

Abstract
Oxidative stress-mediated destruction of normal parenchymal cells during hepatic inflammatory responses contributes to the pathogenesis of immune-mediated hepatitis and is implicated in the progression of acute inflammatory liver injury to chronic inflammatory liver disease. The transcription factor NF-E2-related factor 2 (Nrf2) regulates the expression of a battery of antioxidative enzymes and Nrf2 signaling can be activated by small-molecule drugs that disrupt Keap1-mediated repression of Nrf2 signaling. Therefore, genetic and pharmacologic approaches were used to activate Nrf2 signaling to assess protection against inflammatory liver injury. Profound increases in ind of cell death were observed in both Nrf2 wild-type (Nrf2-WT) mice and Nrf2-disrupted (Nrf2-KO) mice 24-hr following intravenous injection of concanavalin A (12.5 mg/kg, ConA), a model for T cell-mediated acute inflammatory liver injury. However, hepatocyte-specific conditional Keap1 null (Alb-Cre:Keap1flox/−, cKeap1-KO) mice with constitutively enhanced expression of Nrf2-regulated antioxidative genes as well as Nrf2-WT mice but not Nrf2-KO mice pretreated with three daily doses of a triterpenoid that potently activates Nrf2 (30 µmole/kg, CDDO-Im) were highly resistant to ConA-mediated inflammatory liver injury. CDDO-Im pretreatment of both Nrf2-WT and Nrf2-KO mice resulted in equivalent suppression of serum pro-inflammatory soluble proteins suggesting that the hepatoprotection afforded by CDDO-Im pretreatment of Nrf2-WT mice but not Nrf2-KO mice was not due to suppression of systemic pro-inflammatory signaling, but instead was due to activation of Nrf2 signaling in the liver. Enhanced hepatic expression of Nrf2-regulated antioxidative genes inhibited inflammation-mediated oxidative stress, thereby preventing hepatocyte necrosis. Attenuation of hepatocyte death in cKeap1-KO mice and CDDO-Im pretreated Nrf2-WT mice resulted in decreased late-phase pro-inflammatory gene expression in the liver thereby diminishing the sustained influx of inflammatory cells initially stimulated by the ConA challenge. Taken together, these results clearly illustrate that targeted cytoprotection of hepatocytes through Nrf2 signaling during inflammation prevents the amplification of inflammatory responses in the liver.
Keywords: Liver inflammation, Nrf2, Keap1, antioxidative enzymes, cytoprotection, triterpenoid
Introduction
While acute inflammatory responses to pathogen infection and tissue injury are necessary for survival, sustained immune responses can result in damage to non-target cells that are neither infected nor damaged by the initial chemical or physical insult. Reactive oxygen species released from activated macrophages and neutrophils can damage cellular macromolecules in uninfected or non-damaged cells leading to necrosis of these cells(Ohshima et al. 2003). Cell necrosis has been demonstrated to be a pro-inflammatory stimulus in that recognition of necrotic cell debris by macrophages leads to activation of pro-inflammatory cytokine signaling and amplification of the initial inflammatory response(Karin and Greten 2005). Also, inflammation-mediated oxidative stress can potentiate inflammatory cytokine signaling since a number of pro-inflammatory signaling proteins can be regulated in a redox-sensitive fashion(Adachi et al. 2004; Kamata et al. 2005).
Inflammation-mediated cell death has been demonstrated to play a role in the pathogenesis of acute liver injury following exposure to agents such as alcohol(Ceccanti et al. 2006), pharmaceuticals(Jaeschke 2005), and hepatotropic viruses(Choi and Ou 2006; Nakamoto and Kaneko 2003). Immune-mediated liver disease is a serious human health problem since acute hepatitis may progress to chronic fibrosis and cirrhosis, a major risk factor for the development of hepatocellular carcinoma. Intravenous administration of ConA to mice has been developed as a model for T cell-mediated acute inflammatory liver injury in which hepatocytes are targeted by activated inflammatory cells(Tiegs et al. 1992) leading to hepatocyte apoptosis and necrosis. While ConA has been shown to be toxic to primary mouse hepatocytes in cell culture (Leist and Wendel, 1996), numerous studies have demonstrated the necessity of activated immune cells in the mechanism of ConA hepatotoxicity in vivo. Athymic mice and mice with severe combined immunodeficiency syndrome have been shown to be resistant to hepatotoxicity following ConA injection(Tiegs et al. 1992). Additionally, administration of ConA to mice following depletion of either natural killer T cells(Takeda et al. 2000), neutrophils(Bonder et al. 2004) or Kupffer cells(Schümann et al. 2000) failed to elicit hepatocyte death. Therefore, intravenous injection of ConA represents a useful animal model for investigating cytoprotective mechanisms against inflammatory liver injury since hepatocyte death is a direct consequence of immune system activation independent of any directly hepatotoxic chemical insult.
The Keap1-Nrf2 signaling pathway regulates the inducible expression of a battery of cytoprotective genes. Under basal conditions, Nrf2 is repressed through an interaction with Keap1 leading to proteasomal degradation of Nrf2(Dinkova-Kostova et al. 2005). However, exposure to both endogenous and exogenous reactive molecules such as reactive oxygen species, 15-deoxy-delta12,14-prostaglandin J2, dithiolethiones and triterpenoids leads to release of Nrf2 from Keap1 following modification of reactive cysteines within Keap1 or activation of pathways leading to phosphorylation of Nrf2 and disruption of the Keap1-Nrf2 interaction(Kobayashi and Yamamoto 2005). Nrf2 can translocate to the nucleus and in combination with other transcription factors enhance the transcription of antioxidative genes, glutathione homeostasis genes and genes important for the production of reducing equivalents that collectively represent an adaptive response leading to protection against toxicity due to oxidative stress(Kwak et al. 2003; Motohashi et al. 2004; Osburn et al. 2006). Therefore, the Nrf2-regulated adaptive response represents a potential target for attenuation of inflammation by protecting against inflammatory oxidative damage and pro-inflammatory redox-sensitive signaling. Indeed, the lack of an active Nrf2 signaling pathway in mice has been shown to result in increased inflammation and inflammation-mediated oxidative damage in a number of lung and colon disease models(Itoh et al. 2004; Khor et al. 2006; Osburn et al. 2007; Rangasamy et al. 2005; Thimmulappa et al. 2006).
Recently, a hepatocyte-specific conditional Keap1 knockout (cKeap1-KO, Alb-Cre::Keap1(Flox/−)) mouse was developed in which the repression of Nrf2 signaling by Keap1 has been disrupted in hepatocytes leading to amplified Nrf2 signaling, constitutively enhanced expression of Nrf2-regulated cytoprotective genes and attenuated acetaminophen hepatotoxicity(Okawa et al. 2006). Therefore, this model represents a genetic tool to investigate the effect of Nrf2-dependent cytoprotection on acute inflammatory liver injury, due to dampened Keap1 repression of Nrf2 signaling, without the post-natal lethality associated with systemic deletion of Keap1(Wakabayashi et al. 2003). Additionally, administration of the synthetic triterpenoid 1-[2-cyano-3,12- dioxooleana-1,9(11)-dien-28-oyl]imidazole (CDDO-Im) represents a complementary pharmacological tool for examining the effect of activation of Nrf2 signaling on acute inflammatory liver injury since CDDO-Im administration has been shown to enhance expression of Nrf2-regulated cytoprotective genes in a number of organ systems(Yates et al. 2007). Therefore, by comparing the severity of immune-mediated liver injury in mice with genetically and pharmacologically amplified Nrf2 signaling one can deduce the efficacy of hepatocyte-directed, Nrf2-dependent cytoprotection in attenuating hepatic inflammation.
In the present study, mice with hepatocyte-disrupted expression of Keap1, as well as Nrf2-WT mice but not Nrf2-KO mice pretreated with CDDO-Im, were resistant to ConA-mediated oxidative stress and hepatocyte necrosis. Genetic and pharmacological amplification of Nrf2 signaling had no effect on ConA-mediated serum pro-inflammatory soluble protein levels but inhibited later-phase hepatic inflammatory gene expression and inflammatory cell infiltration, apparently due to the abrogation of hepatocyte damage in these mice. Taken together, these results demonstrated that amplified Nrf2 signaling provides powerful protection against acute inflammatory liver injury by preventing inflammation-mediated hepatocyte necrosis leading to diminished pro-inflammatory gene expression and a cessation of the chronic inflammatory response to ConA administration.
Materials and Methods
Experimental animals
cKeap1-KO mice were generated by crossing Alb-Cre::Keap1(+/−) mice with Keap1(Flox/−) mice. Alb-Cre::Keap1(+/−) mice, Keap1(Flox/−) mice and Alb-Cre:Keap1(+/+) (cKeap1-WT) mice were generated on a C57BL/6J background(Okawa et al. 2006). While this transgenic strategy results in knockout of Keap1 in hepatocytes, experimental evidence indicates hypomorphic Keap1 expression in other cell types (Melinda Yates, unpublished observation). Nrf2-KO mice on a C57BL/6J background were generated as previously described(Iida et al. 2004). Nrf2-WT mice (C57BL/6J) were obtained from The Jackson Laboratory (Bar Harbor, ME). Genotypes were confirmed using PCR analysis of tail genomic DNA.
Animal treatments
Female cKeap1-KO, cKeap1-WT, Nrf2-KO and Nrf2-WT mice (9–13 wk old) fed AIN-76A diet and water ad libitum were treated with a single dose of ConA (12.5 mg/kg, i.v.) or PBS vehicle. Separate groups of Nrf2-WT and Nrf2-KO mice were treated with 3 doses of CDDO-Im (30 µmole/kg body weight, p.o., once daily, vehicle - 10% DMSO, 10% Cremaphor, 80% PBS) prior to ConA administration. The last CDDO-Im dose was administered 24-hr prior to the ConA dose. Subgroups of animals (N = 3–5) were euthanized by CO2 asphyxiation, 3- and 24-hr after ConA administration. In all groups, blood was collected by cardiac puncture and livers were harvested. The left lobe was sectioned longitudinally and fixed in 10% neutral buffered formalin and embedded in paraffin. Four µm sections of the liver paraffin tissue blocks were stained with hematoxylin and eosin (H&E). The remaining liver was snap-frozen in liquid nitrogen and stored at −80°C until further processing. Serum was collected by centrifugation of blood at 4,000 × g following incubation at room temperature for 2-hr. All animal experiments were performed in accordance with protocols approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions.
Serum alanine aminotransferase (ALT) activity
ALT enzymatic activity of serum samples was determined using a Vet ACE® chemistry system (Alfa Wassermann, West Caldwell, NJ).
Aconitase activity
Whole liver tissue was homogenized at a concentration of 0.1 g/ml in PBS containing 0.05% butylated hydroxytoluene and Complete protease inhibitor cocktail (Roche, Indianapolis, IN,). Aconitase activity of liver homogenates was determined as previously described(Osburn et al. 2007).
Lipid peroxidation
Lipid peroxidation products (malondialdehyde and 4-hydroxyalkenals in combination) in liver homogenates were detected using the Bioxytech LPO-586 kit (Oxis, Foster City, CA) using the methanesulfonic acid protocol according to the manufacturer’s directions.
Global gene expression analysis
Hepatic global gene expression was compared in nine-week old male cKeap1-KO mice and Alb-Cre::Keap1flox/+ (cKeap1-genetic control) mice. Previous experiments have demonstrated that amplification of Nrf2 signaling results in equivalent hepatic Nrf2-dependent antioxidative gene transcription in male and female mice (Melinda Yates, unpublished observation). Total RNA was isolated using Versagene RNA tissue kit (Gentra, Minneapolis, MN). RNA quality was evaluated using an Agilent Bioanalyzer 2100 (Applied Biosystems, Foster City, CA). Purified RNA was used for double-stranded cDNA synthesis. Double-stranded cDNA synthesis was performed using the Superscript Doubled Stranded cDNA synthesis kit (Invitrogen, Carlsbad, CA). cRNA synthesis was then performed using the Bioarray High Yield RNA Transcript Labeling Kit (Enzo, Farmingdale, NY). Both cDNA and cRNA were purified using the Affymetrix GeneChip Sample Cleanup Module (Affymetrix, Santa Clara, CA). cRNA was used for the fragmentation reaction. The entire fragmentation product was then hybridized to the Affymetrix Mouse Genome 430 2.0 array chip for 18 hours. After 18 hours, the chips were removed, washed and stained using an Affymetrix GeneChip Fluidics Station 400. The chips were then scanned using an Affymetrix GeneChip Scanner 3000 7G. Pairwise comparisons of individual mice (n=3/group) were performed, generating 9 comparisons. Signal strength values were log-transformed to generate a normal distribution. Independent 2 sample t-tests were performed on the log-signal strength to determine differential expression between the two groups (p≤0.05). To limit false positives, only genes with a 1.5-fold change or more and a comparison value of 6 or more were included. Affymetrix Analysis Center website was used for annotation of genes. The expression levels of selected genes were validated by quantitative RT-PCR. Microarray data will be deposited in NCBI GEO upon acceptance of the manuscript.
Serum cytokine and chemokine analysis
Cytometric bead array was used to measure the serum levels of soluble pro-inflammatory proteins using the BD Cytometric Bead Array Mouse Inflammation Kit (BD Biosciences, San Jose, CA) according to manufacturer’s directions. Flow cytometric analysis was performed using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA). Data were acquired and analyzed using BD cytometric bead array software. The concentration of interleukin-2 (IL-2) in serum was determined using an IL-2 ELISA Ready-Set-Go kit (eBiosciences, San Diego, CA) according to manufacturer’s directions. Non-detect values were assigned a concentration equal to the lowest detected value divided by the square root of two.
Hepatic macrophage accumulation
Four µm-thick tissue sections cut from formalin-fixed paraffin-embedded tissue blocks were deparaffinized and rehydrated. Sections were blocked with 5% goat serum and 1% bovine serum albumin and incubated with biotinylated Griffonia simplificifolia Lectin I (1:50, Vector Laboratories, Burlingame, CA). Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide and lectin binding was detected using horseradish peroxidase conjugated strepavidin (1:250, Invitrogen, Carlsbad, CA) followed by visualization using SIGMAFAST™ 3,3′-diaminobenzidine (Sigma-Aldrich, St. Louis, MO). Sections were counterstained with hematoxylin, dehydrated through alcohol and mounted. Positively stained macrophages were manually counted at 400X magnification across six random fields.
Hepatic neutrophil accumulation
Four µm-thick tissue sections cut from formalin-fixed paraffin-embedded tissue blocks were deparaffinized and rehydrated. Neutrophils were stained using a naphthol AS-D chloroacetate (Specific Esterase) kit (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s directions. Sections were counterstained with haematoxylin and mounted. Positively stained neutrophils were manually counted at 400X magnification across six random fields.
Cytokine gene expression
Total RNA was extracted from liver tissue using Versagene RNA purification columns (Gentra, Minneapolis, MN) according to manufacturer’s directions. 1 µg of cDNA was synthesized from 1 µg of mRNA using the iScript cDNA synthesis kit (BioRad, Hercules, CA, USA). Quantitative RT-PCR analysis was performed using IQ Sybr Green supermix. Previously published primer pairs for Hypoxanthine-guanine phosphoribosyltransferase (Osburn et al. 2006), interferon γ (IFNγ) (Overbergh et al 1999), tumor necrosis factor α (TNFα) (Overbergh et al. 1999), monocyte chemoattractant protein-1 (MCP-1)(Hagen et al. 2007), and macrophage inflammatory protein-2 (MIP-2)(Fadl et al. 2007) were synthesized and used for specific gene expression analysis. Melting curve analysis was used to assess amplification specificity. Standard curves using serial dilutions of cDNA were generated for each gene in order to determine amplification efficiency and relative quantification was calculated using the method of Pfaffl(Pfaffl 2001).
Statistical analysis
Student’s t-test, for comparison of two groups, or ANOVA with Student-Neuman Kuels multiple comparison analysis, for comparison of three or more groups, was used to evaluate statistically significant differences.
Results
Genetic and pharmacological amplification of Nrf2 signaling in hepatocytes prevents acute inflammatory liver injury
Histological analysis of liver and determination of serum ALT values was performed in order to assess the protective effect of genetic amplification of Nrf2 signaling and CDDO-pretreatment on immune-mediated hepatotoxicity. Microscopic examination of H&E stained liver sections revealed severe and extensive necrosis in livers from cKeap1-WT but not cKeap1-KO mice 24-hr following ConA challenge (Figure 1a). Likewise, markedly increased serum ALT values were detected in cKeap1-WT mice but not cKeap1-KO mice following ConA administration. At the same time, CDDO-Im pretreatment also inhibited ConA-mediated hepatotoxicity, apparently in an Nrf2-dependent fashion. Severe and extensive necrosis was observed in vehicle-pretreated but not CDDO-Im pretreated Nrf2-WT mice 24-hr following ConA challenge (Figure 1b). Similarly, markedly increased serum ALT values were detected in vehicle-pretreated but not CDDO-Im pretreated Nrf2-WT mice following ConA challenge. On the other hand, CDDO-Im pretreatment of Nrf2-disrupted Nrf2-KO mice failed to prevent ConA-mediated hepatotoxicity as severe and extensive necrosis and markedly increased serum ALT values were observed in both CDDO-Im pretreated and vehicle-pretreated Nrf2-KO mice 24-hr following ConA administration (Figure 1c). Although rigorous lethality studies were not performed, mortality was observed in some vehicle-pretreated and CDDO-Im pretreated Nrf2-KO mice only, suggesting increased susceptibility of Nrf2-KO mice to ConA-mediated lethality. Therefore, either genetic amplification or pharmacological activation of Nrf2 signaling effectively prevented ConA-mediated hepatocyte necrosis.
Figure 1.
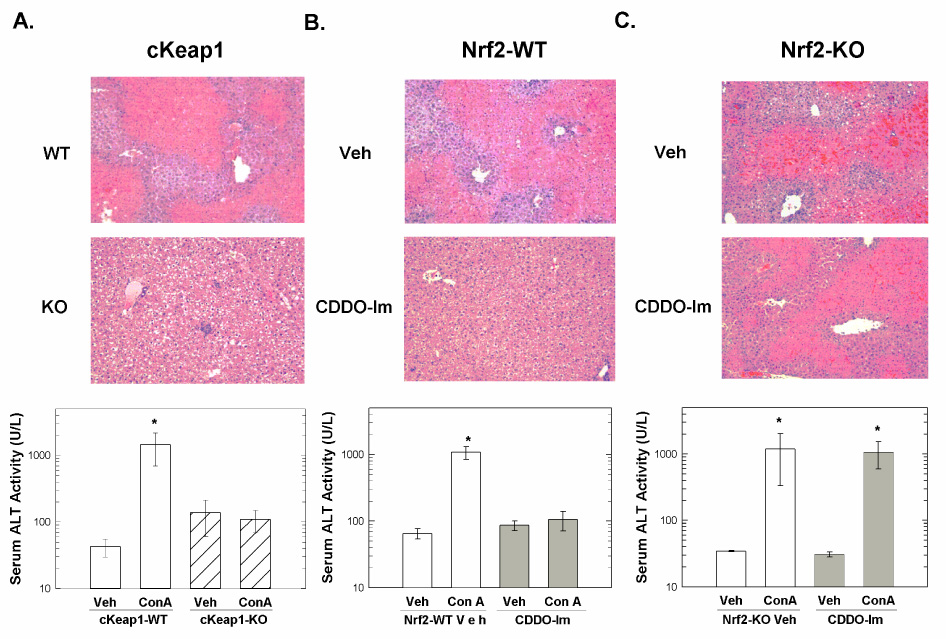
Hepatocyte-specific and systemic amplification of Nrf2 signaling attenuates ConA-mediated hepatotoxicity. Representative images (100X magnification) of liver sections stained with haematoxylin and eosin (top panel) from A) cKeap1-KO and cKeap1-WT mice, B) CDDO-Im pretreated and vehicle-pretreated Nrf2-WT mice, and C) vehicle-pretreated and CDDO-Im pretreated Nrf2-KO mice 24-hr following ConA administration. ALT activities (bottom panel) of serum samples collected from vehicle-and ConA-treated A) cKeap1-KO and cKeap1-WT mice (n = 3), B) CDDO-Im pretreated and vehicle-pretreated Nrf2-WT mice (n = 5), and C) vehicle-pretreated and CDDO-Im pretreated Nrf2-KO mice (n = 4) 24-hr following ConA administration. One mouse died in both vehicle-pretreated and CDDO-Im pretreated Nrf2-KO groups. Serum ALT activities are expressed as U/ml. Values represent the mean ± SEM. * p < 0.001, ANOVA-SNK analysis.
Amplified Nrf2 signaling inhibits ConA-mediated oxidative damage
Since reactive oxygen species released from activated inflammatory cells can contribute to acute inflammatory liver injury, we analyzed the hepatic levels of two indicators of oxidative damage, 24-hr following ConA administration. Aconitase specific activity was measured as an indicator of protein oxidation since exposure of aconitase to reactive oxygen species results in reduction of ferric iron to ferrous iron, release of ferrous iron and subsequent loss of enzyme activity. Loss of hepatic aconitase activity was observed in cKeap1-WT mice but not cKeap1-KO mice and vehicle-pretreated but not CDDO-Im pretreated Nrf2-WT mice, 24-hr following ConA administration (Figure 2a). Next, the hepatic levels of malondialdehyde and hydroxyalkenals, in combination, were assessed as an indicator of lipid peroxidation. Increased lipid peroxidation products were detected in livers from cKeap1-WT mice but not cKeap1-KO mice and vehicle-pretreated but not CDDO-Im pretreated Nrf2-WT mice, 24-hr following ConA administration (Figure 2b). These results indicated that both the genetic amplification and the pharmacological activation of Nrf2 signaling attenuated macromolecular oxidative damage during the inflammatory response triggered by ConA injection.
Figure 2.
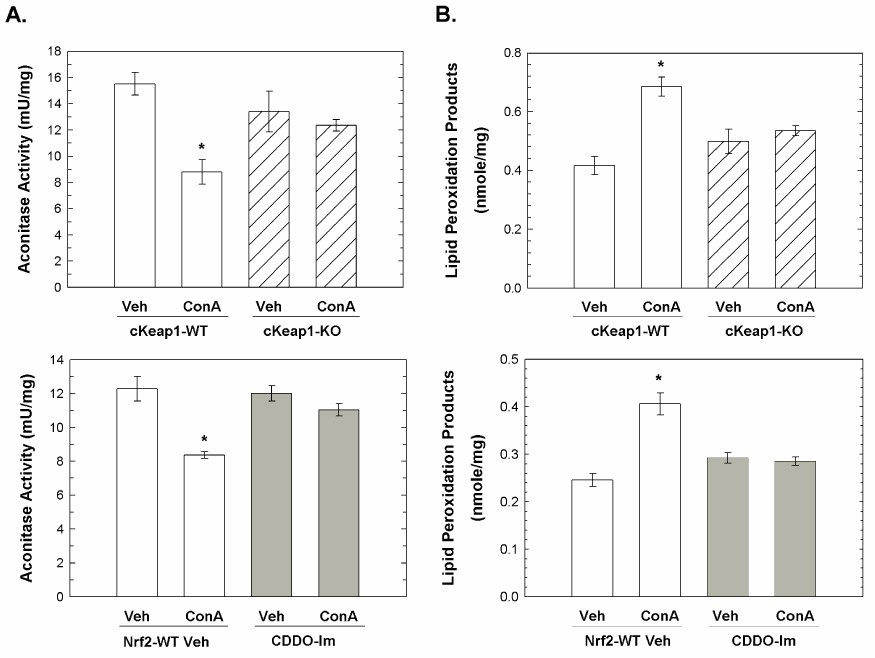
Inhbition of ConA-mediated oxidative damage by hepatocyte-specific and systemic amplification of Nrf2 signaling. Livers were harvested 24-hr following treatment and homogenates were prepared as described in the Materials and Methods section. A) Aconitase activity of liver homogenates from vehicle- and ConA-treated cKeap1-KO mice (N = 3), cKeap1-WT mice (N = 3), vehicle-pretreated Nrf2-WT mice (N = 5) and CDDO-Im pretreated Nrf2-WT mice (N = 5). Aconitase specific activity is expressed as mU activity/mg protein. Values represent the mean ± SEM. * p < 0.001, ANOVA-SNK analysis. B) Lipid peroxidation in liver homogenates from vehicle- and ConA-treated cKeap1-KO mice (N = 3), cKeap1-WT mice (N = 3), vehicle-pretreated Nrf2-WT mice (N = 5) and CDDO-Im pretreated Nrf2-WT mice (N = 5). Data are expressed as nmole/mg protein and Values represent the mean ± SEM. * p < 0.001, ANOVA-SNK analysis.
The decreased oxidative damage observed in cKeap1-KO and CDDO-Im pretreated Nrf2-WT mice was likely due to enhanced expression of Nrf2-regulated antioxidative genes. Nrf2-dependent increased hepatic expression of antioxidative genes has previously been demonstrated in mice following CDDO-Im administration(Yates et al. 2006). Herein, global gene expression was compared between cKeap1-KO and cKeap1-genetic control mice to identify gene changes modulated by constitutive Nrf2 hyperactivation caused by Keap1 deletion in the liver. This analysis identified 590 Nrf2-dependent genes, including Nrf2 (Nfe2l2, fold-change, 1.6). For example, increased basal expression of antioxidative genes, such as Gpx2 (fold-change, 20.9) and Txnrd1 (2.4), as well as xenobiotic metabolism and glutathione homeostasis genes was detected in livers of cKeap1-KO mice relative to control mice (Table 1). Additional gene expression changes can be largely grouped into the following categories: lipid metabolism, cell signaling, cell death, small molecule biochemistry, immune response, cellular growth and proliferation, gene expression, carbohydrate metabolism and cell cycle (data not shown). Despite the broad impact of Keap1 disruption on gene expression, it is likely that enhanced expression of Nrf2-regulated antioxidative genes in cKeap1-KO mice and CDDO-Im pretreated Nrf2-WT mice prevented macromolecular oxidation during ConA-mediated inflammation.
Table 1.
Selected categories from microarray analysis of genes modulated in cKeap1-KO mice compared to cKeap1-genetic control mice
| Description | Symbol | Fold-changea |
|---|---|---|
| Antioxidative genes | ||
| ATX1 (antioxidant protein 1) homolog 1 (yeast) | Atox1 | 1.6 |
| biliverdin reductase B (flavin reductase (NADPH)) | Blvrb | 2.6 |
| ferredoxin reductase | Fdxr | 1.8 |
| glutathione peroxidase 2 | Gpx2 | 20.9 |
| glutathione peroxidase 4 | Gpx4 | 1.5 |
| glutathione reductase 1 | Gsr | 1.6 |
| heme oxygenase (decycling) 1 | Hmox1 | 1.7 |
| sulfiredoxin 1 homolog (S. cerevisiae) | Srxn1 | 4.7 |
| thioredoxin reductase 1 | Txnrd1 | 2.4 |
| phosphogluconate dehydrogenase | Pgd | 2.2 |
|
Glutathione metabolism | ||
| alanyl (membrane) aminopeptidase | Anpep | 0.6 |
| glutamate-cysteine ligase, catalytic subunit | Gclc | 2.3 |
| gamma-glutamyltransferase 6 | Ggt6 | 1.8 |
| glutathione synthetase | Gss | 1.5 |
|
Xenobiotic metabolism | ||
| alcohol dehydrogenase 4 (class II), pi polypeptide | Adh4 | 0.6 |
| aldo-keto reductase family 1, member C19 | Akr1c19 | 2.2 |
| carbonyl reductase 1 | Cbr1 | 5.9 |
| carbonyl reductase 3 | Cbr3 | 117.9 |
| cytochrome P450, family 2, subfamily d, polypeptide 13 | Cyp2d13 | 0.5 |
| cytochrome P450, family 2, subfamily j, polypeptide 5 | Cyp2j5 | 0.6 |
| cytochrome P450, family 2, subfamily j, polypeptide 9 | Cyp2j9 | 0.4 |
| cytochrome P450, family 39, subfamily a, polypeptide 1 | Cyp39a1 | 2.6 |
| cytochrome P450, family 3, subfamily a, polypeptide 44 | Cyp3a44 | 0.1 |
| flavin containing monooxygenase 3 | Fmo3 | 15.4 |
| glutathione S-transferase, alpha 2 (Yc2) | Gsta2 | 5.2 |
| glutathione S-transferase, alpha 4 | Gsta4 | 2.4 |
| glutathione S-transferase, mu 2 | Gstm2 | 3.5 |
| glutathione S-transferase, mu 3 | Gstm3 | 10.1 |
| glutathione S-transferase, mu 4 | Gstm4 | 3.2 |
| glutathione S-transferase, theta 3 | Gstt3 | 2.0 |
| NAD(P)H dehydrogenase, quinone 1 | Nqo1 | 5.1 |
| UDP glucuronosyltransferase 2 family, polypeptide B35 | Ugt2b35 | 1.5 |
Fold-change represents cKeap1-KO/genetic control. Independent 2 sample t-tests were performed on the log-signal strength to determine differential expression between the two groups (p≤0.05).
Amplification of Nrf2 signaling has no effect on early-phase soluble pro-inflammatory protein secretion
A possible explanation for the decreased immune-mediated liver injury in cKeap1-KO mice is that hepatocyte-specific deletion of Keap1, perhaps coupled with hypomorphic knockdown of Keap1 in other tissues, inhibited the initiation of the inflammatory response triggered by ConA injection. To address this possibility, the levels of soluble pro-inflammatory proteins in serum, 3-hr following ConA administration, were assessed to determine whether early-phase ConA-mediated pro-inflammatory cytokine/chemokine profiles were affected by amplification of Nrf2 signaling. Disruption of Keap1 had no effect on ConA-mediated cytokine or chemokine secretion as the serum concentrations of interferon γ IFNγ, tumor necrosis factor α (TNFα), monocyte chemoattractant protein (MCP-1) and interleukin- 2 (IL-2) were similar in cKeap1-WT and cKeap1-KO mice following ConA administration (Figure 3). Therefore, the protection afforded by disruption of Keap1 was directly due to Nrf2-dependent cytoprotection.
Figure 3.
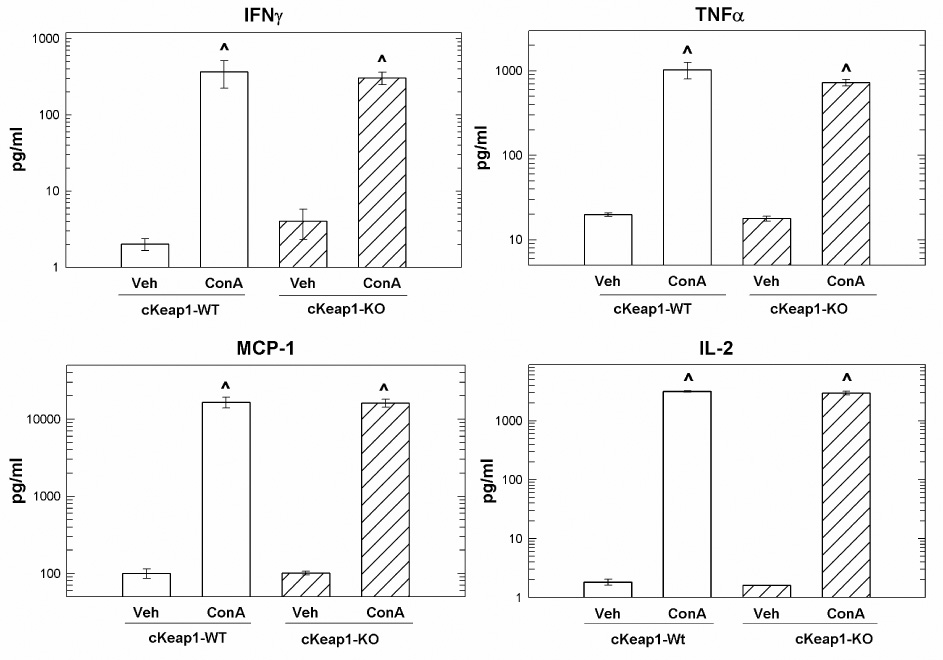
Hepatocyte-specific amplification of Nrf2 signaling has no effect on early-phase serum levels of soluble pro-inflammatory proteins. Concentrations of IFNγ, TNFα, MCP-1 and IL-2 in serum from cKeap1-KO and (N = 3) cKeap1-WT (N = 3) and B) vehicle-pretreated and CDDO-Im pretreated Nrf2-WT (N = 5) and Nrf2-KO (N = 4) mice 3-hr following treatment. Concentrations are expressed as pg/ml. Values represent the mean ± SEM of 3–5 animals. ^ p < 0.001 compared to respective vehicle-treated group, ANOVA-SNK analysis.
Conversely, CDDO-Im pretreatment resulted in decreased serum concentrations (pg/ml, mean ± SEM) of the pro-inflammatory mediators IFNγ (75%, vehicle-pretreated – 708.3 ± 151.3; CDDO-Im pretreated – 175.2 ± 24.4, p < 0.05) and TNFα (51%, vehicle-pretreated – 866.3 ± 53.5; CDDO-Im pretreated – 423.8 ± 36.6, p < 0.05) in CDDO-Im pretreated Nrf2-WT mice compared to vehicle-pretreated Nrf2-WT mice 3-hr following ConA administration (Figure 4). At the same time, CDDO-Im pretreatment had no effect on MCP-1 and IL-2 secretion as the serum concentrations of these proteins were similar in vehicle-pretreated Nrf2-WT and CDDO-Im pretreated Nrf2-WT mice. Likewise, decreased serum concentrations of IFNγ (91%, vehicle-pretreated – 3975.7 ± 1206.5; CDDO-Im pretreated – 329.0 ± 59.2, p < 0.05) and TNFα (57%, vehicle-pretreated – 1574.2 ± 61.2; CDDO-Im pretreated – 686.4 ± 32.7, p < 0.05) were detected in CDDO-Im pretreated Nrf2-KO mice compared to vehicle-pretreated Nrf2-KO mice at 3-hr suggesting that CDDO-Im partially suppressed the initial inflammatory response triggered by ConA injection in both Nrf2-WT and Nrf2-KO mice. At the same time, similar levels of IL-2 and MCP-1 were detected in the serum of vehicle-pretreated and CDDO-Im pretreated Nrf2-KO mice following ConA administration. Therefore, it appeared that CDDO-Im was inhibiting T-cell activation, in an Nrf2-independent manner, since secretion of IFNγ and a downstream target of IFNγ, TNFα, was attenuated by CDDO-Im pretreatment of both Nrf2-WT and Nrf2-KO mice.
Figure 4.
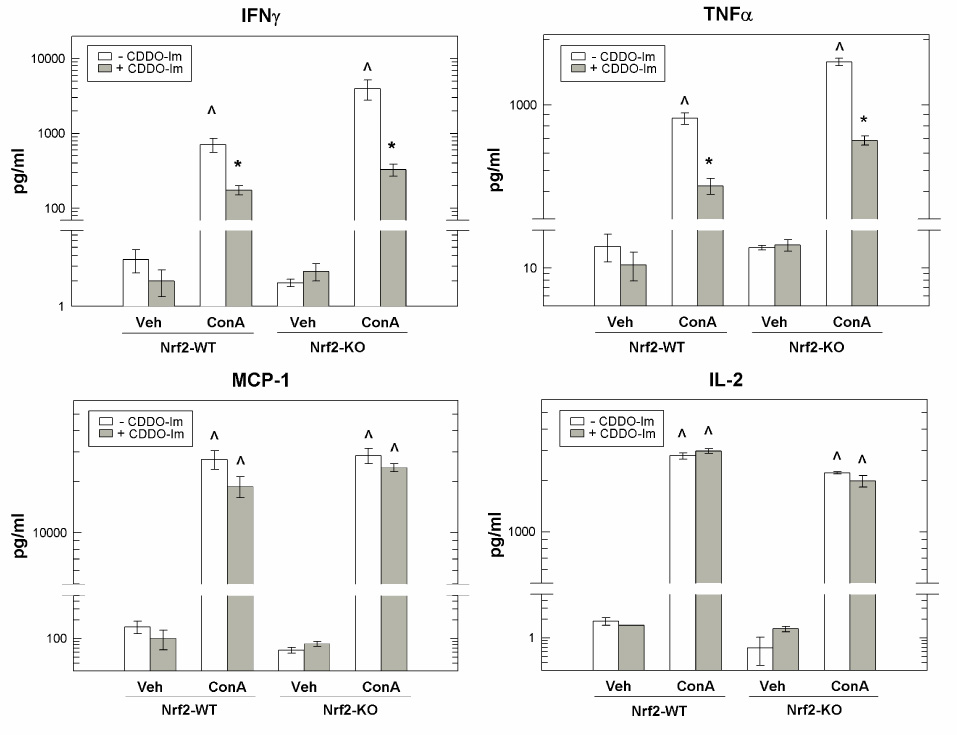
CDDO-Im pretreatment suppresses early-phase serum levels of soluble pro-inflammatory proteins in an Nrf2-independent fashion. Concentrations of IFNγ, TNFα, MCP-1 and IL-2 in serum from vehicle-pretreated and CDDO-Im pretreated Nrf2-WT (N = 5) and Nrf2-KO (N = 4) mice 3-hr following treatment. Gray bars indicate CDDO-Im pretreated groups. Concentrations are expressed as pg/ml. Values represent the mean ± SEM of 3–5 animals. ^ p < 0.001 compared to respective vehicle-treated group, ANOVA-SNK analysis. * p < 0.05 compared to vehicle-pretreated group of the same genotype, ANOVA-SNK analysis.
Late-phase hepatic inflammatory cell infiltration is attenuated by disruption of Keap1 and CDDO-Im pretreatment
The accumulation of hepatic macrophages and neutrophils 24-hr following ConA challenge was quantified to assess whether attenuation of late-phase hepatic inflammatory cell infiltration contributed to the protection afforded by genetic and pharmacological amplification of Nrf2 signaling. Lectin labeling of hepatic macrophages revealed a markedly increased accumulation of hepatic macrophages in cKeap1-WT mice 24-hr following ConA treatment; however, this response was diminished significantly in cKeap1-KO mice (Table 2). Similarly, increased numbers of hepatic macrophages were observed in vehicle-pretreated Nrf2-WT mice following ConA challenge and CDDO-Im pretreatment strikingly blunted this response. Chloroacetate esterase staining of liver sections revealed increased infiltration of neutrophils in livers from cKeap1-WT but not cKeap1-KO mice and vehicle-pretreated but not CDDO-Im pretreated Nrf2-WT mice 24-hr following ConA treatment (Table 2). Therefore, genetic and pharmacological amplification of Nrf2 signaling attenuated late-phase macrophage accumulation and inhibited neutrophil infiltration following ConA administration despite the presence of an early-phase inflammatory response triggered by ConA injection.
Table 2.
Disruption of Keap1 and CDDO-Im pretreatment inhibits hepatic inflammation following ConA administrationa.
| Genotype or Pre-treatment | Treatment | Macrophagesb | Neutrophilsb | Ifnγc | Tnfαc | Mip-2c | Mcp-1c |
|---|---|---|---|---|---|---|---|
| cKeap1-WT | Veh | 2.6 ± 0.2 | 1.6 ± 0.1 | 16.0 ± 5.2 | 10.2 ± 0.6 | 102.3 ± 56. 7 | 29.1 ± 3.9 |
| ConA | 19.2 ± 1.9 * | 14.5 ± 0.5 * | |||||
| cKeap1-KO | Veh | 2.6 ± 0.2 | 1.7 ± 0.2 | 1.4 ± 0.4 ** | 2.8 ± 1.3 ** | 1.4 ± 0.7 ** | 2.7 ± 1.2 ** |
| ConA | 8.1 ± 0.2 ^ | 2.4 ± 0.4 | |||||
| Nrf2-WT + Veh | Veh | 3.2 ± 0.2 | 1.0 ± 0.1 | 18.2 ± 5.4 | 26.0 ± 3.3 | 286.6 ± 87.5 | 66.1 ± 10.6 |
| ConA | 16.3 ± 1.2 * | 8.1 ± 0.7 * | |||||
| Nrf2-WT + CDDO-Im | Veh | 2.5 ± 0.3 | 0.9 ± 0.1 | 1.5 ± 0.4 ¶ | 2.5 ± 0.6 ¶ | 1.1 ± 0.2 ¶ | 2.4 ± 0.3 ¶ |
| ConA | 4.8 ± 0.3 ^ | 1.1 ± 0.2 | |||||
Livers were harvested 24-hr following ConA administration. Values represent the mean ± SE (Nrf2-WT, N = 5; cKeap1-KO/ cKeap1-WT; N = 3)
Data are expressed as cells/field
Gene transcript levels are expressed as relative fold-change (ConA/Veh)
p < 0.05 compared to all groups within genotype, ANOVA-SNK multiple comparisons
p < 0.05 compared to ConA treated group within genotype, ANOVA-SNK multiple comparisons
p < 0.05 compared to cKeap1-WT, Student’s t-test
p < 0.05 compared to Nrf2-WT + Veh, Student’s t-test
Genetic and pharmacological amplification of Nrf2 signaling inhibits hepatic cytokine and chemokine gene expression
We assessed hepatic cytokine and chemokine gene transcription 24-hr following ConA administration as a possible explanation for the reduction of inflammatory cell infiltration observed in cKeap1-KO and CDDO-pretreated Nrf2-WT mice. Markedly increased levels of Ifnγ, Tnfα, Mip-2 and Mcp-1 gene transcripts were detected in livers from cKeap1-WT mice compared to cKeap1-KO mice following ConA administration (Table 2). Similarly, markedly increased gene transcription of Ifnγ, Tnf-α, Mip-2 and Mcp-1 was detected in livers from vehicle-pretreated Nrf2-WT mice compared to CDDO-Im pretreated Nrf2-WT mice following ConA administration. Therefore, genetic amplification of Nrf2 signaling and CDDO-Im pretreatment effectively attenuated hepatic late-phase transcription of pro-inflammatory genes, thereby diminishing the pro-inflammatory stimulus in the liver resulting in decreased infiltration of inflammatory cells.
Discussion
Investigation of protective mechanisms against immune-mediated cell death can identify opportunities for the development of preventive strategies to block the progression of acute hepatitis to chronic hepatic inflammatory disease. This study demonstrated that amplification of Nrf2 signaling in hepatocytes is sufficient for attenuation of acute inflammatory liver injury. Enhanced Nrf2 signaling in hepatocytes led to elevated expression of antioxidative genes, thereby abrogating inflammation-associated oxidative stress and inhibiting hepatocyte necrosis. Interestingly, even though disruption of Keap1 had no effect on early-phase serum levels of soluble pro-inflammatory proteins; late-phase hepatic pro-inflammatory gene expression was substantially lower in cKeap1-KO mice compared to cKeap1-WT mice resulting in decreased accumulation of macrophages and neutrophils in livers following ConA challenge. Therefore, genetic amplification of Nrf2 signaling in hepatocytes had no effect on the initial immune response triggered by ConA injection, but instead resulted in striking protection against the subsequent inflammation-mediated hepatotoxicity and attendant additional hepatic inflammation.
Conversely, CDDO-Im pretreatment attenuated early-phase serum levels of soluble pro-inflammatory proteins in both Nrf2-WT mice and Nrf2-KO mice. However, this partially suppressed inflammatory response was nonetheless sufficiently robust to result in substantial hepatotoxicity in mice lacking functional Nrf2 signaling. Yates, et al (Yates et al. 2006) have previously demonstrated that pretreatment of Nrf2-WT mice but not Nrf2-KO mice results in increased expression of antioxidative genes in the liver. Therefore, the protective effect of CDDO-Im pretreatment can be attributed to enhanced expression of Nrf2-regulated cytoprotective genes in hepatocytes rather than impairment of T-cell activation and signaling. CDDO-Im pretreatment of Nrf2-WT mice also inhibited hepatic late-phase pro-inflammatory gene expression and inflammatory cell infiltration. Therefore, pharmacological activation of Nrf2 signaling mirrored genetic amplification by also effectively attenuating inflammation-mediated hepatotoxicity and preventing additional hepatic inflammation.
One plausible explanation for these observed outcomes in these two models of Nrf2 manipulation is the absence of inflammation-mediated hepatocyte necrosis in cKeap1 and CDDO-Im pretreated Nrf2-WT mice resulted in decreased activation of pro-inflammatory gene expression in macrophages and subsequent diminution of inflammatory cell infiltration in the liver. The presence of necrotic cell debris activates inflammatory signaling in macrophages. Maeda, et al(Maeda et al. 2005) reported that exposure of primary macrophages to a necrotic cell supernatant activated the pro-inflammatory transcription factor nuclear factor-kB (NF-kB). Additionally, high mobility group box 1 protein, a chromatin binding protein released from necrotic cells, has been shown to bind to Toll-like receptors 2 and 4 on macrophages resulting in NF-kB activation(Park et al. 2006) and TNFα release(Scaffidi et al. 2002). NF-kB also regulates the expression of MCP-1(Ueda et al. 1997), a macrophage chemokine, and MIP-2(Lee et al. 2005), a neutrophil chemokine, in macrophages. The hepatic expression of these chemokines was diminished in the current study in cKeap1-KO mice and CDDO-Im pretreated Nrf2-WT mice. Therefore, we speculate that up-regulation of Nrf2-regulated cytoprotective enzymes enhanced hepatocyte survival resulting in decreased production of necrotic cell debris and subsequent recognition by macrophages. This effect would serve to reduce inflammatory cell infiltration in the liver, thereby blocking a continued inflammatory response to the initial ConA challenge.
Attenuation of hepatic inflammatory responses through direct modulation of the immune system has been shown to inhibit acute inflammatory liver injury. Depletion of neutrophils(Bonder et al. 2004) and Kupffer cells(Schümann et al. 2000) provided substantial protection against ConA-mediated liver injury. Diminished acute inflammatory liver injury has also been demonstrated in mice lacking T cells(Tiegs et al. 1992) and natural killer T cells(Takeda et al. 2000). Additionally, the administration of immunomodulatory agents, such as pentoxifylline(Fukuda et al. 2005) and anti-TNFα(Fukuda et al. 2005), has been shown to provide protection against ConA-mediated inflammatory liver injury in mice. Current therapeutic options for treating acute inflammatory liver injury in humans involve the administration of immunomodulatory agents. However, the use of these agents is associated with a number of adverse effects due to the non-specific nature of their mechanism of action(Fantini et al. 2006). The current study highlights an approach that acts independently of immunomodulation but instead relies on activation of cellular defenses in parenchymal cells to combat non-specific inflammation-mediated destruction of normal tissue. Therefore, this approach might be expected to result in fewer side effects than traditional immunomodulatory therapies since the blunting of inflammation can be attributed to blocking the stimulus that is prolonging the inflammatory response without directly affecting initial pro-inflammatory signaling.
In summary, the current study clearly demonstrated the importance of Nrf2 signaling for protection against acute inflammatory liver injury. Nrf2-dependent up-regulation of cytoprotective enzymes in the target organ of the ConA-mediated inflammatory response resulted in profound protection against immune-mediated hepatitis by preventing hepatocyte necrosis and subsequent amplification of the inflammatory response. Inflammatory liver injury represents a serious burden to human health in that progression of acute inflammatory liver injury to chronic inflammatory liver disease increases risk of liver cancer. In fact, hepatocellular carcinoma due to cirrhosis, an end-stage liver disease that may develop from chronic hepatitis due to either chronic alcohol consumption or hepatitis B or C infection, accounts for nearly 80% of all liver cancer cases(Kensler et al. 2003). Therefore, the development of strategies designed to block the progression of acute inflammatory liver injury to chronic inflammatory diseases would prove beneficial to ameliorating the burden of liver cancer in human populations. Collectively, the current study identifies targeting the Nrf2 signaling pathway with inducers such as triterpenoids to bolster the cell survival defenses of uninfected or non-damaged cells as a potentially powerful mechanism for attenuation of acute inflammatory injury that may complement current therapeutic strategies of diminishing pro-inflammatory signaling.
Acknowledgements
The authors would like to thank the Becton Dickinson Immune Function Laboratory for assisting with the cytometric bead array analysis and Dr. Baktiar Karim for assistance with histological analysis.
Funding Funding for this research was provided by National Institutes of Health (R01CA39416, R01CA94076, P30ES03819, R01CA78814 and T32ES07141), National Foundation for Cancer Research, Reata Pharmaceuticals, and ERATO-JST.
References
- Adachi T, Pimentel D, Heibeck V, Hou X, Lee Y, Jiang B, Ido Y, Cohen R. S-Glutathiolation of Ras Mediates Redox-sensitive Signaling by Angiotensin II in Vascular Smooth Muscle Cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- Bonder C, Ajuebor M, Zbytnuik L, Kubes P, Swain M. Essential role for neutrophil recruitment to the liver in concanavalin A-induced hepatitis. J Immunol. 2004;172:45–53. doi: 10.4049/jimmunol.172.1.45. [DOI] [PubMed] [Google Scholar]
- Ceccanti M, Attili A, Balducci G, Attilia F, Giacomelli S, Rotondo C, Sasso G, Xirouchakis E, Attilia M. Acute alcoholic hepatitis. J Clin Gastroenterol. 2006;40:833–841. doi: 10.1097/01.mcg.0000225570.04773.5d. [DOI] [PubMed] [Google Scholar]
- Choi J, Ou J. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am J Physiol Gastrointest Liver Physiol. 2006;290:G847–G851. doi: 10.1152/ajpgi.00522.2005. [DOI] [PubMed] [Google Scholar]
- Cook G, Campbell J, Carr C, Boyd K, Franklin I. Transforming growth factor b from multiple myeloma cells inhibits proliferation and IL-2 responsiveness in T lymphocytes. J Leukoc Biol. 1999;66:981–988. doi: 10.1002/jlb.66.6.981. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of keap1 in cellular protective responses. Chem. Res. Toxicol. 2005;18:1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- Fadl A, Galindo C, Sha J, Zhang F, Garner H, Wang H, Chopra A. Global transcriptional responses of wild-type Aeromonas hydrophila and its virulence-deficient mutant in a murine model of infection. Microb Pathog. 2007;42:193–203. doi: 10.1016/j.micpath.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Fantini M, Becker C, Kiesslich R, Neurath M. Drug insight: novel small molecules and drugs for immunosuppression. Nat Clin Pract Gastroenterol Hepatol. 2006;3:633–644. doi: 10.1038/ncpgasthep0611. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Mogami A, Hisadome M, Komatsu H. Therapeutic administration of Y-40138, a multiple cytokine modulator, inhibits concanavalin A-induced hepatitis in mice. Eur J Pharmacol. 2005;523:137–142. doi: 10.1016/j.ejphar.2005.08.060. [DOI] [PubMed] [Google Scholar]
- Hagen M, Fagan K, Steudel W, Carr M, Lane K, Rodman D, West J. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1473–L1479. doi: 10.1152/ajplung.00197.2006. [DOI] [PubMed] [Google Scholar]
- Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, Shimazui T, Akaza H, Yamamoto M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004;64:6424–6431. doi: 10.1158/0008-5472.CAN-04-1906. [DOI] [PubMed] [Google Scholar]
- Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, Kelly V, Sekizawa K, Uchida K, Yamamoto M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2) Mol Cell Biol. 2004;24:36–45. doi: 10.1128/MCB.24.1.36-45.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H. Role of inflammation in the mechanism of acetaminophen-induced hepatotoxicity. Expert Opin Drug Metab Toxicol. 2005;1:389–397. doi: 10.1517/17425255.1.3.389. [DOI] [PubMed] [Google Scholar]
- Ji Y, Lee H, Goodman C, Uskokovic M, Liby K, Sporn M, Suh N. The synthetic triterpenoid CDDO-imidazolide induces monocytic differentiation by activating the Smad and ERK signaling pathways in HL60 leukemia cells. Mol Cancer Ther. 2006;5:1452–1458. doi: 10.1158/1535-7163.MCT-06-0136. [DOI] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFa-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten F. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Kawasuji A, Hasegawa M, Horikawa M, Fujita T, Matsushita Y, Matsushita T, Fujimoto M, Steeber D, Tedder T, Takehara K, Sato S. L-selectin and intercellular adhesion molecule-1 regulate the development of Concanavalin A-induced liver injury. J Leukoc Biol. 2007;79:696–705. doi: 10.1189/jlb.0905527. [DOI] [PubMed] [Google Scholar]
- Kensler T, Qian G, Chen J, Groopman J. Translational strategies for cancer prevention in liver. Nat Rev Cancer. 2003;3:321–329. doi: 10.1038/nrc1076. [DOI] [PubMed] [Google Scholar]
- Khor T, Huang M, Kwon K, Chan J, Reddy B, Kong A. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium–induced colitis. Cancer Res. 2006;66:11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Kruisbeek A. Unit 3.1: Isolation of Mouse Mononuclear Cells. In: Coligan J, Bierer B, Margulies D, Shevach E, Strober W, editors. Current Protocols in Immunology. John Wiley & Sons, Inc; 2000. [DOI] [PubMed] [Google Scholar]
- Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- Lee K, Lee Y, Kwon H, Kim D. Sp1-associated activation of macrophage inflammatory protein-2 promoter by CpG-oligodeoxynucleotide and lipopolysaccharide. Cell Mol Life Sci. 2005;62:188–198. doi: 10.1007/s00018-004-4399-y. [DOI] [PubMed] [Google Scholar]
- Leist M, Wendel A. A novel mechanism of murine hepatocyte death inducible by concanavalin A. J Hepatol. 1996;25:948–959. doi: 10.1016/s0168-8278(96)80301-1. [DOI] [PubMed] [Google Scholar]
- Liby K, Honda T, Williams C, Risingsong R, Royce D, Suh N, Dinkova-Kostova A, Stephenson K, Talalay P, Sundararajan C, Gribble G, Sporn M. Novel synthetic derivatives of betulinic acid, with diverse cytoprotective, anti-proliferative, or pro-apoptotic activities. Mol Can Ther. 2007;6:2113–2119. doi: 10.1158/1535-7163.MCT-07-0180. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo J, Leffert H, Karin M. IKKb couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Motohashi H, Katsuoka F, Engel JD, Yamamoto M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci U S A. 2004;101:6379–6384. doi: 10.1073/pnas.0305902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto Y, Kaneko S. Mechanisms of viral hepatitis induced liver injury. Curr Mol Med. 2003;3:537–544. doi: 10.2174/1566524033479591. [DOI] [PubMed] [Google Scholar]
- Ohshima H, Tatemichi M, Sawa T. Chemical basis of inflammation-induced carcinogenesis. Arch Biochem Biophys. 2003;417:3–11. doi: 10.1016/s0003-9861(03)00283-2. [DOI] [PubMed] [Google Scholar]
- Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler T, Yamamoto M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun. 2006;339:79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- Osburn W, Karim B, Dolan P, Li Q, Yamamoto M, Huso D, Kensler T. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J CancerInt J Cancer. 2007;121:1883–1891. doi: 10.1002/ijc.22943. [DOI] [PubMed] [Google Scholar]
- Osburn W, Wakabayashi N, Misra V, Nilles T, Biswal S, Trush M, Kensler T. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch Biochem Biophys. 2006;454:7–15. doi: 10.1016/j.abb.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbergh L, Valckx D, Waer M, Mathieu C. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine. 1999;11:305–312. doi: 10.1006/cyto.1998.0426. [DOI] [PubMed] [Google Scholar]
- Park J, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim J, Strassheim D, Sohn J, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–C924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- Pfaffl M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy T, Guo J, Mitzner W, Roman J, Singh A, Fryer A, Yamamoto M, Kensler T, Tuder R, Georas S, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi M. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Schümann J, Wolf D, Pahl A, Brune K, Papadopoulos T, Rooijen Nv, G T. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am J Pathol. 2000;157:1671–1683. doi: 10.1016/S0002-9440(10)64804-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh N, Roberts A, Reffey SB, Miyazono K, Itoh S, Dijke Pt, Heiss E, Place A, Risingsong R, Williams C, Honda T, Gribble G, Sporn M. Synthetic triterpenoids enhance transforming growth factor beta/Smad signaling. Cancer Res. 2003;63:1371–1376. [PubMed] [Google Scholar]
- Takeda K, Hayakawa Y, Kaer LV, Matsuda H, Yagita H, Okumura K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A. 2000;97:5498–5503. doi: 10.1073/pnas.040566697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa R, Lee H, Rangasamy T, Reddy S, Yamamoto M, Kensler T, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiegs G, Hentschel J, Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest. 1992;90:196–203. doi: 10.1172/JCI115836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda A, Ishigatsubo Y, Okubo T, Yoshimura T. Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-kappaB sites and NF-kappaB/Rel subunit specificity. J Biol Chem. 1997;272:31092–31099. doi: 10.1074/jbc.272.49.31092. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop D, Harada T, Engel J, Yamamoto M. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- Yates M, Kwak M, Egner P, Groopman J, Bodreddigari S, Sutter T, Baumgartner K, Roebuck B, Liby K, Yore M, Honda T, Gribble G, Sporn M, Kensler T. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 2006;66:2488–2494. doi: 10.1158/0008-5472.CAN-05-3823. [DOI] [PubMed] [Google Scholar]
- Yates M, Tauchi M, Katsuoka F, Flanders K, Liby K, Honda T, Gribble G, Johnson D, Johnson J, Burton N, Guilarte T, Yamamoto M, Sporn M, T K. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6:154–162. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]