

ABSTRACT
Genetic mutations underlying thrombophilia are often recognized in patients with thromboembolic episodes. However, the clinical and therapeutic implications of such findings often remain unclear. We report the first case of a dural arteriovenous fistula (DAVF) in a patient with a combined factor II and factor V Leiden mutation. A 40-year-old man presented with a large left temporal and intraventricular hemorrhage. An initial angiogram showed thrombosis of the left sigmoid sinus but no evidence of a vascular malformation. One year after the hemorrhage, an angiographic study showed the appearance of a right DAVF. During the follow-up period, the patient was found to harbor heterozygosity for a mutation of factor V and a mutation of factor II. Recognition of the patient's thrombophilia led to prolonged oral anticoagulation therapy to reduce the risk of a recurrent thrombotic episode. Despite the increased risk of bleeding, the therapy was considered justified. DAVFs may occur after sinus thrombosis in patients with combined factor II and factor V mutations. This observation indicates the association of multiple hematological disorders with DAVFs in individual patients. Moreover, it raises the clinical conundrum of how to manage patients with thrombophilia, intracranial hemorrhage, and DAVFs.
Keywords: Factor V Leiden mutation, G20210A mutation, arteriovenous malformation, anticoagulation, hypercoagulable state
Dural arteriovenous fistulas (DAVFs), uncommon direct artery-to-venous sinus shunts, constitute ~12% of all arteriovenous malformations (AVMs).1 Depending on the location and hemodynamics of the lesion, bruit, focal neurological deficits, and visual symptoms represent the most common modes of presentation.2 Although rare, intracranial hemorrhage can also occur.1
Originally, DAVFs were thought to arise congenitally.3,4 Recent studies have emphasized the role of sinus thrombosis and impaired venous outflow.5,6,7,8,9 The elevated sinus pressure related to sinus thrombosis may favor the development of dysplastic dural vessels within the sinus. The dysplastic vessels establish a direct artery-to-sinus communication and cause the preexisting microarteriovenous fistulas in the sinus walls to open.6,8 Sinus thrombosis may result from a large variety of causes, either acquired or congenital.10,11 Among congenital factors, a single point mutation in the gene coding for coagulation factor V has been associated with sinus thrombosis in several reports.10,11,12,13,14 This mutation, also known as factor V Leiden mutation, results in a form of factor V that is resistant to degradation by activated protein C and leads to a hypercoagulable state.14,15,16,17,18,19 Similarly, a single point mutation in the prothrombin (factor II) genes, known as G20210A mutation, has been found to be among the most common independent risk factors for venous thrombosis.20,21 The combination of the two mutations has also been described.22,23 Patients with combined defects have a higher risk of venous thrombosis than those with individual defects.22,23 Both mutations have also been found in some patients with DAVFs.12,24,25 In a recent study, the frequency of prothrombin gene 20210A mutation was higher in patients with DAVF compared with the general population, whereas the incidence of factor V Leiden mutation was not.25
To our knowledge, the present case is the first documented report of the formation of a DAVF after sinus thrombosis and intracranial hemorrhage in a patient with a combined factor II G20210A and factor V Leiden mutation. This observation adds a further element to the association of DAVFs and sinus thrombosis caused by hematological disorders. Moreover, it raises the problem of clinical management in patients with a hypercoagulable state, intracranial hemorrhage, and DAVFs.
CASE REPORT
A 40-year-old man was transferred to our clinic after a severe cephalgic episode associated with acute aphasia. His neurological examination indicated sensory aphasia and quadrantanopsia. He had no other focal neurological deficits. Computed tomography (CT) and magnetic resonance imaging (MRI) of the head showed a large intracerebral and intraventricular hemorrhage involving the left temporal lobe (Fig. 1A). Cerebral angiography showed no findings suggestive of a vascular malformation. However, thrombosis of the left sigmoid sinus was found (Fig. 1B–D). The hematoma was evacuated through a left temporal craniotomy. Postoperatively, the patient recovered and his aphasia resolved over time. Histopathological examination of the specimen showed no finding indicative of an AVM or angiopathy. Routine cardiovascular, radiological, and hematological studies and drug screening performed during the patient's hospitalization showed no source of thrombosis, thromboembolism, or factors other than the sinus thrombosis that might have contributed to the intracranial hemorrhage. Routine anticoagulation therapy (with heparin) was started.
Figure 1.
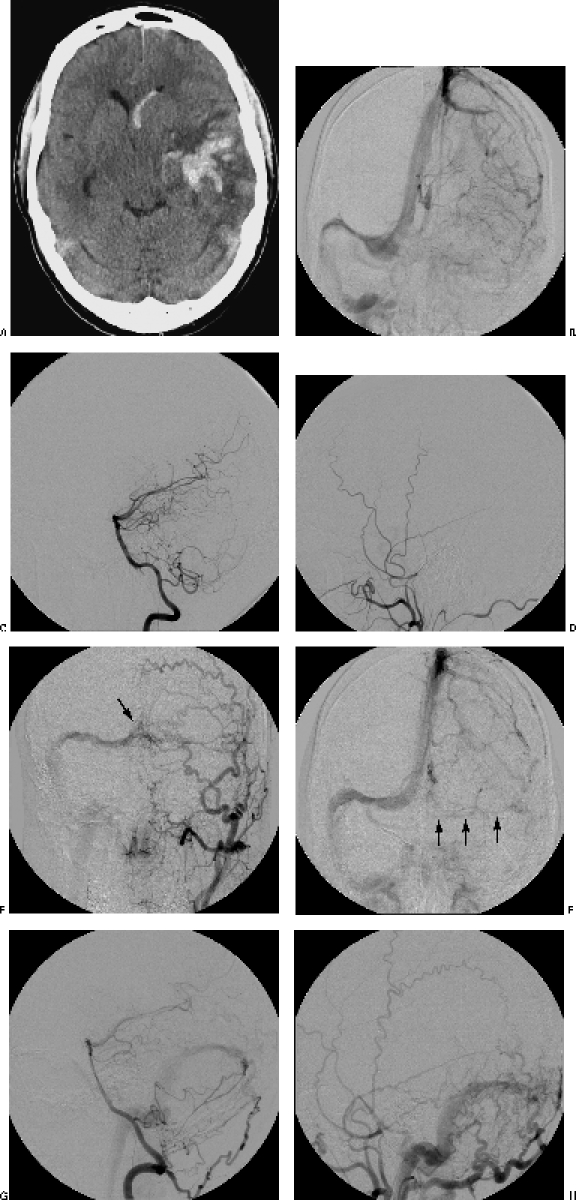
(A) Preoperative computed tomographic image shows the left intracerebral and intraventricular hemorrhage. (B) Preoperative angiogram shows thrombosis of the left sigmoid sinus. However, the lateral projections of (C) the posterior circulation and (D) the left external carotid angiogram show no DAVF. Control angiograms 1 year after treatment show (F) partial patency of the left sigmoid sinus and (E,G,H) the formation of a DAVF between tentorial branches of the (E) left external carotid artery, (G) right vertebral artery, and (H) meningeal branches of the external carotid artery to (E) the right transverse sinus and confluens (arrow).
During the follow-up period, the patient underwent a second hematological screening that revealed both a factor V Leiden mutation with activated protein C-resistance and a G20210A mutation of the prothrombin gene. Anticoagulation therapy was then pursued with warfarin (international normalized ratio [INR], between 2 and 3). Control MRI studies were negative for vascular malformations or new hemorrhage. The patient's only complaint was bilateral pulsatile tinnitus, which developed almost a year after his hemorrhage. Follow-up angiography 12 months after the first angiogram showed a DAVF between meningeal branches of the external carotid artery, tentorial branches of the left internal carotid artery, and the right vertebral artery to the right transverse sinus (Fig. 1E–G). Based on the patient's venous anatomy, the DAVF was defined as a benign, Cognard type IIa lesion, with absence of cortical venous drainage.26,27 On the left side, the patient's sigmoid sinus was partially recanalized (Fig. 1E). The decision was made to withhold treatment of the fistula until the patient became clinically symptomatic.
Despite the risk of hemorrhage, oral anticoagulation was continued because we believe that the underlying combined factor II and factor V Leiden mutations increase the patient's risk of a thrombotic complication.
DISCUSSION
The mechanisms underlying the formation of DAVFs have been long debated. In 1979 Houser et al7 suggested that sinus thrombosis was involved in the development of AVFs, although stenosis or occlusion of the sinus lumen was not always present in these fistulas.28 More recently, animal models have confirmed that sinus thrombosis and elevated sinus pressure may result in the formation of DAVFs.6,29,30,31
The formation of a DAVF was documented angiographically in our patient 1 year after the acute episode with which he presented. At that time, his venous sinus was partially patent again (Fig. 2). On the basis of the absence of cortical venous drainage in our case, the DAVF was regarded as benign (Cognard type IIa).26,27 In their series of 117 patients with DAVFs, Satomi and colleagues27 found that patients with DAVFs without cortical venous drainage (Cognard type I and IIa lesions) can be managed conservatively. Our patient tolerated the DAVF well; therefore, no treatment was performed. Because there is a risk that cortical venous drainage will develop,27 the patient is followed by clinical and radiological controls with both angiographic MRI and conventional angiography. Treatment of the fistula would be indicated if the patient's clinical or radiological status changes.
Figure 2.

The flowchart shows the mechanism proposed to underlie the development of the patient's complex clinical picture. The genetic process begins with the mutation that caused the thrombosis and hemorrhage (1). Increased venous pressure (2) leads to the DAVF (3). Treatment with anticoagulation is a dilemma because of its beneficial effects on thromboembolic events and its adverse effects on hemorrhage. The dotted lines indicate the possible positive effects on recanalization of the thrombosed sinus but the increased risk of hemorrhage and development of the DAVF. (With permission from Barrow Neurological Institute.)
Sinus Thrombosis
Sinus thrombosis is an uncommon condition that may have numerous causes.10,11,13 It usually occurs in young, healthy people.13 The most common sites of thrombosis are the transverse and sigmoid sinuses.13 Sinus thrombosis can lead to brain edema, infarction, or intracranial hemorrhage, as in our patient, and also to pulmonary embolism.11,13
There is a high rate of misdiagnosis, which can preclude prompt and aggressive intervention.13 In fact, our patient was transferred to our clinic several days after he initially developed symptoms, thereby delaying diagnosis and treatment of the sinus thrombosis.
In the 1940s, heparin was suggested for the treatment of sinus thrombosis. In 1991 Einhaupl et al32 confirmed that dose-adjusted intravenous heparin was an effective treatment for sinus thrombosis and showed that intracranial hemorrhage is not a contraindication to heparin treatment.11,13 The development of active thrombolytic therapies has widened the spectrum of management options. The ideal therapy for sinus thrombosis, however, remains controversial.11,13 Different treatments are available: systemic anticoagulation therapies with heparin alone or in association with endovascular chemical thrombolysis or mechanical endovascular thrombectomy.
A recent retrospective study of 31 cases of dural sinus thrombosis compared different treatment strategies. Soleau et al13 confirmed that observation alone is insufficient and that anticoagulation therapy may lead to improvement in 75% of the cases. Chemical thrombolysis or mechanical thrombectomy led to improvement in 60% and 88% of their cases, respectively. Particularly, chemical or mechanical thrombolysis may lead to a more rapid recanalization of the sinus than heparin alone and thus may help reduce venous pressure.11,13
However, these methodologies are also burdened by a higher rate of hemorrhagic complications (with high morbidity and mortality), especially in patients with a preexisting intracranial hemorrhage or in patients referred for surgery, as in our patient. Conversely, no new hemorrhagic episodes were identified after anticoagulation therapy alone.13 Although a more aggressive intervention may have prevented the venous hypertension and associated DAVF in our patient, it also may have resulted in hemorrhagic complications. Moreover, treatment of a primary cause, when identified, should be part of the comprehensive management of sinus thrombosis. However, despite extensive investigations in cases of sinus thrombosis, no cause may be found in as many as 20% of the cases.11
Thrombophilia Due to Factor V Leiden and Factor II Mutations
Two common genetic risk factors for venous thrombosis have now been identified. In the early 1990s, investigators in Sweden and the Netherlands reported a novel pathogenetic risk factor for thrombosis: inherited activated protein C-resistance, also referred to as factor V Leiden mutation.14,15,16,33 Since then several clinical studies have indicated that this mutation is associated with an increased risk for primary and recurrent venous thrombosis.17
In most cases, activated protein C-resistance is caused by a single point mutation in the gene coding for factor V (G to A transition at nucleotide position 1691). As a result, a single amino acid, glutamine, is replaced for arginine at position 506 in the activated protein C cleavage site (Arg(R)506 to Gln(Q)).34,35 Activated protein C degrades factor V. The mutated form, FV Leiden or FV:Q506, is less efficiently degraded than the normal FV. Thrombin generation then increases and a hypercoagulable state follows.34 Factor V Leiden mutation is highly prevalent in the general population, especially in Europeans.36 It may occur in 20 to 60% of patients with venous thrombosis.14,34
Heterozygosity for FV:Q506 is associated with a 5- to 10-fold increased risk of thrombosis. By comparison, homozygous cases have a 50- to 100-fold increased risk of thrombosis.34 Our patient was found to be heterozygous for factor V Leiden mutation. This finding is consistent with a previous report that found DAVFs in three of seven patients with heterozygosity for factor V Leiden mutation.24
The factor V Leiden mutation represents the most common genetic risk factor for venous thrombosis.14,15,16,37 However, as many as 15% of the cases with activated protein C-resistance are not caused by factor V Leiden mutation. Rather, they are due to acquired, phenotypic-activated protein C-resistance in conditions such as pregnancy or systemic lupus erythematosus.23,37 Some authors have emphasized that activated protein C-resistance should be suspected during the acute phase in all patients with sinus thrombosis even in absence of a personal or family history of venous thrombosis.10 For patients with negative results, they recommend another search after the acute phase has resolved.10 During the acute phase, as in our case, the results of the investigations may transiently be falsely negative.10
Prothrombin (factor II) is a precursor of thrombin that plays a critical role in clotting.21 Poort and colleagues20 identified that substitution of adenine for guanine at position 20210 (G20210A) in the noncoding 3′ terminal end of the prothrombin gene is associated with high levels of prothrombin. It produces a hypercoagulable state. Similar to factor V Leiden mutation, prothrombin G20210A mutation increases the risk of venous thromboembolism.20,21,38,39 This mutation is also predominant in the European population.20,36 Among genetic causes of venous thrombosis, prothrombin G20210A mutation represents, after the factor V Leiden mutation, the second most common independent risk factor.23,39 In particular, the prevalence of the prothrombin G20210A mutation is higher in patients with cerebral vein thrombosis (20%) than in healthy controls (3%).40
A further increase in the risk of recurrence after a first thrombotic episode in carriers of the G20210A mutation alone has not been found in all studies.41 Thus, it has been suggested that those carriers should be treated for venous thrombosis no longer than patients with a normal genotype.41 However, other studies have shown that the association of both prothrombin G20210A and factor V Leiden mutation increases not only the risk of developing venous thrombotic episodes23,38,40 but also the risk of recurrences.22,39 In fact, de Stefano et al22 found that patients who were heterozygous for both factor V Leiden and prothrombin G20210A had a 2.6-fold higher risk of recurrent thrombosis than carriers of factor V Leiden alone.
Management of thrombophilia related to factor II and V Leiden mutations is still debated.18,20,42 Although maintaining an INR of 2 to 3 with oral anticoagulation therapy seems highly effective in preventing thrombotic recurrences, this benefit is partially offset by the risk of major bleeding.20,42 Short-term prophylaxis has been recommended during high-risk situations, especially in homozygous patients.18,20 However, a decision to prolong the duration of the anticoagulation therapy must be tailored to the individual patient's risk of recurrent thrombosis in the absence of treatment and to the risk of bleeding.39,42 Our case was particularly challenging because the patient presented with an intracranial hemorrhage and because of the appearance of his DAVF. However, these pathologies were probably related to venous thrombosis caused by the underlying complex hematological disorder. Consequently, we pursued treatment with anticoagulation therapy to prevent further thrombotic episodes. Further studies to assess the management of patients with the combination of these genetic mutations and a DAVF are needed. These common mutations may result in a hypercoagulable state that can be associated with spontaneous venous thrombosis (Fig. 3). As confirmed in our case and recommended previously in the literature,25 in cases of spontaneous intracranial sinus thrombosis of unknown cause and development of a DAVF, screening for thrombophilic genetic polymorphism may be indicated. The need for long-term, even perhaps lifelong, anticoagulant therapy in these individuals must be considered in the management.
Figure 3.

Schematic representation of a proposed mechanism that explains the appearance of the fistula and its implications for management. (1) Sinus occlusion was associated with increased venous pressure (2) and intracerebral hemorrhage. (3) Appearance of the DAVF. The numbers correspond to the numbers in Fig. 2. (With permission from Barrow Neurological Institute.)
CONCLUSIONS
Sinus thrombosis, intracranial hemorrhage, and DAVF represent separate clinical entities that are pathologically related in some patients. In the present case, these three different pathologies may all have been initiated by an unrecognized hypercoagulable state caused by an underlying combined prothrombin and factor V Leiden mutation. These common mutations may result in a hypercoagulable state that can be associated with spontaneous venous thrombosis. When a venous sinus is affected, the increased pressure in the venous system may induce intracranial hemorrhage and the appearance of large DAVFs. Hematological screening for factors mutations may produce false-negatives during the acute phase and should be repeated at a later time.
REFERENCES
- Lanzino G, Jensen M E, Kongable G L, Kassell N F. Angiographic characteristics of dural arteriovenous malformations that present with intracranial hemorrhage. Acta Neurochir (Wien) 1994;129:140–145. doi: 10.1007/BF01406493. [DOI] [PubMed] [Google Scholar]
- Halbach V V. In: Valavanis A, editor. Interventional Neuroradiology. Berlin/New York: Springer-Verlag; 1993. Embolization of dural arteriovenous malformations. pp. 35–54.
- Gordon I J, Shah B L, Hardman D R, Chameides L. Giant dural supratentorial arteriovenous malformation. AJR Am J Roentgenol. 1977;129:734–736. doi: 10.2214/ajr.129.4.734. [DOI] [PubMed] [Google Scholar]
- Newton T H, Cronqvist S. Involvement of dural arteries in intracranial arteriovenous malformations. Radiology. 1969;93:1071–1078. doi: 10.1148/93.5.1071. [DOI] [PubMed] [Google Scholar]
- Awad I A, Little J R, Akarawi W P, Ahl J. Intracranial dural arteriovenous malformations: factors predisposing to an aggressive neurological course. J Neurosurg. 1990;72:839–850. doi: 10.3171/jns.1990.72.6.0839. [DOI] [PubMed] [Google Scholar]
- Herman J M, Spetzler R F, Bederson J B, Kurbat J M, Zabramski J M. Genesis of a dural arteriovenous malformation in a rat model. J Neurosurg. 1995;83:539–545. doi: 10.3171/jns.1995.83.3.0539. [DOI] [PubMed] [Google Scholar]
- Houser O W, Campbell J K, Campbell R J, Sundt T M., Jr Arteriovenous malformation affecting the transverse dural venous sinus: an acquired lesion. Mayo Clin Proc. 1979;54:651–661. [PubMed] [Google Scholar]
- Mullan S. Reflections upon the nature and management of intracranial and intraspinal vascular malformations and fistulae. J Neurosurg. 1994;80:606–616. doi: 10.3171/jns.1994.80.4.0606. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Miyasaka Y, Tanaka R, Kurata A, Fujii K. Dural-pial arteriovenous malformation after sinus thrombosis. Stroke. 1998;29:1721–1724. doi: 10.1161/01.str.29.8.1721. [DOI] [PubMed] [Google Scholar]
- Deschiens M A, Conard J, Horellou M H, et al. Coagulation studies, factor V Leiden, and anticardiolipin antibodies in 40 cases of cerebral venous thrombosis. Stroke. 1996;27:1724–1730. doi: 10.1161/01.str.27.10.1724. [DOI] [PubMed] [Google Scholar]
- Kimber J. Cerebral venous sinus thrombosis. QJM. 2002;95:137–142. doi: 10.1093/qjmed/95.3.137. [DOI] [PubMed] [Google Scholar]
- Kraus J A, Stuper B K, Nahser H C, Klockgether T, Berlit P. Significantly increased prevalence of factor V Leiden in patients with dural arteriovenous fistulas. J Neurol. 2000;247:521–523. doi: 10.1007/s004150070150. [DOI] [PubMed] [Google Scholar]
- Soleau S W, Schmidt R, Stevens S, Osborn A, MacDonald J D. Extensive experience with dural sinus thrombosis. Neurosurgery. 2003;52:534–544. doi: 10.1227/01.neu.0000047815.21786.c1. [DOI] [PubMed] [Google Scholar]
- Svensson P J, Dahlback B. Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med. 1994;330:517–522. doi: 10.1056/NEJM199402243300801. [DOI] [PubMed] [Google Scholar]
- Dahlback B, Carlsson M, Svensson P J. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA. 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster T, Rosendaal F R, de Ronde H, Briet E, Vandenbroucke J P, Bertina R M. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet. 1993;342:1503–1506. doi: 10.1016/s0140-6736(05)80081-9. [DOI] [PubMed] [Google Scholar]
- Price D T, Ridker P M, Factor V. Leiden mutation and the risks for thromboembolic disease: a clinical perspective. Ann Intern Med. 1997;127:895–903. doi: 10.7326/0003-4819-127-10-199711150-00007. [DOI] [PubMed] [Google Scholar]
- Rosendaal F R, Koster T, Vandenbroucke J P, Reitsma P H. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance) Blood. 1995;85:1504–1508. [PubMed] [Google Scholar]
- Sun X, Evatt B, Griffin J H. Blood coagulation factor Va abnormality associated with resistance to activated protein C in venous thrombophilia. Blood. 1994;83:3120–3125. [PubMed] [Google Scholar]
- Poort S R, Rosendaal F R, Reitsma P H, Bertina R M. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88:3698–3703. [PubMed] [Google Scholar]
- Ridker P M, Hennekens C H, Miletich J P. G20210A mutation in prothrombin gene and risk of myocardial infarction, stroke, and venous thrombosis in a large cohort of US men. Circulation. 1999;99:999–1004. doi: 10.1161/01.cir.99.8.999. [DOI] [PubMed] [Google Scholar]
- De Stefano V, Martinelli I, Mannucci P M, et al. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med. 1999;341:801–806. doi: 10.1056/NEJM199909093411104. [DOI] [PubMed] [Google Scholar]
- Zoller B, Garcia D F, Hillarp A, Dahlback B. Thrombophilia as a multigenic disease. Haematologica. 1999;84:59–70. [PubMed] [Google Scholar]
- Kraus J A, Stuper B K, Berlit P. Association of resistance to activated protein C and dural arteriovenous fistulas. J Neurol. 1998;245:731–733. doi: 10.1007/s004150050276. [DOI] [PubMed] [Google Scholar]
- Gerlach R, Yahya H, Rohde S, et al. Increased incidence of thrombophilic abnormalities in patients with cranial dural arteriovenous fistulae. Neurol Res. 2003;25:745–748. doi: 10.1179/016164103101202101. [DOI] [PubMed] [Google Scholar]
- Cognard C, Gobin Y P, Pierot L, et al. Cerebral dural arteriovenous fistulas: clinical and angiographic correlation with a revised classification of venous drainage. Radiology. 1995;194:671–680. doi: 10.1148/radiology.194.3.7862961. [DOI] [PubMed] [Google Scholar]
- Satomi J, Dijk J M van, Terbrugge K G, Willinsky R A, Wallace M C. Benign cranial dural arteriovenous fistulas: outcome of conservative management based on the natural history of the lesion. J Neurosurg. 2002;97:767–770. doi: 10.3171/jns.2002.97.4.0767. [DOI] [PubMed] [Google Scholar]
- Hamada Y, Goto K, Inoue T, et al. Histopathological aspects of dural arteriovenous fistulas in the transverse-sigmoid sinus region in nine patients. Neurosurgery. 1997;40:452–456. doi: 10.1097/00006123-199703000-00005. [DOI] [PubMed] [Google Scholar]
- Terada T, Higashida R T, Halbach V V, et al. Development of acquired arteriovenous fistulas in rats due to venous hypertension. J Neurosurg. 1994;80:884–889. doi: 10.3171/jns.1994.80.5.0884. [DOI] [PubMed] [Google Scholar]
- Lawton M T, Jacobowitz R, Spetzler R F. Redefined role of angiogenesis in the pathogenesis of dural arteriovenous malformations. J Neurosurg. 1997;87:267–274. doi: 10.3171/jns.1997.87.2.0267. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Lawton M T, Du R, et al. Expression of hypoxia-inducible factor-1 and vascular endothelial growth factor in response to venous hypertension. Neurosurgery. 2006;59:687–696. doi: 10.1227/01.NEU.0000228962.68204.CF. [DOI] [PubMed] [Google Scholar]
- Einhaupl K M, Villringer A, Meister W, et al. Heparin treatment in sinus venous thrombosis. Lancet. 1991;338:597–600. doi: 10.1016/0140-6736(91)90607-q. [DOI] [PubMed] [Google Scholar]
- Bertina R M, Koeleman B P, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- Dahlback B. New molecular insights into the genetics of thrombophilia: resistance to activated protein C caused by Arg506 to Gln mutation in factor V as a pathogenic risk factor for venous thrombosis. Thromb Haemost. 1995;74:139–148. [PubMed] [Google Scholar]
- Simioni P, Prandoni P, Lensing A W, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood. 2000;96:3329–3333. [PubMed] [Google Scholar]
- Rees D C, Chapman N H, Webster M T, Guerreiro J F, Rochette J, Clegg J B. Born to clot: the European burden. Br J Haematol. 1999;105:564–566. [PubMed] [Google Scholar]
- Rodeghiero F, Tosetto A. Activated protein C resistance and factor V Leiden mutation are independent risk factors for venous thromboembolism. Ann Intern Med. 1999;130:643–650. doi: 10.7326/0003-4819-130-8-199904200-00004. [DOI] [PubMed] [Google Scholar]
- Margaglione M, Brancaccio V, Giuliani N, et al. Increased risk for venous thrombosis in carriers of the prothrombin G→A20210 gene variant. Ann Intern Med. 1998;129:89–93. doi: 10.7326/0003-4819-129-2-199807150-00003. [DOI] [PubMed] [Google Scholar]
- Simioni P, Prandoni P, Lensing A W, et al. The risk of recurrent venous thromboembolism in patients with an Arg506-Gln mutation in the gene for factor V (factor V Leiden) N Engl J Med. 1997;336:399–403. doi: 10.1056/NEJM199702063360602. [DOI] [PubMed] [Google Scholar]
- Martinelli I, Sacchi E, Landi G, Taioli E, Duca F, Mannucci P M. High risk of cerebral-vein thrombosis in carriers of a prothrombin-gene mutation and in users of oral contraceptives. N Engl J Med. 1998;338:1793–1797. doi: 10.1056/NEJM199806183382502. [DOI] [PubMed] [Google Scholar]
- De Stefano V, Martinelli I, Mannucci P M, et al. The risk of recurrent venous thromboembolism among heterozygous carriers of the G20210A prothrombin gene mutation. Br J Haematol. 2001;113:630–635. doi: 10.1046/j.1365-2141.2001.02827.x. [DOI] [PubMed] [Google Scholar]
- Bauer K A. Role of thrombophilia in deciding on the duration of anticoagulation. Semin Thromb Hemost. 2004;30:633–637. doi: 10.1055/s-2004-861505. [DOI] [PubMed] [Google Scholar]