

Abstract
The United States Food and Drug Administration recently issued an Exploratory Investigational New Drug Guidance that provides a platform for the evaluation of targeted anticancer agents in small, early phase human clinical trials that can be used to establish the feasibility of proof-of-principle target modulation assays, as well as the preliminary pharmacokinetics, and molecular imaging potential of new anticancer molecules. The exploratory IND allows for reduced requirements for manufacturing and toxicologic assessment. Early clinical trials performed in this fashion have no therapeutic intent. In this series of papers in CCR Focus, the development of this new IND mechanism, its impact on clinical trial design and clinical pharmacodynamics, the ethical implications of non-therapeutic clinical investigations, and the perspective of the pharmaceutical industry on this approach are examined.
Keywords: phase 0, cancer clinical trials, exploratory IND, pharmacodynamics, drug development
Introduction
The focus of oncologic drug discovery has changed markedly over the past five years, based on the initial success of the first generation of molecularly-targeted therapeutic agents and the dramatic increase in our knowledge of the potential signal transduction sites available for therapeutic interference. However, despite this remarkable increase in the breadth of our insights into basic tumor cell biology, the increase in the number of new therapeutic molecules expected to follow from those insights has not fully materialized. Hence, attention has been focused recently on the serious challenges facing oncologic drug development. The leading cause of attrition in clinical trials of novel oncologic therapeutics is now a lack of clinical activity (rather than toxicity); and unfortunately, only five to ten percent of the molecules that are subjects of Investigational New Drug (IND) applications progress beyond early phases of development (1). Furthermore, an increase in development timelines, as well as the lack of predictability of toxicity and effectiveness testing when traditional animal models are employed, have led to increased costs, and risk aversion by a pharmaceutical industry that is undergoing major consolidation (2;3). Oncologic drug development has also been challenged by a paucity of biomarkers that could permit regulatory approvals to advance more rapidly based on validated molecular signals that predict early clinical endpoints (4).
It is remarkable to consider that the basic clinical trials structure for oncologic new drug development, progressing from studies of safety, to efficacy, to comparative therapeutic benefit has been largely unchanged for over thirty-five years (5). Furthermore, despite numerous advances in the biostatistical design of phase I clinical trials over this time frame (6;7), most early phase clinical investigations still do not effectively take advantage of the tools of modern molecular biology to confirm the mechanism(s) of action of the agents under study (8;9).
Despite, or perhaps because of, this broad range of obstacles, there is a pressing need to shorten cancer drug development timelines; to enhance molecular drug discovery; to streamline procedures to assess oncologic drug effects (toxicity, effectiveness, and mechanism of action) very much earlier in the development cycle, thus preventing therapeutic failures during phase III studies; and to provide a rigorous scientific base for a wide range of potential indications for new cancer drugs (10;11). Ultimately, there is a critical requirement to more effectively apply currently-available scientific tools to the cancer drug development process to enhance productivity and innovation, speeding the delivery of new anticancer agents to patients1.
It is in this context that the US Food and Drug Administration (US FDA) issued its call to change the focus of drug development in 2004; the Critical Path Report aimed to provide new opportunities for the translation of basic discovery into novel diagnostic and therapeutic products2. At the same time, the US FDA commenced work on a series of new guidance documents designed to speed the development of novel therapeutic agents; these guidances focused on manufacturing practices for novel small molecules, as well as new approaches to early phase clinical investigations. Working with investigators from academic centers, the US National Cancer Institute (NCI), and the pharmaceutical and biotechnology industries, the FDA promulgated the final form of a new guidance for exploratory IND (ExpIND) studies developed to speed the completion of early phase clinical trials in February of 20063. This document was meant to stimulate earlier human testing of novel imaging agents and therapeutics, as well as the concomitant biomarker discovery that is a critical component of the development of targeted anticancer agents (12).
Over the past two years, there has been substantial discussion regarding the potential value to oncologic therapeutics of the so-called Phase 0 (in contrast to Phase I) clinical trials described in the ExpIND guidance (13–17). However, because the initial first-in-human phase 0 clinical studies performed under the ExpIND are only now coming to fruition, there will be a considerable lag-phase before sufficient data will become available from which to assess the real value of this approach for oncologic drug development (18;19). Thus, while a definitive assessment of this methodology is not currently possible, we are in a position now to examine the progress of the ExpIND approach, and in particular the use of the ExpIND to perform proof-of-principle, pharmacodynamically-driven Phase 0 trials, as the concept moves into the clinic.
Overview
In this issue of Clinical Cancer Research, the Focus is on the development and use of the FDA’s ExpIND in early phase cancer therapeutics. Articles by investigators who have been involved in the elaboration and early application of the ExpIND provide evaluations of this new approach from both academic and pharmaceutical company viewpoints. In addition, the critical role of pharmacodynamics in such trials, and the perspectives of clinical bioethicists and a patient advocate are reviewed to provide insights into the value, as well as risks, inherent in this new cancer clinical trials paradigm.
Hillary Calvert and Ruth Plummer (20) have reviewed the differences in clinical trial design between the various types of Phase I trials, as currently practiced, and the relevance of those designs to pharmacodynamically-guided Phase 0 studies. It is clear that pharmacodynamic endpoints can be used to frame critical questions in the setting of a traditional Phase I study. And there are many similarities in the Phase I or Phase 0 approach: concern over interpatient variability, need to understand the molecular pharmacology of the drug under study, and the assumption that increasing drug dose will increase the chance of observing a desired effect. However, there is considerably more concern with the depth of understanding of the underlying biology interfered with by the agent in question if pharmacodynamic endpoints play a determinative role in either a traditional Phase I dose-escalation study or a Phase 0 trial employing limited drug exposures.
Jacobson-Kram and Mills (21), who were both involved in developing the ExpIND, describe the intention of the FDA to utilize existing regulations to enhance the flexibility available for investigators pursuing early phase cancer therapeutic and imaging clinical trials. As outlined in the FDA guidance3, the ExpIND supports clinical trials, often first-in-human studies, performed on a small number of subjects (usually < 30), that involve limited drug exposures (often no more than 7 days), and have no therapeutic intent. The underlying principle advanced by the ExpIND is that by permitting agents to enter clinical testing earlier in the drug development cycle, prior to formal safety testing, and based on a reduced preclinical genetic and toxicological evaluation, the pharmacology of a compound, or group of analogs, could be examined much earlier. This approach would be especially useful for a molecularly-targeted drug where critical proof-of-principle biochemical, pharmacokinetic, or imaging properties could determine the drug’s ultimate development path, based on early data from the clinic rather than from animal models, obtained at low risk because of limited drug exposure. Such early clinical studies would provide essential information on which to base the final choice of a lead molecule for formal Phase I testing; agents chosen on this stronger scientific base would, in theory, have greater ultimate potential for therapeutic success.
As outlined by Jacobson-Kram and Mills, there are a variety of scenarios available under the ExpIND, all of which aim to speed up the assessment of promising preclinical compounds, whether analyzing several analogs at once, or a series of drugs sequentially, in trials requiring 3–4 rather than 12–18 months each to complete. All of these options require an open, close working relationship with the FDA in the development of the early clinical trials performed under an ExpIND. By decreasing preclinical testing requirements, this new regulatory paradigm should, furthermore, specifically enhance the ability of academic investigators and those working in small biotechnology companies to bring novel molecules to patients.
Murgo, et al. (22) discuss in detail the many issues involved in designing a successful oncologic Phase 0 trial. These issues include the special attention needed for optimal drug selection, as well as the need for a rational transition from preclinical to clinical development. The particular care required in defining an appropriate preclinical model that can be “clinically-qualified” to help guide the subsequent study by modeling in an animal tumor system in vivo is emphasized, as are the requirements for carefully-defined tissue handling and processing standard operating procedures, as well as the qualification of the pharmacodynamic assay performed with tissues obtained under clinical conditions. These are especially important considerations in light of the difficulties that have recently been described in using correlative studies to guide early phase cancer therapeutics (9). Phase 0 studies, because of the small number of patients enrolled, also require novel statistical considerations if meaningful conclusions are to be drawn from the pharmacodynamic data that is developed.
Eliopoulos and colleagues (23) provide an important perspective from the pharmaceutical industry regarding the role of Phase 0 trials in early anticancer drug development. Although only a small number of such studies have been performed (19), they suggest that the information from Phase 0 trials may allow for a more accurate assessment of the risk surrounding the development of a specific drug candidate or class of drugs, and that this risk assessment in an industrial setting can shape the resources made available for future investigations. Phase 0 trials may also help to shift development resources away from agents that do not have favorable pharmacologic properties at an earlier stage of development. Most importantly, such investigations can provide an earlier stream of human data about a new drug that may allow for more rational decision making in the drug development process.
Gutierrez and Collyar (24) review participation in Phase 0 clinical trials from the perspective of the patient. The risks of participation in a Phase 0 study include the potential for harm of any research-related interventions, including tumor biopsies, and the potential for delay in the participation in other clinical trials that may provide therapeutic benefit. These risks are balanced by the limited exposure to the study drug and, hence, limited potential for toxicity in such studies. The most common reasons given for participation in a Phase 0 trial are altruistic in nature and depend on the prior physician-patient relationship; the major reasons that patients give for declining to participate are the non-therapeutic nature of the study, the requirement for tumor biopsies, and the recommendation of the referring oncologist or related health professional. Gutierrez and Collyar also make clear the need to educate the patient community more extensively about the overall process of drug development and the role Phase 0 clinical trials may play in that process.
Abdoler, et al. (25) examine the ethical implications of performing Phase 0 trials in oncology and compare those implications to the well-described ethical issues that surround Phase I investigations. One of the most important considerations is that of risk. In the authors’ view, because of the lower doses and limited duration of drug exposure, Phase 0 trials will, in general, be associated with much lower levels of toxicity compared with Phase I investigations that have the ascertainment of a maximally tolerated dose as their major endpoint. This is not to imply that tumor biopsies, if utilized in Phase 0 studies, are without risk; rather, that risk from administration of the investigational agent itself is much less likely. Because Phase 0 trials are performed without the possibility of providing therapeutic benefit, Abdoler and colleagues discuss the acceptability of exposing research subjects to some risks for the benefit of society, as long as the net risks are not excessive and are justified by the social value that may be gained by the clinical trial. Finally, this paper provides a detailed evaluation of informed consent for Phase 0 trials, and the authors recommend several important strategies to minimize risk and improve the formal understanding by the research subject of the nature of the research process into which they are entering.
These six papers in this issue of Clinical Cancer Research address many ongoing questions in the oncologic community related to the development of the Phase 0 clinical trials paradigm, questions that only now are beginning to be explored in depth (19). It is likely that the next 5 to 10 years will be required to fully understand which agents are most appropriate for Phase 0 proof-of-principle investigations, whether the early investment in additional drug development resources provides a commensurate savings in late stage development time and effectiveness, if and how often false negative results in Phase 0 studies might obscure the development of potentially active agents, and whether or not there will be broad acceptance of this approach by the patient community.
Role of Clinical Pharmacodynamics in Oncologic Phase 0 Trials
A pharmacodynamic (PD) effect is generally understood as a change in a measurable endpoint that is reasonably expected to respond to the drug's mechanism of action; for example, changes in absolute neutrophil count following cytotoxic chemotherapy. A Phase 0 clinical pharmacodynamic trial is intended to demonstrate a desired drug action on its intended molecular target in human malignancy. Because many molecularly-targeted agents are not expected to be clinically effective as single agents in common cancers, conventional Phase I/II trials may be unable to distinguish agents that modulate intended targets from those that do not. This may create a conundrum in which targeted agents are prioritized on the basis of single-agent activity that they are not expected to exhibit. In contrast, a clinical pharmacodynamic trial can potentially identify those investigational agents that deserve full clinical development, even those inactive as single agents, using evidence of target modulation in human malignancy as the basis for this decision. When coupled with measurement of achieved drug level in a tumor biopsy, Phase 0 pharmacodynamic trials can provide important information about investigational agents that fail to modify their intended targets. This may occur by distinguishing those agents that fail to achieve adequate intratumoral levels to affect the target (a pharmaceutical failure), from those that do not affect a drug target in situ despite reaching adequate intratumoral drug levels (a pharmacological failure). Because the purpose of a Phase 0 pharmacodynamic clinical trial is to obtain evidence of drug action on its molecular target in a clinical setting, the results of the pharmacodynamic assessment may become the primary, and sometimes sole, objective of the Phase 0 protocol. Given this primary objective, the reason for participating in the trial is to eliminate inactive agents from the clinical development pipeline, and potentially to enrich for active agents in Phase II clinical trials.
There is also an ethical responsibility to obtain useful results from testing each biopsy specimen from every patient enrolled on a Phase 0 trial. This represents an important paradigm shift from the historical practice of conducting correlative studies in oncology trials, in which clinical pharmacodynamics have been studied in early clinical investigations using existing laboratory assays to probe available tissue specimens for molecular evidence of drug-induced changes. Such studies are often secondary objectives of clinical trial protocols, wherein donation of research tissue specimens is not mandatory for trial participation and completion of the lab assays is often delayed until the trial is over, when specimens are processed as a batch, typically with unknown effects of specimen storage on the endpoint. In contrast, when pharmacodynamic results are primary endpoints of clinical trials, there are higher standards to meet before a biopsy procedure is justified to provide tissue for a laboratory assay. What should be involved in setting these higher standards?
1. Setting and Reaching Higher Standards for Laboratory Assays
The laboratory assay of drug action must be rigorously prepared for its intended use. This assay preparation stage starts with early, close collaboration between applied scientists and the discovery scientists (both those who selected the intended drug action and those who understand the function of its target) to establish the scientific foundations of the measurement. This interactive transfer step informs what measured endpoint will most likely indicate drug action on target, what assay technology that can be validated will be most suitable for this measurement, and what preclinical models (at least two) will be most useful for validating the assay and modeling the Phase 0 trial. After the initial assay development, the fledgling assay can be applied to pilot studies of the investigational agent in preclinical models to determine the feasibility of finding a change in the pharmacodynamic endpoint after treatment with a range of doses relative to the single dose mouse MTD. If results are encouraging, pharmacodynamic assay optimization and validation are pursued using master lots of key assay reagents and calibration standards at defined levels of purity and performance. Required clinical procedures for tissue collection, processing, and storage to obtain valid assay results are established empirically using the validated assay. Assay optimization includes minimizing replicate variability and optimizing sensitivity (i.e., slope in the dynamic range) so that a 30% change in the pharmacodynamic readout is statistically significant; achieving a dynamic range that includes values at or below 10% of the upper limit of quantitation; achieving dilution linearity of the analyte in a relevant biomatrix; and applying this conservatively to a 2–3 mg needle biopsy specimen, which assumes approximately 50% yield. Although these are more ambitious technical goals than are usually applied to correlative laboratory studies, they are driven by the overarching goal that results from this assay will be a primary objective of the Phase 0 trial, and that being able to demonstrate a modest drug effect on its target minimizes the dose of the investigational agent. These assay performance goals are applicable to a wide variety of technology platforms, such as immunosandwich assays, immunofluorescent assays on tissue sections, and RT-qPCR
2. Clinical Readiness of a Pharmacodynamic Assay: Successful Modeling of the Phase 0 Trial, Including Medical Procedures for Collecting Specimens
In addition to pharmacodynamic assay validation and proof that assay analytical performance is adequate for demonstrating the expected effect level on molecular target function, it is also important for the preclinical modeling to replicate the clinical setting in which the assay will be practiced, especially tissue collection and handling procedures that are required for obtaining valid assay results. Using preclinical model(s) to demonstrate that the validated assay and its companion tissue handling standard operating procedures can be practiced in the clinic is the final prerequisite to meet for the laboratory to assert that the assay is ready for clinical application. Biopsy methods have recently been developed for human tumor xenografts, including repeat needle biopsies of the same nodule, to mimic the clinical situation, albeit using general anesthesia. Although not a perfect model, this approach is an improvement over studying highly responsive pharmacodynamic endpoints in tumor samples obtained from necropsy, where necrotic tissues probably contain rapidly waning energy supplies, including the ATP required by many kinases (26). Similar replication of clinical procedures for collecting surrogate tissues for pharmacodynamic assays is also possible, and in the case of peripheral blood leukocytes, can be bridged to the treated patient by ex vivo exposure of whole blood prior to processing for assays, using clinical standard operating procedures (SOPs). However, even the minutes that elapse between placing the guide needle in the lesion and removing the needle biopsy could be enough time for drug response signals to deteriorate (27).
3. Qualifying Drug - Molecular Target Pairs as Suitable for Phase 0 Clinical Trials
Even after validating a high-performance assay capable of demonstrating a 30% change in the PD endpoint using calibration standards, tumor heterogeneity could result in a degree of sampling variability that could exceed any statistically significant change in the PD endpoint to be expected at non-toxic dose levels (Fig. 1). High sampling variation in a pharmacodynamic endpoint will require a large magnitude of drug effect to reach statistical significance. On the other hand, a modest drug-induced change in the pharmacodynamic endpoint may reach statistical significance if there is little sampling variation at baseline. Importantly, therefore, both the targeted therapy and its molecular target must qualify together, as a pair, for suitability for Phase 0 clinical trial evaluation. If non-toxic dose levels of an investigational agent fail to cause a significant change in the pharmacodynamic endpoint, either due to high sampling variability or to weak drug action on target, then either the investigational agent under examination must proceed to a dose-escalation trial where PD endpoints can be studied at higher systemic exposures, or the Phase 0 trial must be changed to examine a more useful endpoint (e.g., a different assay of the current drug target, or a new assay of a different target). Unfortunately, a drug-target pair cannot be qualified as a Phase 0 candidate without first evaluating the pair with a valid, clinically-useful assay and the specimen handling SOPs to obtain the needed data. Programs will need to anticipate that some portion of resources will result in valid pharmacodynamic assays that end up proving that some molecular target endpoints are not suitable for Phase 0 clinical trials.
Fig. 1.
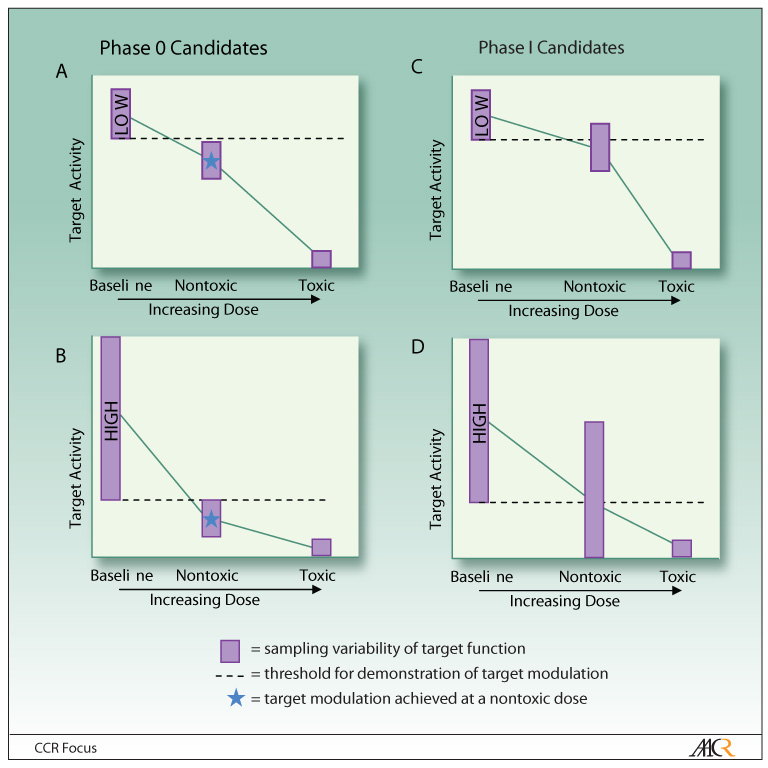
Determining whether the molecular target is a good candidate for evaluation in a Phase 0 clinical trial. The decision to proceed with a Phase 0 rather than Phase I trial to evaluate a drug’s effect on its molecular target depends both on the amount of variability in the target and the therapeutic index of the targeted agent. Sampling variability in the pharmacodynamic endpoint due to tumor heterogeneity, diurnal variation, or other factors can be “low” or “high” for a baseline evaluation. Molecular targets suitable for Phase 0 trials can have either low (A) or high (B) variability at baseline, as long as there is a non-toxic dose of the investigational agent that achieves a degree of target modulation that lies outside the baseline range. Targets are not suitable for Phase 0 trials (C) if target modulation by the investigational drug is never significantly different from baseline, or (D) if a significant difference in target activity can be achieved only at potentially toxic drug doses. In both (C) and (D), the targets would be better approached with a traditional Phase I trial in which the drug dose can be escalated to determine dose limiting toxicity.
Conclusions
Pharmacodynamic and pharmacokinetic data of high quality must be obtained to justify the resources and time associated with the conduct of a Phase 0 study. Pharmacodynamic data obtained without rigorous attention to assay development and specimen acquisition often fails to inform decisions about the clinical development of a novel therapeutic agent. Recent NCI studies have demonstrated that experimentally qualified pharmacodynamic tests, along with requisite collection and handling practices for clinical specimens, can be prepared and implemented in the clinical setting within a 6–12 month time frame, driven primarily by the quality of the discovery science which forms the assays' foundations.
Because these pharmacodynamic endpoints report on drug action at intended molecular targets, the availability of validated assays could potentially propel some of these molecular signals into use as surrogate markers that predict early clinical endpoints (4) and accelerate regulatory approval for a specific indication. However, the complexity and cross-talk of signal transduction pathways in common malignancies suggest that pharmacodynamic responses of intended molecular targets are unlikely to be translated into clinical responses in an individual patient. It seems more reasonable to suggest that a pharmacodynamic effect demonstrated in the setting of an early therapeutic trial will be one of the necessary, but not sufficient, results ultimately required for clinical benefit. Thus, it is important to clearly distinguish pharmacodynamic endpoints from other types of biomarkers that may predict individual patient outcome or stage of disease.
Most likely, the value of pharmacodynamic assays in the earliest cancer therapeutics trials will be as accurate indicators of molecularly targeted treatments that do not affect their targets in human disease, and therefore do not deserve further Phase I/II clinical development. The time frame covering the period from assay preparation to demonstration of target modulation in patients is, in the best case, approximately 12 months. However, this time estimate depends, in part, on the accuracy of the conclusions about target function and drug action that are available when assay development is initiated, so that the pharmacodynamic assay specifically and accurately measures these effects and informs the decision to advance the new drug into full clinical development.
If the use of the ExpIND mechanism assists in the prediction/elimination of future clinical drug failures through the performance of small studies at the very beginning of clinical development that are pharmacokinetically and pharmacodynamically informative, while providing enhanced molecular assays for later stage clinical investigations, this new regulatory guidance will have been an important achievement. However, the ultimate utility of the ExpIND will be determined over time based on the frequency of its application to a broad spectrum of novel oncologic therapies, and whether or not a more focused approach to proof-of-principle investigations in oncologic drug development shortens the timeline for the introduction of new, and more effective, anticancer agents.
Footnotes
This paper was written with the support of the Division of Cancer Treatment and Diagnosis and the Center for Cancer Research of the National Cancer Institute. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400.
References
- 1.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84:1424–1431. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garber K. Sugen falls as casualty of Pfizer--Pharmacia merger. Nat Biotechnol. 2007;7:722–723. doi: 10.1038/nbt0703-722. [DOI] [PubMed] [Google Scholar]
- 4.Park JW, Kerbel RS, Kelloff GJ, et al. Rationale for biomarkers and surrogate end points in mechanism-driven oncology drug development. Clin Cancer Res. 2004;10:3885–3896. doi: 10.1158/1078-0432.CCR-03-0785. [DOI] [PubMed] [Google Scholar]
- 5.Goldin A, Carter SK. Screening and evaluation of antitumor agents. In: Holland JF, Frei E III, editors. Cancer Medicine. Philadelphia: Lea & Febiger; 1973. pp. 605–628. [Google Scholar]
- 6.Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC. Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst. 1997;89:1138–1147. doi: 10.1093/jnci/89.15.1138. [DOI] [PubMed] [Google Scholar]
- 7.O'Quigley J, Pepe M, Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics. 1990;46:33–48. [PubMed] [Google Scholar]
- 8.Parulekar WR, Eisenhauer EA. Phase I trial design for solid tumor studies of targeted, non-cytotoxic agents: theory and practice. J Natl Cancer Inst. 2004;96:990–997. doi: 10.1093/jnci/djh182. [DOI] [PubMed] [Google Scholar]
- 9.Goulart BH, Clark JW, Pien HH, Roberts TG, Finkelstein SN, Chabner BA. Trends in the use and role of biomarkers in phase I oncology trials. Clin Cancer Res. 2007;13:6719–6726. doi: 10.1158/1078-0432.CCR-06-2860. [DOI] [PubMed] [Google Scholar]
- 10.Kummar S, Gutierrez M, Doroshow JH, Murgo AJ. Drug development in oncology: classical cytotoxics and molecularly targeted agents. Br J Clin Pharmacol. 2006;62:15–26. doi: 10.1111/j.1365-2125.2006.02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomaszewski JE, Doroshow JH. Preclinical development of molecularly targeted agents in oncology. In: Kaufman HL, Wadler S, Antman K, editors. Cancer Drug Discovery and Development: Molecular Targeting in Oncology. Totowa, NJ: Humana Press; 2008. pp. 703–718. [Google Scholar]
- 12.Workman P, Aboagye EO, Chung YL, et al. Minimally invasive pharmacokinetic and pharmacodynamic technologies in hypothesis-testing clinical trials of innovative therapies. J Natl Cancer Inst. 2006;98:580–598. doi: 10.1093/jnci/djj162. [DOI] [PubMed] [Google Scholar]
- 13.Kummar S, Kinders R, Rubinstein L, et al. Compressing drug development timelines in oncology using phase '0' trials. Nat Rev Cancer. 2007;7:131–139. doi: 10.1038/nrc2066. [DOI] [PubMed] [Google Scholar]
- 14.Marchetti S, Schellens JH. The impact of FDA and EMEA guidelines on drug development in relation to Phase 0 trials. Br J Cancer. 2007;97:577–581. doi: 10.1038/sj.bjc.6603925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sparreboom A. Unexplored pharmacokinetic opportunities with microdosing in oncology. Clin Cancer Res. 2007;13:4033–4034. doi: 10.1158/1078-0432.CCR-07-0540. [DOI] [PubMed] [Google Scholar]
- 16.Kimmelman J. Ethics at phase 0: clarifying the issues. J Law Med Ethics. 2007;35:727–733. doi: 10.1111/j.1748-720X.2007.00194.x. [DOI] [PubMed] [Google Scholar]
- 17.Hill TP. Phase 0 trials: are they ethically challenged? Clin Cancer Res. 2007;13:783–784. doi: 10.1158/1078-0432.CCR-06-2365. [DOI] [PubMed] [Google Scholar]
- 18.Kummar S, Kinders R, Gutierrez M, et al. Inhibition of poly(ADP-ribose) polymerase (PARP) by ABT-888 in patients with advanced malignancies: Results of a phase 0 trial. J Clin Oncol. 2007;25 Suppl. 1:142s. doi: 10.1200/JCO.2008.19.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson WT. Innovative Early Development Regulatory Approaches: expIND, expCTA, Microdosing. Clin Pharm Ther. 2008;83:358–360. doi: 10.1038/sj.clpt.6100461. [DOI] [PubMed] [Google Scholar]
- 20.Calvert AH, Plummer R. The development of Phase I cancer trial methodologies - the use of pharmacokinetic and pharmacodynamic endpoints sets the scene for phase 0 cancer clinical trials. Clin Cancer Res. 2008;14 doi: 10.1158/1078-0432.CCR-07-4559. in press. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson-Kram D, Mills G. Leveraging exploratory INDs to speed drug development. Clin Cancer Res. 2008;14 doi: 10.1158/1078-0432.CCR-07-4558. in press. [DOI] [PubMed] [Google Scholar]
- 22.Murgo AJ, Kummar S, Rubinstein L, et al. Designing phase 0 cancer clinical trials. Clin Cancer Res. 2008;14 doi: 10.1158/1078-0432.CCR-07-4560. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliopoulos H, Giranda V, Carr R, Tiehen R, Leahy T, Gordon G. Phase 0 trials: an industry perspective. Clin Cancer Res. 2008;14 doi: 10.1158/1078-0432.CCR-07-4586. in press. [DOI] [PubMed] [Google Scholar]
- 24.Gutierrez M, Collyar D. Patient perspectives on phase 0 clinical trials. Clin Cancer Res. 2008;14 doi: 10.1158/1078-0432.CCR-07-4561. in press. [DOI] [PubMed] [Google Scholar]
- 25.Abdoler E, Taylor H, Wendler D. The ethics of phase 0 oncology trials. Clin Cancer Res. 2008;14 doi: 10.1158/1078-0432.CCR-08-0876. in press. [DOI] [PubMed] [Google Scholar]
- 26.Kinders RJ, Hollingshead M, Parchment RE, et al. Preclinical modeling of a phase 0 clinical trial protocol. J Clin Oncol. 2007;25 Suppl. 1:616s. doi: 10.1158/1078-0432.CCR-08-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker AF, Dragovich T, Ihle NT, Williams R, Fenoglio-Preiser CM, Powis G. Stability of phospho-protein as a biological marker of tumor signaling. Clin Cancer Res. 2005;11:4338–4340. doi: 10.1158/1078-0432.CCR-05-0422. [DOI] [PubMed] [Google Scholar]