Quantitative analysis of both protein expression and serine/threonine post-translational modifications through stable isotope labeling with dithiothreitol. Proteomics. 2005;5:388–398. doi: 10.1002/pmic.200401066. [DOI] [PubMed] [Google Scholar]
- 37.Myers MG, Jr., Mendez R, Shi P, Pierce JH, Rhoads R, White MF. The COOH-terminal tyrosine phosphorylation sites on IRS-1 bind SHP-2 and negatively regulate insulin signaling. J. Biol. Chem. 1998;273:26908–26914. doi: 10.1074/jbc.273.41.26908. [DOI] [PubMed] [Google Scholar]
- 38.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 39.Greene MW, Morrice N, Garofalo RS, Roth RA. Modulation of human insulin receptor substrate-1 tyrosine phosphorylation by protein kinase Cδ. Biochem. J. 2004;378:105–116. doi: 10.1042/BJ20031493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mothe I, Van Obberghen E. Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J. Biol. Chem. 1996;271:11222–11227. doi: 10.1074/jbc.271.19.11222. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Soos TJ, Li X, Wu J, Degennaro M, Sun X, Littman DR, Birnbaum MJ, Polakiewicz RD. Protein kinase C θ inhibits insulin signaling by phosphorylating IRS1 at Ser1101. J. Biol. Chem. 2004;279:45304–45307. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.