

Abstract
Retinoic acids and their derivatives potentiate anti-cancer effects in breast cancer cells. The aberrant expression of telomerase is critical to the continued proliferation of most cancer cells. Thus, telomerase is an attractive target for chemo-prevention and treatment of breast cancer. 9cUAB30 is a novel synthetic retinoid X receptor-selective retinoic acid (RA) that effectively reduces the tumorigenic phenotype in mouse breast carcinoma with lower toxic effects than natural retinoid treatments. We have assessed 9cUAB30 retinoic acid treatment of human breast cancer cells to determine the potential of this drug as an effective telomerase inhibitor and its application to cancer therapy. 9cUAB30 was found to decrease DNA methyltransferase and telomerase expression in MDA-MB-361, T-47D, and MCF-7 human breast cancer cells and to inhibit the proliferation of these cells. This low-toxicity retinoid also reduced colony formation in soft agar assays in each of these cell types. Combination treatments of 9cUAB30 and all-trans RA proved to be synergistically more effective than either RA alone, further suggesting a possible general epigenetic mechanism that contributes to the anti-telomerase activity of the retinoids. Therefore, the novel retinoid, 9cUAB30, is effective in inhibiting the growth of human breast cancer cells, its anti-cancer effects appear to be related to telomerase inhibition and the mechanism for this process could be mediated through epigenetic modifications.
Keywords: retinoids, breast cancer, 9-cis UAB30 retinoic acid, telomerase
Introduction
Telomerase is the enzyme responsible for solving the end-replication problem in linear chromosomes and hTERT, the catalytic component of telomerase, is an attractive molecular target for anti-cancer therapies due to its differential expression in cancer and normal cells (reviewed in ref. 1). There is significant interplay between genetic and epigenetic regulatory mechanisms at the hTERT promoter, hence the need to elucidate the complex mechanisms of telomerase control and the stable silencing of telomerase that occurs during the normal course of cellular differentiation. Retinoic acids (RAs), derived from vitamin A, are important mediators of developmental, growth, and morphogenic processes in vivo (2) and have been shown to down-regulate telomerase activity in a number of cancers (3-7). Responsiveness to RA is determined by retinoic acid receptor (RAR) and retinoid X receptor (RXR) expression. These two classes of steroid receptor-related nuclear proteins each have, α, β and γ isoforms (reviewed in ref. 8) that further enhance the cell-specific nature of retinoid sensitivity. RA derivatives change the expression of cyclins, cdks, CDKIs, E2F1, and c-Myc (9,10) in breast cancer cells, making these cells an attractive model system for promoter analysis and RA treatment. Although a direct relationship between RA treatments and estrogen has not yet been found, RA responsiveness appears to be greatest in estrogen receptor (ER)-positive breast cancer cells that express high levels of RARα (11,12). Both 13-cis RA and all-trans RA (ATRA) have been shown to decrease tritiated thymidine incorporation in ER-positive MCF-7 cells (13). ER-negative cells have decreased levels of RARα and are resistant to retinoids (12). In ER-negative MDA-MB-231 cells, which are RA-resistant and express neither RARα nor RARβ, the induction of an expression vector encoding RARα restores sensitivity to ATRA and causes growth inhibition suggesting that RARα mediates the effects of retinoids (12). When cells lack RARα, the RXR pathway ensues and activates RARβ (14). The tumor suppressor RARβ likely inhibits the action of AP-1 (15) and is absent in most ER-positive breast cancer cells such as MCF-7 and MDA-MB-361 cells (11,16-19). RA therapy can induce the expression of RARβ in some types of cancer (20).
RAs have been shown to down-regulate telomerase activity in breast cancer, prostate cancer, and leukemia (3-5). Although the hTERT promoter lacks a retinoid response element and gene repression is a downstream effect of retinoid treatment, studies have shown that certain RAs significantly down-regulate hTERT gene expression in MCF-7 and MCF-10A cells (3,4). The various molecular targets of retinoid signaling are still being discovered. It has been demonstrated that retinoids repress AP-1-responsive gene expression by inhibiting MKK6/p38 and MEK/ERK signaling pathways (5). This repression is reversed, however, if c-fos and c-jun are overexpressed, implicating c-jun/c-fos as retinoid targets.
RAs specific for RAR (such as ATRA) cause RAR/RXR heterodimers only. RXR can form homodimers or heterodimers with other receptors such as vitamin D3, Nur-77, thyroid hormone, and RAR (21,22), making it more specific and more selective than RAR. Ligands specific for RXR are much less toxic than RAR-selective retinoids (23,24). 9-cis RA binds RAR and RXR receptors. ATRA binds only RAR receptors (14). 9-cis UAB30 (9cUAB30) RA [(2E,4E,6Z,8E)-8-(3′,4′-dihydro-1′(2′H)-napthalen-1′-ylidene)-3,7-dimethyl-2,4,6-octatrienoic acid)] is a synthetic analog of 9-cis RA with reduced RAR-binding activity relative to ATRA and 9-cis RA (25). 9cUAB30 also binds RXRα with higher affinity than the natural ligand. Several isoforms of the UAB class of RAs have been synthesized, but 9cUAB30 is the most potent and selective for activating RXRs (25). 9cUAB30 is effective in the prevention of N-methyl-N-nitrosourea-induced murine breast cancer without signs of toxicity (26,27). It is also rapidly taken up in the serum of mice and has a longer half-life than 9-cis RA. Moreover, this RA does not accumulate in the serum, alter estrus cycles or uterine or ovarian weights, or increase liver retinyl palmitate levels (27). 9cUAB30 may have antagonistic RARα binding activity, but can still transactivate RARβ and γ with milder effects and maintain the effectiveness of RA treatment. Retinoids often result in high levels of toxicity, thereby rendering therapy an impractical option. However, new synthetic analogs of natural RAs such as 9cUAB30 offer great potential because they are more selective, equally effective, and seem to generate lower numbers of undesired side effects. It has been previously suggested that the RAR- and RXR-selective combination might be the most potent in inhibiting tumorigenesis (28-30).
The exact mechanism of action for RA-mediated inhibition of hTERT has not yet been elucidated, but epigenetic mechanisms may explain many aspects of hTERT regulation and, consequently, effects that retinoids have at the hTERT promoter. DNA methyltransferases (DNMTs), the mediators of DNA methylation that normally silence genes, are overexpressed in tumors (31). Additional studies have shown elevation of the gene expression of all three major DNMTs during tumorigenesis (32). Paradoxically, the hTERT promoter has been shown to be highly methylated in most tumor cell types, with the exception of cancers of embryonic origin, rendering hTERT active (33). The tumor-specific differences in methylation status of the hTERT promoter and telomerase activity may be due to the selection of cells with site-specific changes in transcriptional activator binding characteristics. To examine the role of DNA methylation in the retinoid-induced down-regulation of hTERT, we investigated DNMT expression during the course of treatment of T47D breast cancer cells.
We have previously shown significant down-regulation of hTERT expression with RA treatment in human terato-carcinoma (HT) cells (34). This study examines the effects of 9cUAB30 on MCF-7, T47D, and MDA-MB-361 cells, all of which are ER-positive breast cancer cell lines that vary in RAR and RXR expression and retinoid sensitivity. Combination treatments with RXR-selective and RAR-selective RAs hold the greatest potential as effective cancer therapies.
Materials and methods
Cell culture, treatments, and proliferation analysis
T47D breast ductal carcinoma cells (ATCC HTB-133) were grown in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 25 mM HEPES, 1X amphotericin B, penicillin, and streptomycin (APS, Mediatech), and L-glutamine. MCF-7 cells (ATCC HTB-22) and MDA-MB-361 cells (ATCC HTB-27) were grown in 1X DMEM (Dulbecco’s modified Eagle’s medium, Mediatech) supplemented with 10% FBS, 1.0 mM sodium pyruvate, 4.5 g/ml glucose, 0.1 mM non-essential amino acids, 1X APS (Mediatech), and L-glutamine. All subculture of cells was carried out in a humidified incubator at 37°C in 5% CO2. 9cUAB30 is a novel retinoid prepared by Dr Donald Muccio and colleagues (25). ATRA (Sigma) and 9cUAB30 were prepared in 100% ethanol, infused with nitrogen, aliquotted, and stored in the dark at -80°C. The appropriate culture media was supplemented with varying concentrations of RAs, ranging from 0.1-10 μM. Dose-response analysis was conducted (data not shown) and the retinoids were used at optimal concentrations for each indicated cell type. Cells were treated with RAs two days after being plated. Untreated cells fed complete medium and grown simultaneously and in parallel with treated cells served as controls. Growth medium was replaced every third day of culture. All cell culture work and handling of the retinoids was carried out under yellow light to maintain RA stability. To gauge proliferation, treated cells and non-treated controls were harvested with 1X trypsin-EDTA (0.25% trypsin/2.21 mM EDTA in HBSS) and counted using trypan blue exclusion staining on a standard hemacytometer.
RT-PCR of hTERT and DNMTs
Following subculture and treatment with retinoids, cells were harvested with trypsin and washed with PBS. RNA was extracted according to the manufacturer’s instructions with the RNeasy kit (Qiagen). Total RNA (2-4 μg) was converted to cDNA with oligo-dT primers using the SuperScript First Strand Synthesis System (Invitrogen). cDNA was amplified by PCR with 2X PCR Master Mix (Promega) under conditions published previously (35-37).
For each primer set, the number of amplification cycles was predetermined to ensure that readouts were taken in the exponential phase. PCR products (10 μl) were subjected to electrophoresis in 2-3% agarose gels prestained with ethidiumbromide (Sigma) and visualized with UV light. Gels were analyzed with Kodak ID 3.6.1 Digital Science Software and standard error of the means (SEMs) calculated from quantified data in triplicate experiments.
TRAP in RA-treated cells
The telomeric repeat amplification protocol (TRAP) assay was performed with the TRAPeze XL telomerase detection kit (Serologicals). Briefly, 1×106 cells per sample were harvested and lysed with 200 μl of CHAPS buffer and centrifuged at 12,000 × g at 4°C for 30 min to extract protein. Protein samples were quantified with the Bradford-based Bio-Rad protein assay. Protein (40 μg) from each sample was used to detect telomerase activity in the extracts, evidenced by a laddering in 6-base pair increments from telomeric repeats added to the oligonucleotide substrate in the reaction. Extended substrates were amplified with PCR using fluorescein-labeled Amplifluor® primers for 36 cycles with an annealing temperature of 59°C, followed by a final extension of 55°C for 25 min as described previously (34). Samples were visualized after being subjected to electrophoresis through a 10% non-denaturing polyacrylamide gel stained with SYBR-Green (Molecular Probes). Fluorescence emitted by the bands and the height of the ladder were proportional to the amounts of product generated and band intensity was analyzed with Kodak ID 3.6.1 Digital Science software.
Soft agar assay
Solid bottom layers consisted of 0.66% agar noble in sodium bicarbonate-free 1X RPMI-1640 supplemented with 20% FBS, L-glutamine, and 1X ampicillin/penicillin/streptomycin (APS) for T47D cells or 0.5% agar noble in 2X DMEM supplemented with 20% FBS for MDA-MB-361 cells (38). The semi-solid top layer of 0.33% noble agar contained RPMI and 1×104 T47D or 7.5×104 MDA-MB-361 cells. ATRA or 9cUAB30 was added to the semisolid top layer along with cells at concentrations ranging from 0.1-10 μM. Untreated cells were grown in parallel as a control. Fresh aliquots of 1 ml control or retinoid-containing media were added to the top layers every 4 days. At 21 and 28 days, colonies consisting of more than 25-30 cells were counted and photographed with a Nikon 900 Coolpix digital camera (38). SEMs are the result of triplicate experiments.
Apoptosis assays
The ApopTag peroxidase assay (Serologicals) was used for TUNEL-based labeling of apoptotic nuclei of control or 10 μM-treated (9cUAB30 or ATRA) T47D cells after 3 and 6 days. Cells (1×105) were fixed in a solution of 1% paraformaldehyde for 10 min at room temperature. Endogenous peroxidase was quenched with 3% H2O2 in PBS. Cells were incubated with terminal deoxynucleotidyl transferase (TdT) for 1 h at 37°C in a humidified chamber. Labeled substrates were incubated with anti-digoxigenin peroxidase antibody for 30 min at room temperature. Nuclei were counterstained with methyl green (0.5%, Sigma) and photographs were taken with a Nikon 900 Coolpix digital camera. Apoptosis was detected after 12 days in treated and untreated MDA-MB-361 and MCF-7 cells by fluorescence-activated cell sorting with the Vybrant™ apoptosis assay kit No. 2 (Molecular Probes) as described previously (39). Cells with phosphatidylserine on the membrane undergoing early apoptosis were bound by the phospholipid-binding Annexin-V labeled with Alexa Fluor 488 and visualized as green fluorescence by fluorescence-activated cell sorting analysis (FACScalibur, Becton-Dickinson). Cells in later stages of cell death (necrosis) were sorted with propidium iodide-staining of dead cells with red fluorescence. Live, non-apoptotic cells showed little or no fluorescence. CellQuest software (Becton-Dickinson) was used to determine the percentage of cells present at each stage of the cell cycle.
Results
9cUAB30 effectively inhibits breast cancer cellular proliferation
9cUAB30 was compared to ATRA in triplicate experiments on T47D, MDA-MB-361, and MCF-7 human breast cancer cells for 3, 6, 9, and 12 days with the concentrations and combination treatments discussed previously. Changes in proliferation for each cell line with and without RA are shown in Fig. 1. Each drug was an effective inhibitor of proliferation in all cell types. In MDA-MB-361 cells, 9cUAB30 induced an approximate 5-fold decrease in growth by day 12 with respect to untreated cells. ATRA and combination treatments were more effective and decreased cell growth by 25- and 35-fold, respectively (Fig. 1B). Human MCF-7 cells treated with 9cUAB30 decreased proliferation by 4.5-fold by day 12. ATRA had a weaker effect in MCF-7 cells, showing a lower degree of inhibition on cellular proliferation at day 12 than effects seen with 9cUAB30 (Fig. 1C). Collectively, these studies show for the first time that 9cUAB30, a novel low-toxicity retinoid effective in mouse breast cancer, is also effective in human breast cancers and has efficacy comparable to ATRA depending upon cell type. Combination treatments with 9cUAB30 and ATRA at reduced concentrations appeared to have synergistic effects in MDA-MB-361 cells that were assessed for combined therapy.
Figure 1.
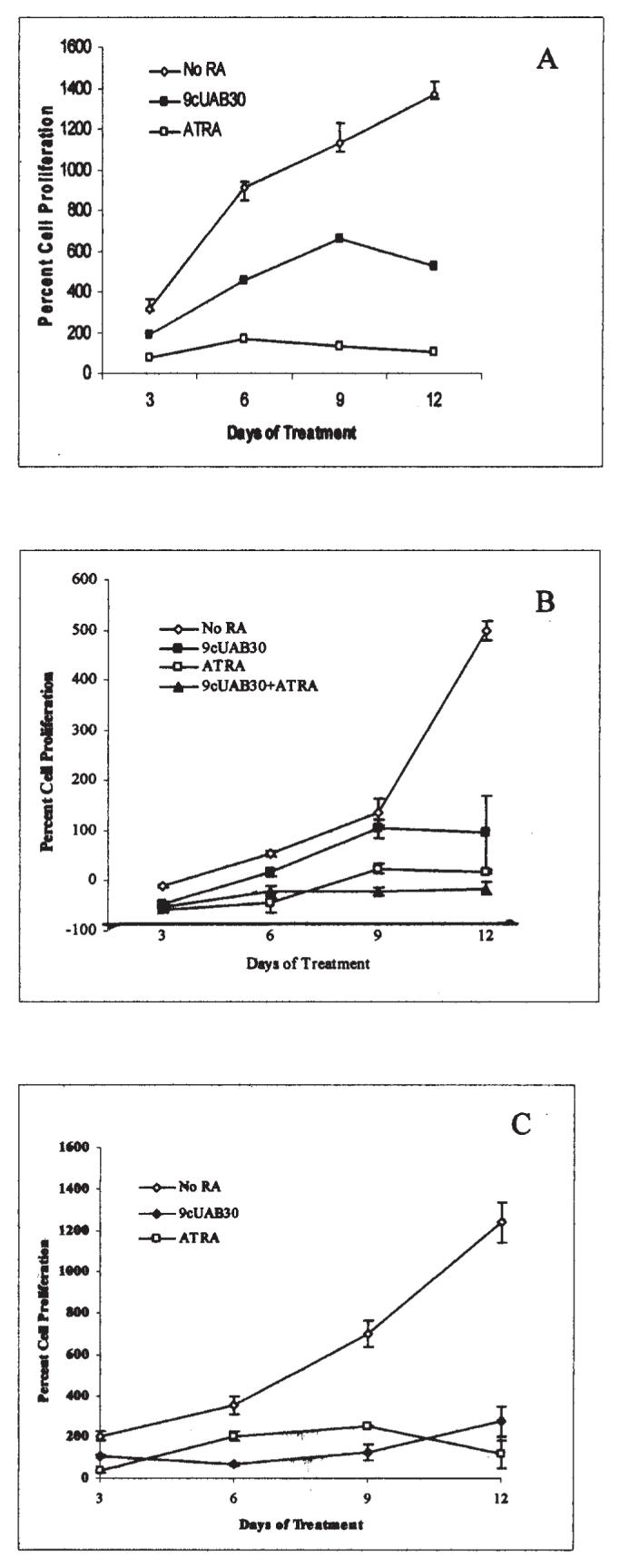
Novel retinoid treatment decreases the proliferation rates of human breast cancer cells. (A) Human T47D breast carcinoma cells were grown in untreated control media (◇) or either 2 μM ATRA (□) or 10 μM 9cUAB30 (◆) for 12 days of concurrent culture. (B) Human MDA-MB-361 cells remained untreated (◇) or were treated with 10 μM 9cUAB30 (■), 2 μM ATRA (□) or a combination of 5 μM UAB-30 and 1 μM ATRA (▲). (C) Human MCF-7 cells were untreated (◇), treated with 10 μM 9cUAB-30 (◆), or treated with 2 μM ATRA (□). Proliferation is expressed as the percentage of the ratio of live cells harvested at days 3, 6, 9, and 12 of treatment to the number of live cells originally plated. Average cell numbers obtained by trypan blue exclusionary staining performed with a standard hemacytometer are presented with SEMs from triplicate culturing of each treatment group.
hTERT expression reduced with RA treatments
Fig. 2 shows the changes in hTERT expression in RA-treated cells. Untreated control cells were used to represent 100% of hTERT mRNA levels. RT-PCR of hTERT mRNA expressed in T47D cells showed that ATRA and 9cUAB30 treatment for 3 days reduced expression by approximately 40% (Fig. 2A). By day 6, ATRA and 9cUAB30 treatment resulted in a reduction of hTERT mRNA levels to 15% of those seen in control cells and these levels continued through days 9 and 12 of treatment. Cells were incubated with 10 μM 9cUAB30 or ATRA, which were the optimal concentrations of these retinoids in the T47D cells based on dose response analysis (data not shown). hTERT mRNA expression was also assessed in MDA-MB-361 cells after treatment with 10 μM 9cUAB30, 2 μM ATRA or a combination of 5 μM 9cUAB30/1 μM ATRA (Fig. 2B). These optimal concentrations of retinoids for MDA-MB-361 cells were also based on dose response analysis (data not shown). 9cUAB30 down-regulated hTERT as early as day 3 relative to control untreated cells and demonstrated almost complete inhibition of hTERT mRNA production by day 9, showing a 98% decrease thereby demonstrating for the first time that 9cUAB30 has anti-telomerase activity. Surprisingly, ATRA was less potent than 9cUAB30, with hTERT mRNA levels decreasing from 30% of controls to 18% by day 12. Combination treatments were slightly more effective than ATRA alone, but not as effective as 9cUAB30. Combination treatments were also slower to act, decreasing hTERT mRNA levels to 41% of untreated controls at day 3 and to 12% by day 12. MCF-7 cells showed less inhibition of hTERT with RA compared to the MDA-MB-361 cells (Fig. 2C). Triplicate experiments showed only a 30% down-regulation of hTERT mRNA with 10 μM 9cUAB30 at day 12. Duplicate experiments with ATRA demonstrated 28% inhibition by day 12, but treatments caused a greater decrease in expression to approximately 60% of untreated values by day 9.
Figure 2.
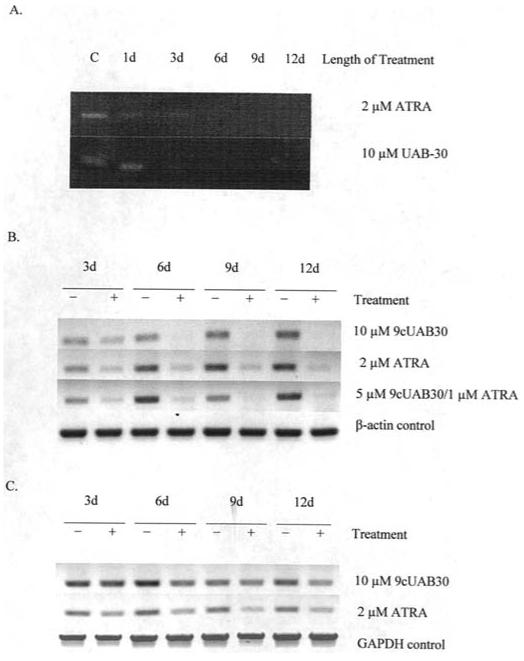
hTERT mRNA expression decreases with retinoic acid treatment. (A) RT-PCR amplification of a 219-bp segment of hTERT mRNA in T47D human breast carcinoma cells treated with either 10 μM RA or 9cUAB30 over 12 days of concurrent culture. (B) hTERT expression in MDA-MB-361 cells. +, samples treated with retinoic acid; -, untreated samples; d, days. (C) hTERT cDNA in MCF-7 cells with and without retinoid treatment. β-actin and GAPDH served as controls to normalize the loading and quality of RNA. The data shown are representative of triplicate gels from three individual experiments.
mRNA levels of epigenetic modulators DNMTs decreased with retinoids
Changes in DNMT expression for T47D cells with and without RAs are shown in Fig. 3. DNMT levels were generally down-regulated in T47D cells after 12 days of exposure. With ATRA, DNMT1 expression decreased to 7.5% of control cells with 12 days of treatment (Fig. 3A). DNMT3a and DNMT3b declined to 6 and 33% of control DNMT3a and DNMT3b levels, respectively, by 12 days of treatment in response to ATRA. 9cUAB30 treatment led to a gradual decrease of DNMT1 to 7% of control levels by day 12 in the T47D cells (Fig. 3B). DNMT3a was also significantly down-regulated by 12 days of 9cUAB30 treatment. DNMT3b levels declined to 11% of control levels by day 12. GAPDH concentration was used as a control and did not show significant changes with treatment. These studies therefore show that the novel retinoid, 9cUAB30, as well as ATRA are capable of altering DNMT expression in human breast cancer cells.
Figure 3.
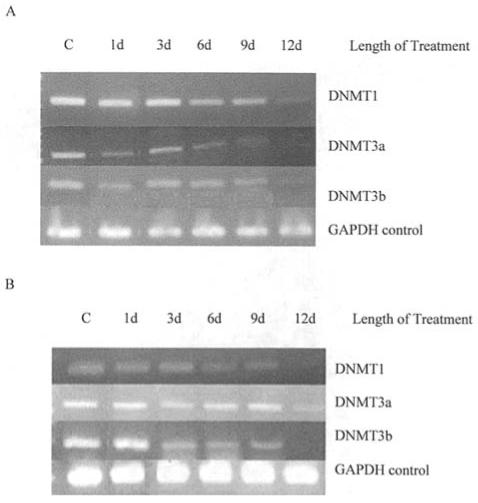
DNMT mRNA expression is altered with retinoid treatment. (A) DNA methyltransferase (DNMT) expression in T47D cells exposed to 10 μM ATRA over 12 days of concurrent culture. Product bands are shown for DNMT1, DNMT3a, and DNMT3b and the ubiquitous housekeeping enzyme GAPDH. (B) RT-PCR amplification of DNMT mRNAs in T47D breast cancer cells treated with 10 μM 9cUAB30 over 12 days of concurrent culture. Product bands are shown for DNMT1, DNMT3a, and DNMT3b and GAPDH. d, days.
Telomerase activity altered with RA treatments in a cell type-specific manner
Telomere Repeat Amplification Protocol (TRAP) analyses assessing telomerase activity for each cell type with and without RA treatments are displayed in Fig. 4. We show for the first time that the low-toxicity novel retinoid, 9cUAB30, inhibits telomerase activity comparable to ATRA. The telomerase enzyme is known to have a long half-life and can maintain activity for at least 24 h or more after down-regulation of hTERT mRNA production (34). ATRA has a stronger effect in lower concentrations with a reduction in enzyme activity to 20% of controls in T47D cells treated with 0.1 μM ATRA for 6 days as compared to 10% in cells grown in 0.1 μM of 9cUAB30 (Fig. 4A1). T47D breast cancer cells treated with 10 μM ATRA for 1 day showed only a 10% reduction relative to control levels (Fig. 4A2). By day 6 of ATRA treatment, however, telomerase activity decreased to 65% of control levels. ATRA treatment reduced telomerase activity to 23% by day 9 and to 15% of control levels by day 12. Similar reduction in telomerase activity was seen with 9cUAB30 in T47D cells (Fig. 4A3). 9cUAB30 may act as an anti-telomerase agent with efficacy similar to ATRA over longer periods of treatment in T47D cells.
Figure 4.
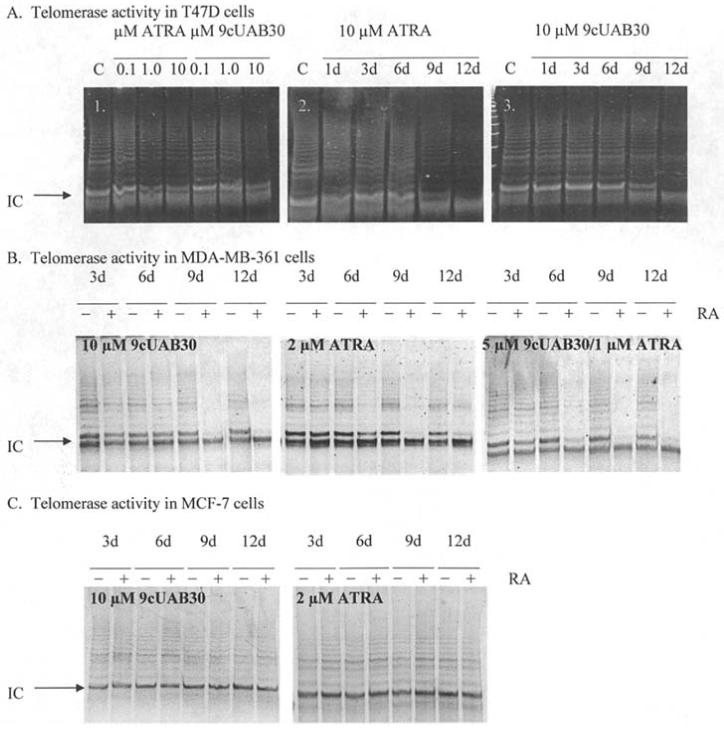
Analysis of telomerase activity in breast cancer cells in response to retinoids. (A1) Dose-responsive inhibition of telomerase activity with ATRA and 9cUAB30. DNA products from the TRAP assay are presented for samples treated with 0.1, 1, and 10 μM ATRA or 9cUAB30 for 6 days of concurrent treatment. (A2) Inhibition of telomerase over 12-day treatment with 10 μM ATRA. TRAP assay results are presented for T47D breast cancer cells exposed to 10 μM ATRA for 12 days of concurrent treatment. (A3) Inhibition of telomerase over 12 days of treatment with 9cUAB30. C, untreated control cell samples. (B) Retinoid-induced down-regulation of telomerase activity in MDA-MB-361 cells. Cells were treated with 10 μM 9cUAB30, 2 μM ATRA, or a combination of 5 μM 9cUAB30 and 1 μM ATRA. (C) Telomerase activity in MCF-7 cells following treatment with retinoids. Treatments were 10 μM 9cUAB30 or 2 μM ATRA. +, treated samples; and -, untreated samples. In all gels shown a 61-bp internal control (IC) included in the reaction mixture for TRAP assays was used to standardize loading. The ladder formed above the IC represents 6-bp increments of TTAGGG telomeric repeats. The height of the ladder formed and the intensity of fluorescence are proportional to telomerase activity in the sample.
Telomerase activity in response to RA treatments was also examined in MDA-MB-361 and MCF-7 cells. There was significant efficacy in telomerase inhibition in MDA-MB-361 cells (Fig. 4B). Visual differences in band intensities between treated and untreated cells were observed as early as day 6 for all RAs, with almost complete disappearance of banding by day 9. Triplicate experiments show that the 9cUAB30/ATRA combination was most effective in MDA-MB-361 cells, with 82% inhibition by day 9 and 71% inhibition at day 12. 9cUAB30 treatments at 10 μM final concentration revealed 72% telomerase inhibition at day 12 compared to untreated cells and this inhibition was in a more steady, stepwise pattern compared to effects seen with the combination treatments. ATRA alone only down-regulated telomerase to 40% by day 12 in MDA-MB-361 cells. In MCF-7 cells, telomerase inhibition was less than the other cell lines with all RA treatments and there were no significant differences in telomerase activities in these cells through 12 days of treatment in these cells (Fig. 4C). Therefore the new 9cUAB30 retinoid has significant anti-telomerase activity in specific types of breast cancer cells comparable to the effects of the more toxic ATRA.
Dose-dependent decrease of anchorage-independent growth observed with RAs
The abilities to grow independently of a basal substrate and to overcome contact inhibition are hall-marks of tumorigenic growth and can be used as indicators of the efficacy of agents designed for anti-cancer therapy. Since 9cUAB30 and ATRA were shown to have significant anti-telomerase effects in T47D and MDA-MB-361 cells, we asked whether the tumorigenesis of these cells was reduced in response to these compounds. Fig. 5 shows the results for the soft agar analysis of retinoid-treated T47D and MDA-MB-361 cells. Soft agar analysis was performed on T47D cells exposed to 0.1, 1, and 10 μM ATRA or 9cUAB30. ATRA-treated plates showed a reduction of colony growth in soft agar even at low concentrations to 11% of control growth with 0.1 μM ATRA (Fig. 5B). 9cUAB30 also inhibited colony formation of these human cells, with 0.1 μM treatment reducing growth to 62% of control and 10 μM reducing growth to only 12% of control levels. 9cUAB30 may, therefore, be similar to ATRA in its anti-tumor effect when used at adequate concentrations in T47D cells thereby showing for the first time the efficacy of this novel compound in human breast cancer cells (Fig. 5B). Since MCF-7 cells proved resistant to retinoid treatment, the effects of retinoid exposure on the tumorigenicity of MDA-MB-361 cells were investigated. Triplicate soft agar experiments were performed with ATRA and 9cUAB30 on MDA-MB-361 cells (Fig. 5C). MDA-MB-361 cells were grown in 0.33% agar with either no RA, 10 μM 9cUAB30, 2 μM ATRA, or combination treatments for 21 days. Untreated cells generated an average of 96 colonies of 25 cells or greater per plate. 9cUAB30 treated cells formed approximately 88% fewer colonies (around 12 colonies). ATRA decreased tumorigenicity by ∼98% (averaging only 2 colonies). Combination treatments were the most effective and virtually eliminated this aspect of the tumor phenotype in treated MDA-MB-361 cells (Fig. 5C).
Figure 5.
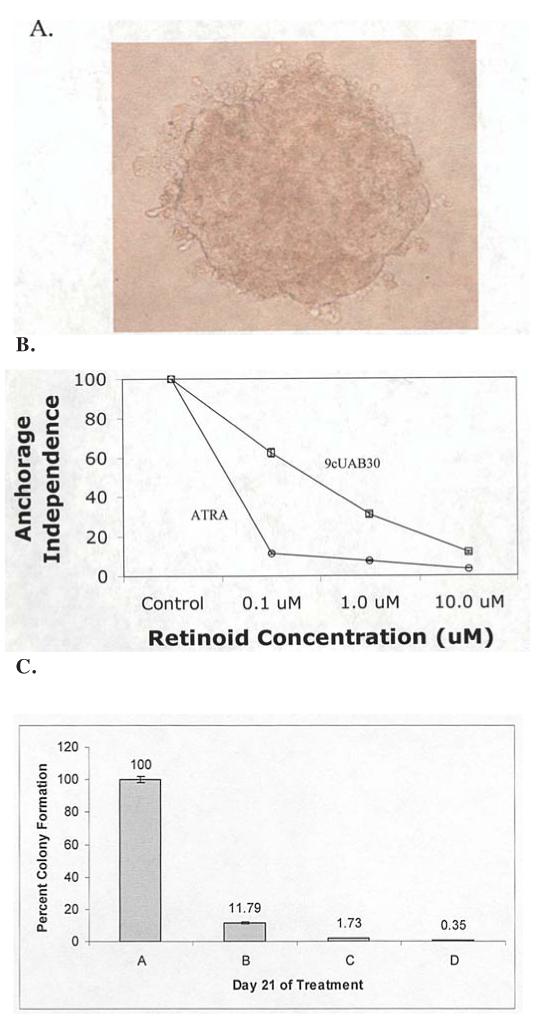
Soft agar analysis of retinoid-treated T47D and MDA-MB-361 cells. (A) Representative photograph of an MDA-MB-361 colony of greater than 25 cells that were counted at each time point taken at x400 magnification. (B) Examination of contact inhibition growth requirements was performed using soft agar analysis in untreated control T47D cells and cells exposed to 0.1, 1.0, or 10.0 μM ATRA or 9cUAB30 for 28 days. Colony counting results for both groups are summarized as a percent of untreated control cell colony counts. (C) Levels of tumorigenicity of MDA-MB-361 cells following 10 μM 9cUAB30 (green horizontal stripes), 2 μM ATRA (red diagonal stripes), and 5 μM 9cUAB30 and 1 μM ATRA combination (yellow dots) treatments compared to untreated MDA-MB-361 cells (blue vertical stripes). Level of tumorigenicity is expressed as a ratio of the percentage of treated colonies to the number of untreated colonies.
Significant apoptosis induced with 9cUAB30
Apoptosis is an important mechanism to clear malignant cells from the body and is commonly induced with retinoid therapies. Detection of apoptosis in T47D cells was evident at day 3 and 6 of ATRA and 9cUAB30 treatment (Fig. 6A-E). Day 6 of ATRA exposure led to the staining of entire clusters of cells (Fig. 6D). 9cUAB30 treatment caused apoptosis, but to a lesser extent (Fig. 6E). ATRA and 9cUAB30 were tested for their abilities to induce apoptosis in MDA-MB-361 and MCF-7 cells (Fig. 6F and G). MDA-MB-361 cells treated with 9cUAB30 exhibited 3.5 times the amount of total apoptosis of untreated cells (Fig. 6F). Most of the apoptotic cells were in the later stages of cell death (necrosis). ATRA was only 43% as effective as 9cUAB30 in apoptosis, but caused levels of necrosis similar to 9cUAB30. Combination treatments were the most effective, generating 8 times as much apoptosis as controls (Fig. 6F). In MCF-7 cells, 9cUAB30 plus ATRA and 9cUAB30 alone induced similar levels of apoptosis with 2.5 times the apoptosis of untreated cells (Fig. 6G). At day 12, more 9cUAB30-treated cells were in the early stages of cell death. Cells treated with combination therapy were undergoing later stages of apoptosis. ATRA was the least effective in MCF-7 cells as well, resulting in 1.4 times the apoptosis of untreated cells. None of the retinoid treatments appeared to significantly alter the percentage of dead MCF-7 cells present compared to cells not exposed to RAs (Fig. 6G). Comparative analyses of apoptosis in response to ATRA and 9cUAB30 for MDA-MB-361 and MCF-7 cells are shown in Fig. 6H.
Figure 6.
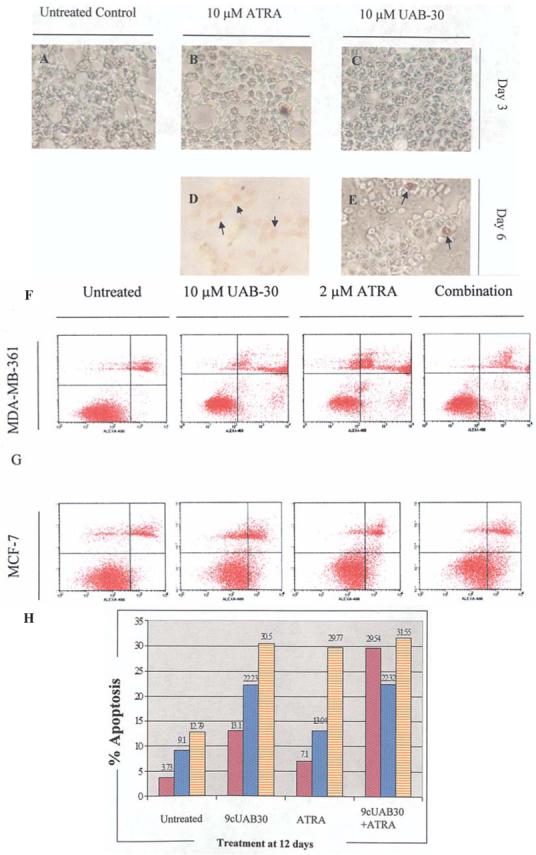
Treatment with retinoids significantly affected apoptosis in breast cells. (A-E) TUNEL-based labeling of apoptotic DNA fragments. Terminal deoxynucleotidyl transferase (TdT)-anti-peroxidase conjugation was performed using the ApopTag analysis (Serologicals) in untreated control cells and 10 μM ATRA- and 9cUAB30-treated T47D cells through 6 days of culture. (A) Control, (B) day 3 ATRA-treated, and (C) day 3 9cUAB30-treated breast cancer cells stained with peroxidase substrate. (D and E) TUNEL-staining of retinoid-exposed cells from 6 days of culture. Arrows indicate the presence of brown peroxidase-stained cells while green background is indicative of normal nuclei labeled with methyl green counterstain. (F and G) Fluorescence-activated cell sorting (FACS) analysis of apoptosis and death of MDA-MB-361 and MCF-7 cells in response to 12 days of retinoid exposure. Early apoptotic cells (lower right quadrants) were detected by FACS analysis as cells that incorporated Alexa®Fluor 488 (Alexa-488)-labeled Annexin V into the plasma membrane. Late apoptosis/necrosis (upper right quadrant) was detected as those cells that stained with both Alexa-488 and propidium iodide (PI). The lower left quadrant includes cells that did not incorporate either Alexa-488 or PI and represent live cells not undergoing apoptosis. The upper left quadrant displays cells that stained with PI and represent dead cells. Cell sorting was performed using FACScalibur, and analysis was achieved utilizing CellQuest software. (H) Comparative analysis of apoptosis induction in breast cancer cells. MDA-MB-361 cells (purple) and MCF-7 cells (blue dots) were treated for 12 days with 10 μM 9cUAB30, 2 μM ATRA, and a combination of 5 μM 9cUAB30 and 1 μM ATRA (orange).
Discussion
In this study, we have shown for the first time that the novel low-toxicity retinoid, 9cUAB30, is efficacious in human breast cancers and down-regulates telomerase in a number of breast cancer cells suggesting that this process could be linked to an inhibition in DNA methyltransferase gene expression in these cells. Retinoids are morphogens that induce the establishment of embryonic patterning, loss of proliferation, cellular differentiation, and apoptosis (40,41) and allow for the selective targeting of highly proliferative cell populations. Epigenetic mechanisms established during differentiation are thought to play a role in the maintenance of telomerase silencing observed in most somatic cells (42,43). The effects seen in this study, such as the loss of proliferation and induced apoptosis, are independent of the induction of a differentiated morphology, since T47D cells are a highly differentiated cancer cell line. The association of many human cancer cell lines with high hTERT promoter methylation in differentiated primary source tissues (43) suggests that retinoid agents are likely capable of interacting with telomerase regulatory pathways even in cells in which differentiating mechanisms are not induced.
Subsequent to hTERT transcriptional silencing, loss of telomerase activity follows several days after hTERT mRNA is down-regulated (Figs. 2 and 4) due to the extended stability of the enzyme. 9cUAB30 appears to have less effect on enzyme activity because of chemical modifications from ATRA that alter binding to RARα (25). As a candidate for a less toxic cancer therapeutic agent (41), the novel synthetic retinoid 9cUAB30 could be an alternative to traditional retinoid chemo-therapies when used in conjunction with endogenous RAs to generate the telomerase silencing, induction of apoptosis, and loss of proliferation seen in this study. Telomerase silencing may occur in association with antagonistic binding of RARα, a characteristic of 9cUAB30 suggested by retinoid receptor affinity studies (25). Synthetic retinoids lacking RARα agonist activity may thus be a promising strategy for anti-telomerase effects as indicated in this study with 9cUAB30.
DNMT1, important for the maintenance of altered gene expression patterns in cancers, is known to be upregulated in human tumors (32). The down-regulation of DNMT1 in retinoid-treated samples seen in this study suggests a general loss of tumorigenic potential (Fig. 3). DNMT3a and 3b are known to be overexpressed in a cell line-specific manner and possess coordinated regulatory functions (31,37), making them the likely effectors of gene regulatory events during the retinoid treatments of this study. The down-regulation of the DNMTs as shown in this study is not only consistent with the reduced tumorigenicity of the cancer cells in response to the retinoids, but also suggests a possible link to the RA-induced decrease in hTERT observed in this investigation. For example, most cancer cells are known to have high levels of methylation of the hTERT promoter that paradoxically contribute to hTERT gene expression (33). Inhibition of DNA methylating activity in these cells with 5-aza-2′-deoxycyditine (5azadC) down-regulates the expression of hTERT (43) and it is feasible that the retinoid-induced reduction in DNMT gene expression in human breast cancer cells as found in this study could provide a new mechanism for decreased hTERT activity in the treated cells. Further studies will be required to establish a relationship between these two processes, but our findings are very suggestive of a potential novel epigenetic effect in the mechanisms for down-regulation of hTERT in the retinoid-treated cancer cells through modulation of the DNMTs.
Concentrations used for combination treatments in this study were half that of individual RAs, but caused a 7-fold greater reduction in proliferation than 9cUAB30 alone and abolished tumorigenicity. Combination treatments and 9cUAB30 alone both reduced telomerase activity by approximately 90%. The RXR-selective RA was 6 times more effective in down-regulating hTERT mRNA in our study, confirming studies that suggested the importance of RXR-selective RAs (44). We found that ATRA and 9cUAB30 combination treatments were twice as successful compared to singular treatments in inducing apoptosis in MDA-MB-361 cells. Down-regulation of hTERT induces apoptosis independently of telomerase activity in breast cancer by intermolecular actions with p53 (45) and the inhibition of telomerase induces apoptosis within a relatively short time independently of telomere shortening (45,46).
MDA-MB-361 cells were more sensitive to retinoids than MCF-7 cells, even though both cell lines lacked RARβ expression. The greater resistance to RAs in MCF-7 cells may be due to the additional lack of RARα2 expression in these cells. MDA-MB-361 cells express high levels of RARα in general (11,18). This study supports other findings that implicate RAR, and more specifically, RARα 2 as the receptor involved in RA-induced growth inhibition and apoptosis (12). If RARβ is the primary receptor involved in these effects, retinoid therapy would be sufficient to induce RARβ expression in MDA-MB-361 cells, but not MCF-7 cells. MCF-7 cells have a hypermethylated RARβ promoter that is not inducible by RAs alone (20). A study that supported RARβ as a component of the anti-proliferative and proapoptotic effects in MCF-7 cells following the induction of its expression by a combination of a demethylating agent and retinoid therapy (30) further supports this hypothesis. If estrogen receptor status determines RA sensitivity, loss of this receptor would generate a decrease in responsiveness and explain observations in this study. ATRA and natural 9-cis RA induced down-regulation of ERs in MCF-7 cells (47). Since estrogen activates c-Myc expression in ER-positive cells, the decrease in ERs could generate a down-regulation of c-Myc, which is the most important activator of hTERT (48). Mad1, a repressor that competes with c-Myc at E-box sites in the hTERT core promoter, would then be free to bind to the promoter of hTERT and repress it through histone deacetylase (HDAC) recruitment (48). This suggested mechanism from our studies is supported by the recent findings that HDAC inhibitors enhance the retinoid response in breast cancer cell lines (49).
9cUAB30 was more effective than 2 μM of ATRA in MCF-7 cells, comparable to effects of this drug seen in MDA-MB-361 cells. MCF-7 cells lack both RARβ and RARα2, but express RXRα which can selectively bind 9cUAB30 (16,18). 9cUAB30 may generate stronger effects because there are more receptors for 9cUAB30 present in these cells and proliferation, consequently, would occur through activation of the RXR pathway. Both RAs showed similar effectiveness in MCF-7 cells with hTERT being down-regulated by day 12, but ATRA appeared to cause the greatest decrease across the duration of exposure. ATRA-induced hTERT down-regulation in acute promyelocytic leukemia and the RAR pathway was reportedly responsible for these effects (4). If endogenous ATRA, even at very low levels, worked in synergy with administration of 9cUAB30, telomerase activity could be inhibited due to crosstalk between RARα and RXRs and their respective ligands. This crosstalk has been proposed to cause strong down-regulation of hTERT (50). Combination treatments were found to have the greatest effect in our study, so ATRA alone may not be sufficient to cause significant inhibition of telomerase until later treatment stages. Future studies that extend the treatment time of RAs on MCF-7 cells may be beneficial in further understanding the effects of these drugs.
This investigation, therefore, provides a detailed analysis of the effects of the novel retinoid 9cUAB30 as well as ATRA on the apoptosis and cell proliferation of breast cancer cells and indicates that down-regulation of telomerase may be an important mechanism through which the retinoids mediate their anti-cancer effects. Moreover, the efficacy of the low-toxicity 9cUAB30 retinoid has not previously been demonstrated in human cancer cells. We also show for the first time that the DNA methyltransferases are decreased in these retinoid-treated cells. Inhibition of DNA methylation with 5azadC is known to paradoxically down-regulate telomerase (43). Although our studies suggest an interesting and potentially important possible role of epigenetics in the telomerase inhibition and anti-cancer effects of 9cUAB30 and ATRA, future investigations will be required to establish a more direct link between the inhibition of DNMT and hTERT expression in response to retinoids.
Acknowledgments
Appreciation is expressed to Dr Donald Muccio and colleagues for providing 9cUAB30 and helpful discussions. This work was supported in part by grants from the American Association for Cancer Research, the National Cancer Institute, the Susan G. Komen Breast Cancer Foundation, and the UAB Breast SPORE.
Abbreviations
- RA
retinoid acid
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- ER
estrogen receptor
- ATRA
all-trans retinoic acid
- DNMT
DNA methyltransferase
- HT
human terato-carcinoma
References
- 1.Kelland LR. Overcoming the immortality of tumor cells by telomere and telomerase based cancer therapeutics - current status and future prospects. Eur J Cancer. 2005;41:971–979. doi: 10.1016/j.ejca.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 2.Gudas L. Retinoids and vertebrate development. J Biol Chem. 1994;269:15399–15402. [PubMed] [Google Scholar]
- 3.Choi SH, Kang HK, Im EO, et al. Inhibition of cell growth and telomerase activity in breast cancer cells in vitro by retinoic acids. Int J Oncol. 2000;17:971–976. doi: 10.3892/ijo.17.5.971. [DOI] [PubMed] [Google Scholar]
- 4.Pendino F, Flexor M, Delhommeau F, Bute D, Lanotte M, Segal-Bendirdjian E. Retinoids down-regulate telomerase and telomere length in a pathway distinct from leukemia cell differentiation. Proc Natl Acad Sci USA. 2001;98:6662–6667. doi: 10.1073/pnas.111464998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dedieu S, Lefebvre P. Retinoids interfere with the AP1 signalling pathway in human breast cancer cells. Cell Signal. 2006;18:889–898. doi: 10.1016/j.cellsig.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Xiao X, Sidorov IA, Gee J, Lempicki RA, Dimitrov DS. Retinoic acid-induced downmodulation of telomerase activity in human cancer cells. Exp Mol Pathol. 2005;79:108–117. doi: 10.1016/j.yexmp.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Pendino F, Hillion J, Dudognon C, et al. Telomerase targeting by retinoids in cells from patients with myeloid leukemias of various subtypes, not only APL. Leukemia. 2006;20:599–603. doi: 10.1038/sj.leu.2404127. [DOI] [PubMed] [Google Scholar]
- 8.Sussman F, De Lera AR. Ligand recognition by RAR and RXR receptors: binding and selectivity. J Med Chem. 2005;48:6212–6219. doi: 10.1021/jm050285w. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Q, Stetler-Stevenson M, Steeg PS. Inhibition of cyclin D expression in human breast carcinoma cells by retinoids in vitro. Oncogene. 1997;15:107–115. doi: 10.1038/sj.onc.1201142. [DOI] [PubMed] [Google Scholar]
- 10.Sheikh MS, Shao ZM, Chen JC, Ondonez JV, Fontana JA. Retinoid modulation of c-myc and max gene expression in human breast carcinoma. Anticancer Res. 1993;13:1387–1392. [PubMed] [Google Scholar]
- 11.Roman SD, Clarke CL, Hall RE, Alexander IE, Sutherland RL. Expression and regulation of retinoic acid receptors in human breast cancer cells. Cancer Res. 1992;52:2236–2242. [PubMed] [Google Scholar]
- 12.Schneider SM, Offterdinger M, Huber H, Grunt TW. Activation of retinoic acid receptor alpha is sufficient for full induction of retinoid responses in SK-BR-3 and T47D human breast cancer cells. Cancer Res. 2000;60:5479–5487. [PubMed] [Google Scholar]
- 13.Czeczuga-Semeniuk E, Wolczynski S, Dzieciol J, Dabrowska M, Anchim T, Tomaszewska I. 13-cis retinoic acid and all-trans retinoic acid in the regulation of the proliferation and survival of human breast cancer cell line MCF-7. Cell Mol Biol Lett. 2001;6:925–939. [PubMed] [Google Scholar]
- 14.Paik J, Blaner WS, Sommer KM, Moe R, Swisshelm K. Retinoids, retinoic acid receptors, and breast cancer. Cancer Invest. 2003;21:304–312. doi: 10.1081/cnv-120016425. [DOI] [PubMed] [Google Scholar]
- 15.Lin F, Xiao D, Kolluri SK, Zhang X. Unique anti-activator protein-1 activity of retinoic acid receptor beta. Cancer Res. 2000;60:3271–3280. [PubMed] [Google Scholar]
- 16.Yang L, Tin UC, Wu K, Brown P. Role of retinoid receptors in the prevention and treatment of breast cancer. J Mamm Gland Biol Neopl. 1999;4:377–388. doi: 10.1023/a:1018718401126. [DOI] [PubMed] [Google Scholar]
- 17.Dietze EC, Caldwell LE, Marcom K, et al. Retinoids and retinoic acid receptors regulate growth arrest and apoptosis in human mammary epithelial cells and modulate expression of CBP/p300. Microsc Res Tech. 2002;59:23–40. doi: 10.1002/jemt.10174. [DOI] [PubMed] [Google Scholar]
- 18.Farias EF, Arapshian A, Bleiweiss IJ, Waxman S, Zelent A, Mira-y-Lopez R. Retinoic acid receptor α2 growth suppressor epigenetically silenced in MCF-7 human breast cancer cells. Cell Growth Differ. 2002;13:335–341. [PubMed] [Google Scholar]
- 19.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumors: an update. Breast Cancer Res Treat. 2004;83:249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- 20.Wu Q, Dawson MI, Zheng Y, et al. Inhibition of trans-retinoic acid-resistant human breast cancer cell growth by retinoid X receptor-selective retinoids. Mol Cell Biol. 1997;17:6598–6608. doi: 10.1128/mcb.17.11.6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toma S, Isnardi L, Rafo P, et al. RARα antagonist RA 41-5253 inhibits proliferation and induces apoptosis in breast-cancer cell lines. Int J Cancer. 1998;78:86–94. doi: 10.1002/(sici)1097-0215(19980925)78:1<86::aid-ijc14>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 22.Vivat-Hannah V, Bourguet W, Gottardis M, Gronemeyer H. Separation of retinoid X receptor homo- and heterodimerization functions. Mol Cell Biol. 2003;23:7678–7688. doi: 10.1128/MCB.23.21.7678-7688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller VA, Benedetti FM, Rigas JR, et al. Initial clinical trial of a selective retinoid X receptor ligand LGD1069. J Clin Oncol. 1997;15:790–795. doi: 10.1200/JCO.1997.15.2.790. [DOI] [PubMed] [Google Scholar]
- 24.Shen Q, Brown PH. Novel aspects for the prevention of breast cancer: targeting transcription factors and signal transduction pathways. J Mamm Gland Biol Neoplasia. 2003;8:45–73. doi: 10.1023/a:1025783221557. [DOI] [PubMed] [Google Scholar]
- 25.Muccio DM, Brouillette WJ, Breitman TR, et al. Conformationally defined retinoic acid analogues: 4 potential new agents for promyelocytic and juvenile myelomonocytic leukemias. J Med Chem. 1998;41:1679–1687. doi: 10.1021/jm970635h. [DOI] [PubMed] [Google Scholar]
- 26.Atigadda VR, Vines KK, Grubbs CJ, et al. Conformationally defined retinoic acid analogues. 5. Large-scale synthesis and mammary cancer chemopreventive activity for (2E,4E,6Z,8E)-8-(3′,4′-dihydro-1′(2′H)-naphthalen-1′-ylidene)-3,7-dimethyl-2,4,6-octatrienoic acid (9cUAB30) J Med Chem. 2003;46:3766–3769. doi: 10.1021/jm030095q. [DOI] [PubMed] [Google Scholar]
- 27.Grubbs CJ, Hill DL, Bland KI, et al. 9cUAB30, an RXR specific retinoid, and/or tamoxifen in the prevention of methyl-nitrosourea-induced mammary cancers. Cancer Lett. 2003;201:17–24. doi: 10.1016/s0304-3835(03)00461-0. [DOI] [PubMed] [Google Scholar]
- 28.Fitzgerald P, Chandratna RA, Heyman RA, Allegretto EA. Retinoic acid receptor alpha expression correlates with retinoid-induced growth inhibition of human breast cancer cells regardless of estrogen receptor status. Cancer Res. 1997;57:2642–2650. [PubMed] [Google Scholar]
- 29.Wu S, Zhang D, Donigan A, Dawson MI, Soprano DR, Soprano KJ. Effects of conformationally restricted synthetic retinoids on ovarian tumor cell growth. J Cell Biochem. 1998;68:378–388. doi: 10.1002/(sici)1097-4644(19980301)68:3<378::aid-jcb8>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 30.Pratt MA, Niu M, White D. Differential regulation of protein expression, growth and apoptosis by natural and synthetic retinoids. J Cell Biochem. 2003;90:692–708. doi: 10.1002/jcb.10682. [DOI] [PubMed] [Google Scholar]
- 31.Lopatina N, Haskell JF, Andrews LG, Poole JC, Saldanha S, Tollefsbol TO. Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J Cell Biochem. 2002;84:324–334. doi: 10.1002/jcb.10015. [DOI] [PubMed] [Google Scholar]
- 32.Casillas MA, Jr, Lopatina N, Andrews LG, Tollefsbol TO. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol Cell Biochem. 2003;252:33–43. doi: 10.1023/a:1025548623524. [DOI] [PubMed] [Google Scholar]
- 33.Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer. 2002;101:335–341. doi: 10.1002/ijc.10593. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Saldanha SN, Pate MS, Andrews LG, Tollefsbol TO. Epigenetic regulation of human telomerase reverse transcriptase promoter activity during cellular differentiation. Genes Chromosomes Cancer. 2004;41:26–37. doi: 10.1002/gcc.20058. [DOI] [PubMed] [Google Scholar]
- 35.Elmore LW, Rehder CW, Di X, et al. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–33515. doi: 10.1074/jbc.M205477200. [DOI] [PubMed] [Google Scholar]
- 36.Dietze EC, Troch MM, Bowie ML, Yee L, Bean GR, Seewaldt VL. CBP/p300 induction is required for retinoic acid sensitivity in human mammary cells. Biochem Biophys Res Commun. 2003;302:841–848. doi: 10.1016/s0006-291x(03)00266-3. [DOI] [PubMed] [Google Scholar]
- 37.Mizuno S, Chijiwa T, Okamura T, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97:1172–1179. doi: 10.1182/blood.v97.5.1172. [DOI] [PubMed] [Google Scholar]
- 38.Casillas MA, Brotherton SL, Andrews G, Ruppert JM, Tollefsbol TO. Induction of endogenous telomerase (hTERT) by c-Myc in WI-38 fibroblasts transformed with specific genetic elements. Gene. 2003;316:57–65. doi: 10.1016/s0378-1119(03)00739-x. [DOI] [PubMed] [Google Scholar]
- 39.Mittal A, Pate MS, Wylie RC, Tollefsbol TO, Katiyar SK. EGCG down-regulates telomerase in human breast carcinoma MCF-7 cells, leading to suppression of cell viability and inducing apoptosis. Int J Oncol. 2004;24:703–710. [PubMed] [Google Scholar]
- 40.Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- 41.Eckhardt S. Recent progress in the development of anticancer agents. Curr Med Chem Anti-Cancer Agents. 2002;2:419–439. doi: 10.2174/1568011024606389. [DOI] [PubMed] [Google Scholar]
- 42.Ahmed A, Tollefsbol T. Telomeres and telomerase: basic science implications for aging. J Am Geriatr Soc. 2001;49:1105–1109. doi: 10.1046/j.1532-5415.2001.49217.x. [DOI] [PubMed] [Google Scholar]
- 43.Guilleret I, Benhattar J. Demethylation of the human telomerase catalytic subunit (hTERT) gene promoter reduced hTERT expression and telomerase activity and shortened telomeres. Exp Cell Res. 2003;289:326–334. doi: 10.1016/s0014-4827(03)00281-7. [DOI] [PubMed] [Google Scholar]
- 44.Pendino F, Dudognon C, Delhommeau F, et al. Retinoic acid receptor α and retinoid-X receptor-specific agonists synergistically target telomerase expression and induce tumor cell death. Oncogene. 2003;22:9142–9150. doi: 10.1038/sj.onc.1207093. [DOI] [PubMed] [Google Scholar]
- 45.Cao Y, Li H, Mu F-T, Ebisui O, Funder JW, Liu J. Telomerase activation causes vascular smooth muscle cell proliferation in genetic hypertension. FASEB J. 2001;16:96–98. doi: 10.1096/cj.01-0447fje. [DOI] [PubMed] [Google Scholar]
- 46.Saretzski G, Ludwig A, von Zglinicki T, Runnebaum IB. Ribozyme-mediated telomerase inhibition induces immediate cell loss but not telomere shortening in ovarian cancer cells. Cancer Gene Ther. 2001;8:827–834. doi: 10.1038/sj.cgt.7700383. [DOI] [PubMed] [Google Scholar]
- 47.Rubin M, Fenig E, Rosenauer A, et al. 9-cis retinoic acid inhibits growth of breast cancer cells and down-regulates estrogen receptor RNA and protein. Cancer Res. 1994;54:6549–6656. [PubMed] [Google Scholar]
- 48.Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev. 2002;66:407–425. doi: 10.1128/MMBR.66.3.407-425.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emionite L, Galmozzi F, Grattarola M, Boccardo F, Vergani L, Toma S. Histone deacetylase inhibitors enhance retinoid response in human breast cancer cell lines. Anticancer Res. 2004;24:4019–4024. [PubMed] [Google Scholar]
- 50.Cao Y, Li H, Deb S, Liu J. TERT regulates cell survival independent of telomerase enzymatic activity. Oncogene. 2002;21:3130–3138. doi: 10.1038/sj.onc.1205419. [DOI] [PubMed] [Google Scholar]