

Abstract
We have shown previously that there is a temporal increase in the levels of CXCL10 and CXCR3 mRNA during spontaneous murine colitis. We now show that CXCL10 is significantly expressed by mucosal CD4+ T cells, natural killer (NK) cells, and NKT cells, but not by dendritic cells (DCs), during chronic murine colitis. CXCL10 blockade alleviated chronic colitis and attenuated the associated increase in serum amyloid A (SAA), interleukin-12 (IL-12)p40, tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), IL-1α, and IL-1β Levels as well as in the number of CD4+ T, NKT, and NK cells that express CXCL10 and CXCR3, compared with groups treated with control antibody (Ab). After CXCL10 blockade, the number of CXCR3+ DCs in the mesenteric lymph nodes (MLNs) and Peyer’s patches (PPs) were increased to levels found before the onset of colitis. In contrast, the numbers of splenic and intestinal lamina propria (LP) CXCR3+ DCs were reduced after anti-CXCL10 Ab treatment, compared with controls. Ex vivo antigen and CXCL10 stimulation of mucosal cells caused an increase in MHC class II, CD40, and CD86 as well as a decrease in CD30 ligand (CD30L) expression by DCs. This study provides insights into CXCL10 expression during inflammatory bowel disease (IBD) and the cellular and molecular mechanisms of CXCL10-mediated colitis. Our data also support the premise that CXCL10 blockade can attenuate chronic colitis by preventing the activation and recruitment of CXCR3+ leukocytes during IBD.
INTRODUCTION
The etiology and pathogenesis of the two major forms of inflammatory bowel disease (IBD), Crohn’s disease (CD) and ulcerative colitis (UC), are poorly understood (Podolsky 2002). It is widely held that human IBD is multifactorial and caused by immunologic, environmental, and genetic factors. It has been suggested that colitis may be the result of massive cellular infiltrates and is associated with abnormalities in the immune system and normal gut flora (MacDonald and Pettersson 2000; Singh and others 2007a) or an overall autoimmune dysregulation/imbalance in T cells (Kuhn and others 1993; Mombaerts and others 1993; Hollander and others 1995).
Under conventional housing conditions, interleukin-10−/− (IL-10−/−) mice develop spontaneous colitis that has similarities to human CD. However, this murine model differs from human CD in that the colitis in IL-10−/− mice does not yield focal granulomatous or transmural inflammation. There is a consensus that the mucosa of CD patients is dominated by T cells producing inflammatory cytokines (Fiocchi 1998). We have shown that CXCL9, CXCL10, and CXCL11 are upregulated at the sites of murine colitis and clinical IBD (Singh and others 2003b, 2007b). It is well established that CD4+ Th1 cells mediate the chronic inflammation observed in the colons of IL-10−/− mice and that interferon-γ (IFN-γ) plays a major role during the initial phase of inflammation. The primary cellular source of early IFN-γ production appears to be the natural killer (NK) cells, which have been shown to be involved in the differentiation of naïve CD4+ T cells into Th1 cells (Romagnani 1992). To this end, the number of CD4+ CXCR3+ T cells in the intestinal lamina propria (LP) has been shown to be higher in IBD patients than in normal, healthy donors (Yuan and others 2001).
One of the ligands for CXCR3, CXCL10, is an immediate-early gene that is induced by IFN-γ and expressed by epithelium, fibroblasts, keratinocytes, NK cells, and monocytes (Luster and Ravetch 1987; Farber 1997). Whereas we and others have shown that blocking CXCL10 expression prevents the development of asymptomatic colitis (Singh and others 2003a; Hyun and others 2005), the present study demonstrates that CXCL10 inhibition also leads to remission of chronic colitis in IL-10−/− mice. The present study also shows that CD4+ T cells, NK cells, and NKT cells of the mucosa produce CXCL10, which correlates with higher numbers of CXCR3+ Th1 cells and CXCR3+ DCs as well as supports increases in MHC class II and costimulatory molecule expression by DCs.
MATERIALS AND METHODS
Animals
Female IL-10−/− mice, on a B6 background, age 8–12 weeks, were purchased from Jackson Laboratories (Bar Harbor, ME), and 4–5-month-old female New Zealand rabbits (Myrtle’s Rabbitory, Thompson Station, TN) were used to generate anti-CXCL10 antibodies (Ab). The animals were housed and maintained in microisolator cages under conventional housing conditions. Experimental groups consisted of 5 mice, and each study was repeated three times. The guidelines proposed by the committee for the Care of Laboratory Animal Resources Commission of Life Sciences of the National Research Council were followed to minimize animal pain and distress.
Cell isolation
Spleens and mesenteric lymph nodes (MLNs) from mice were mechanically dissociated, and red blood cells (RBCs) were disrupted with lysis buffer. The single-cell suspensions of spleen and MLN cells were passed through a sterile wire screen (Sigma, St. Louis, MO) and cells from intestinal LP and Peyer’s patches (PPs) were isolated as described previously (Lillard and others 2001, 2003). Thereafter, lymphocytes were maintained in complete medium, which consisted of RPMI 1640 supplemented with 10 mL/L nonessential amino acids (Mediatech, Washington, DC), 1 mM sodium pyruvate (Sigma), 10 mM HEPES (Mediatech), 100 U/mL penicillin, 100 μg/mL streptomycin, 40 μg/mL gentamicin (Elkins-Sinn, Inc., Cherry Hill, NJ), 50 μM mercaptoethanol (Sigma), and 10% fetal bovine serum (FBS) (Atlanta Biologicals, Atlanta, GA).
Immunogens and Abs
The potential level of endotoxin contamination of immunogens was quantified by the chromogenic Limulus amebocyte lysate assay (Cape Cod, Inc., Falmouth, MA) to be < 5 EU/mg. Chicken egg ovalbumin (OVA) and bovine serum albumin (BSA) were purchased from Sigma. Rat antimouse CXCL10 monoclonal antibody (mAb) was obtained from R&D Systems (Minneapolis, MN). Rabbit antimouse CXCR3 Ab was purchased from Zymed Laboratories Inc. (San Francisco, CA). Anti-CXCL10 and anti-CXCR3 Abs were conjugated to allophycocyanin (APC) (Prozyme, San Leandro, CA). The detailed methods of anti-CXCL10 Ab generation, purification, and cross-reactivity determination for subsequent in vivo neutralization of CXCL10 were described previously (Singh and others 2003a). In brief, CXCL10 plus complete Freund’s or incomplete Freund’s adjuvants (Sigma) were used to generate anti-CXCL10 Ab titers of ~1:2 × 106 such that 10 μL anti-CXCL10 Ab neutralized 20 ng CXCL10 by ELISA (R&D Systems) and chemotaxis assay (Chemicon, Temecula, CA) using CXCR3+ T cells. This Ab was titrated by direct ELISA, and no cross-reactivity was detected when tested against other CXCR3 ligands (CXCL9, CXCL11), chemokines (CCL2, CCL3, CCL4, CCL5, CCL7, CCL8, CCL11, CXCL6, CXCL8, and CXCL13), cytokines (IL-2, IL-4, IL-5, IL-6, IL-10, IL-12, and tumor necrosis factor-α [TNF-α]), CCR5, CXCR4, and CCR3 transfectants. The Th cell-derived cytokines (IL-1α, IL-1β, IL-2, IL-12p40, IFN-γ, and TNF-α) in serum were determined using a Beadlyte mouse multicytokine detection system kit (Bio-Rad, Hercules, CA). The serum amyloid A (SAA) level in mouse sera was determined by ELISA (Biosource International, Camarillo, CA).
Flow cytometry analysis
Cells from the spleen, MLNs, PPs, and LP were freshly isolated, as described, for flow cytometry analysis. Cells were treated with Fc blocker for 15 min at 4°C, washed with FACS staining buffer, and then stained with CY-conjugated, FITC-conjugated or PE-conjugated anti-CD3, anti-CD4, anti-CD8, anti-CD11b, CD11c, anti-NK1.1 Abs or with respective isotype controls (BD-PharMingen, San Diego CA) for 30 min with shaking. The cells were then washed two times with FACS staining buffer and resuspended in BD Cytofix/Cytoperm solution for 20 min (BD-PharMingen). Next, the cells were washed two times with BD perm/wash solution for 10 min at 4°C. The brefeldin A-treated, fixed, and permeabilized cells were stained using APC-conjugated anti-CXCL10, anti-TNF-α, anti-IFN-γ, anti-IL-12p40, and/or anti-CXCR3 Abs for 30 min at 4°C in the dark. The lymphocytes were washed with FACS staining buffer (phosphate-buffered saline [PBS] with 1% BSA) and analyzed by flow cytometry (Becton Dickinson, Mountain View, CA).
Anti-CXCL10 Ab treatment of chronic colitis
The body weight of each mouse was monitored twice a week. We monitored SAA and IL-6 levels every week and determined that the onset of asymptomatic colitis occurred when these levels exceeded 200 μg/mL and 500 pg/mL, respectively. For the study of chronic colitis, SAA and IL-6 levels were allowed to reach 300 μg/mL and 5.0 ng/mL, respectively, with at least a 10%–15% decrease from original body weight. Ten weeks after the onset of asymptomatic colitis, mice received an intraperitoneal injection of 200 μL of the anti-CXCL10 Ab or preimmune Ab solution every 3 days until they were killed (Fig. 1).
FIG. 1.
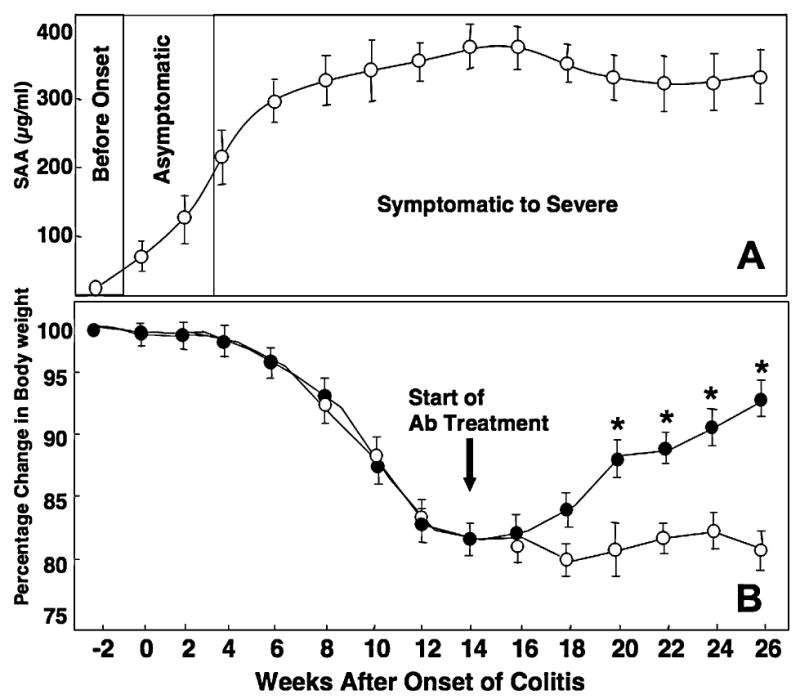
IL-10−/− mice received 200 μL control Ab (open circles) or antimouse CXCL10 Ab (closed circles) every 3 days starting 14 weeks after the onset of symptomatic colitis, when mice had lost about 10%–15% of their initial body weight and attained a peak in SAA levels, and continued until the mice were killed at week 26. The level of SAA ± SEM (A) and body weight (B) of the IL-10−/− mice were recorded every week, and the change from initial body weight was expressed as a percentage of the weight before the onset of colitis (week –2) minus the weight at subsequent weeks divided by the weight before the onset of colitis (±SEM). Data represent the mean of three independent experiments involving 5 mice per group. *Statistically significant differences (p < 0.01) between anti-CXCL10 Ab-treated and control Ab-treated groups.
Cytokine, chemokine, and receptor mRNA expression
For RT-PCR primer design, mouse mRNA sequences for TNF-α, IFN-γ, CXCL10, IL-12p40, and CXCR3 mRNAs as well as 18S rRNA were obtained from the NIH-NCBI genebank, accession numbers NM013693, K00083, M33266, M86671, AF045146, and X00686.1, respectively. Primers were designed using the Beacon 2.0 computer program to generate 100, 98, 95, 102, 96, and 149 base pair (bp) fragments of TNF-α, IFN-γ, CXCL10, IL-12p40, CXCR3, and 18S rRNA, respectively. Thermodynamic analysis of the primers was conducted using the following computer programs: Primer Premier and MIT Primer III (Boston, MA). The resulting primer sets were compared against the entire mouse genome to confirm specificity and to ensure that they flanked mRNA splicing regions.
Potential genomic DNA contamination was removed from these samples by treating them with 10 U/μL RNase-free DNase (Invitrogen, San Diego, CA) for 15 min at 37°C. RNA was then precipitated and resuspended in RNA Secure (Ambion, Austin, TX). cDNA was generated by reverse transcribing 1.5 μg total RNA using Taqman reverse transcription reagents (Applied Biosystems, Foster City, CA), according to the manufacturer’s protocols. Subsequently, cDNAs were amplified with specific cDNA primers (IDT, Coraville, IA) using SYBR Green PCR master mix reagents (Applied Biosystems), according to the manufacturer’s protocol. The number of mRNA copies in each tissue sample was evaluated by RT-PCR analysis using the Bio-Rad Icycler and software.
CXCL10 and OVA stimulation of DO11.10 leukocytes
MLN Th (CD3+ CD4+) cells and DCs (CD11b+ CD11cHi) were isolated from DO11.10 mice by FACS sorting. CD4+ T cells and DCs were subsequently added at a ratio of 5:1, respectively, at a density of 106 cells/mL in complete medium containing 0, 1, 10, 100, or 1000 ng/mL CXCL10. A class II-restricted OVA peptide containing amino acids 323–339 (1 mg/mL) was used to indirectly activate DCs in a T cell-dependent fashion. After incubation for 2 days, the cells were stained with FITC-conjugated, PE-conjugated, Cy5-conjugated, and/or APC-conjugated rat antimouse I-Ab/I-Eb, CD80, CD86, CD30L, CD11b, and CD11c mAbs as well as respective isotype controls (PharMingen) for 30 min with shaking. The lymphocytes were washed with FACS buffer (PBS with 1% BSA), fixed in 2% paraformaldehyde in PBS, and analyzed by flow cytometry (Becton Dickinson).
Histology
Intestinal tissues were preserved using Streck fixative (Streck Laboratories, LaVista, NE) and embedded in paraffin. Fixed tissues were sectioned at 6 μm and stained with hematoxylin and eosin (H&E) for microscopic examination. The histologic evaluation methods were performed as described previously (Singh and others 2003a).
Statistics
The data are expressed as the mean ± standard error mean (SEM) and compared using a two-tailed paired Student’s t-test or an unpaired Mann-Whitney U-test. The results were analyzed using the Statview II statistical program (Abacus Concepts, Inc., Berkeley, CA) and Microsoft Excel (Microsoft, Seattle, WA) for Macintosh computers. Kolmogorov-Smirnov (K-S) two-sample test using CELL Quest Software (BD-PharMingen) for Macintosh computers was used to compute the statistical significance between histograms. Results were considered statistically significant if p values were < 0.01.
RESULTS
Characteristics of chronic colitis progression in IL-10−/− mice
Chronic colitis in IL-10−/− mice corresponded with an increase in SAA levels (>300 μg/mL) (Fig. 1A) and with a 10%–15% reduction in the body weight of the mice compared with their initial body weight (Fig. 1B). CXCL10 blockade in mice with chronic colitis alleviated weight loss when compared with the weight loss experienced by IL-10−/− mice with chronic colitis treated with control Ab.
IFN-γ, IL-12p40, TNF-α, CXCL10, and CXCR3 gene expression after anti-CXCL10 Ab treatment
Significant increases in the expression of TNF-α and IL-12p40 mRNA were noted in the MLNs and colons of IL-10−/− mice with chronic colitis compared with anti-CXCL10 Ab-treated mice (Fig. 2). CXCL10 mRNA expression by the colon and MLNs was also significantly elevated during chronic colitis in IL-10−/− mice treated with control Ab compared with anti-CXCL10 Ab-treated mice. IFN-γ Levels were reduced in the MLNs of mice with severe colitis following anti-CXCL10 Ab treatment compared with control Ab treatment; however, this Th1-associated cytokine was below detectable levels in the colons of both groups. CXCR3 mRNA expression was significantly reduced in the colons of IL-10−/− mice with colitis after CXCL10 inhibition, but its level in MLNs was not diminished during the same treatment compared with control Ab-treated mice.
FIG. 2.

After development of chronic colitis, IL-10−/− mice received 200 μL of either control Ab (solid bars) or anti-CXCL10 Ab (hatched bars) every 3 days starting 14 weeks after the onset of symptomatic colitis, when mice had lost about 15% of their initial body weight, or no additions in normal wild type (WT) mice (open bars). After the mice were sacrificed, total RNA was isolated from the colons and MLNs of mice untreated or treated with either control Ab or anti-CXCL10 Ab. The levels of IFN-γ, CXCL10, TNF-α, IL-12p40, and CXCR3 mRNA expression were ascertained by RT-PCR analysis that was capable of detecting > 20 copies of transcribed cDNA. Log10 copies of transcripts are expressed relative to actual copies of 18S rRNA ± SEM. Data represent the mean of three independent experiments involving 5 mice per group. *Statistically significant differences (p < 0.01) between anti-CXCL10 and control Ab-treated groups.
Anti-CXCL10 Ab ameliorates serum Th1 and inflammatory cytokine increases associated with chronic colitis
In confirmation of the RT-PCR analysis, anti-CXCL10 Ab treatment decreased IFN-γ and IL-12p40 serum levels in IL-10−/− mice with chronic colitis (Fig. 3). Serum IL-2, TNF-α, IL-1α, and IL-1β Levels also declined after CXCL10 blockade in IL-10−/− mice with chronic colitis compared with the control Ab-treated mice. These data indicate that CXCL10 blockade caused the reduction of SAA, IL-6, IL-12p40, IFN-γ, IL-2, TNF-α, IL-1α, and IL-1β serum levels of the IL-10−/− mice with chronic colitis.
FIG. 3.

IL-10−/− mice received 200 μL of either control Ab (open circles) or anti-CXCL10 Ab (closed circles) every 3 days, starting 14 weeks after the onset of symptomatic colitis, which continued through week 26. Before killing, levels of serum cytokines at week 26 were determined by ELISA capable of detecting > 10 pg/mL of IL-12p40, IL-2, TNF-α, IFN-γ, IL-1α, and IL-1β. Data presented are the mean concentrations ± SEM. *Statistically significant differences (p < 0.01) between the two groups. Experimental groups consisted of 5 mice, and experiments were repeated three times. Data represent the mean of three independent experiments.
Changes in colitis severity following CXCL10 blockade
The mice that received anti-CXCL10 Ab showed a significant reduction in intestinal inflammation. An increase in leukocyte infiltrates (Fig. 4A) and distortion of glandular architecture (Fig. 4B) were observed in the intestines during chronic colitis. Anti-CXCL10 Ab reduced the lymphocyte infiltration and partially restored the glandular and goblet cell architecture (Fig. 4C), which also coincided with lengthening of intestinal crypts (Fig. 4D). The mean histologic scores of IL-10−/− mice with severe colitis that received control Ab were significantly higher than the scores of mice treated with anti-CXCL10 Ab (Table 1). Similarly, SAA levels correlated with the severity of colitis as determined by histologic analysis. Pathologic changes included leukocyte infiltrates in the LP of the colon of control Ab-treated IL-10−/− mice, with the number of these infiltrates being reduced after CXCL10 blockade. Taken together, the results show a marked improvement in the characteristic intestinal inflammation associated with chronic colitis after CXCL10 blockade.
FIG. 4.

Histopathology of the colons from IL-10−/− mice with chronic colitis treated with either (A and B) control Ab or (C and D) anti-CXCL10 Ab as before. Sections were examined by light microscopy. Experimental groups consisted of 5 mice, and experiments were repeated three times.
Table 1.
Histologic Evaluation of Control and Anti-CXCL10 Ab-Treated IL-10−/− Mice with Chronic Colitisa
| Treatment/group | Number of mice | Colitis disease score (0–12) | SAA (μg/mL) |
|---|---|---|---|
| Anti-CXCL10 Ab | 15 | 4.17 ± 0.40* | 113.6 ± 16* |
| Control Ab | 15 | 8.01 ± 0.91 | 387.3 ± 17 |
| Untreated WT C57BL/6 | 2 | 0 | 13.4 ± 0.6 |
The persistence or improvement of chronic/severe colitis was monitored by evaluating SAA levels and histopathologic changes in the coons of IL-10−/− mice that received 200 μL of either control or anti-CXCL10 Ab every 3 days, starting at 14 weeks after the establishment of asymptomatic colitis (or 10 weeks after symptomatic disease). After the mice were killed, intestines were fixed, sectioned at 6 μm, and stained with H&E. The sections were examined microscopically at magnifications of 40× and 100× and scored for the severity of colitis.
Statistically significant differences (p < 0.01) between control Ab-treated and anti-CXCL10 Ab-treated groups.
Characteristics of Th cells during chronic colitis and changes after CXCL10 inhibition
Flow cytometry analysis revealed that CD4+ T cells from the MLNs express CXCL10 during chronic colitis. CXCL10 blockade in IL-10−/− mice with chronic colitis reduced the percentage of total CXCL10+ CD4+ and CXCR3+ CD4+ cells in the MLNs and the number of similar cells in the LP (Fig. 5A). Moreover, the number of CD4+ T cell infiltrates expressing CXCL10 and CXCR3 in the spleen, MLNs, PPs, and LP of IL-10−/− mice with chronic colitis was significantly reduced following CXCL10 blockade compared with similar mice treated with control Ab. We also observed a significant decline in the total number of splenic, MLN, PP, and LP CD4+ T cells expressing IFN-γ or TNF-α or both in mice treated with anti-CXCL10 Ab compared with mice treated with control Ab (Fig. 5B).
FIG. 5.
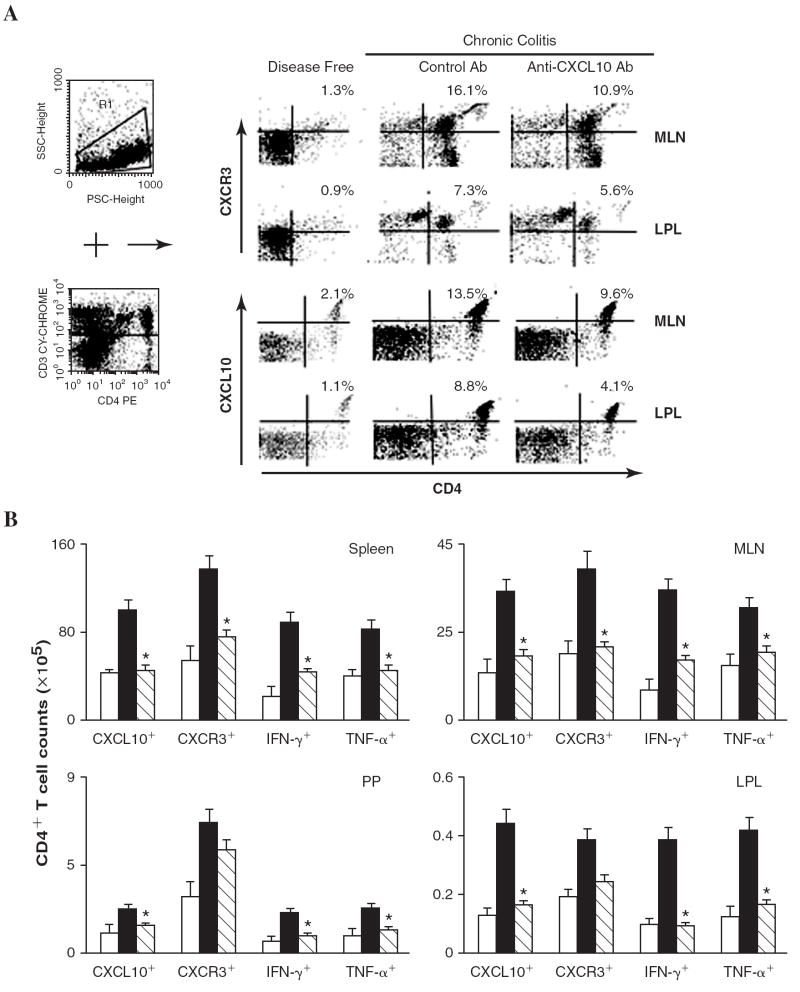
Splenic, MLN, PP, and LP lymphocytes were isolated from IL-10−/− mice before the onset of colitis (disease free) or during chronic colitis (26 weeks after the onset of symptomatic colitis) treated with either anti-CXCL10 Ab or control Ab. (A) MLN or LP-derived CD3+ lymphocytes (R1+R4) were characterized for CD4 and CXCR3 expression by flow cytometry. The numbers in the top right quadrant indicate the percentage of total CXCR3+ or CXCL10+ CD4+ T cells. Similar analysis was conducted for CD4+ T cells from the spleen, MLN, PP, and LP. (B) Data are presented as the total number of CXCL10+, CXCR3+, IFN-γ+, and/or TNF-α+ CD4+ T cells in each immune compartment from disease-free mice (open bars) and mice with chronic colitis treated with control Ab (solid bars) or anti-CXCL10 Ab (hatched bars). Data represent the total number of cells ± SEM from three independent experiments involving 5 mice per group. Statistically significant differences (p < 0.01) between anti-CXCL10-treated and control Ab-treated groups.
CXCR3, TNF-α, IFN-γ, and CXCL10 expression by NK and NKT cells
The mean fluorescence intensity of NK cells that expressed CXCL10 as well as CXCR3, IFN-γ, and TNF-α was elevated during chronic colitis, but this intensity of expression declined to near background levels after anti-CXCL10 Ab treatment (data not shown). CXCL10 blockade also reduced the number of splenic NK cells that expressed CXCL10, CXCR3, and IFN-γ during chronic colitis (Fig. 6A). The numbers of CXCL10+ and CXCR3+ NK cell populations in MLN did not vary during chronic colitis compared with cell numbers from similar mice before the onset of colitis. Following CXCL10 blockade, more NK cells than other cells from the MLN expressed IFN-γ. The number of TNF-α+-expressing NK cells was also elevated during severe/chronic colitis, but their number was significantly decreased following anti-CXCL10 Ab treatment. Similarly, the increase compared with normal controls in the numbers of CXCL10+, CXCR3+, IFN-γ+, and/or TNF-α+ NK cells from the PP was significantly decreased after CXCL10 blockade.
FIG. 6.
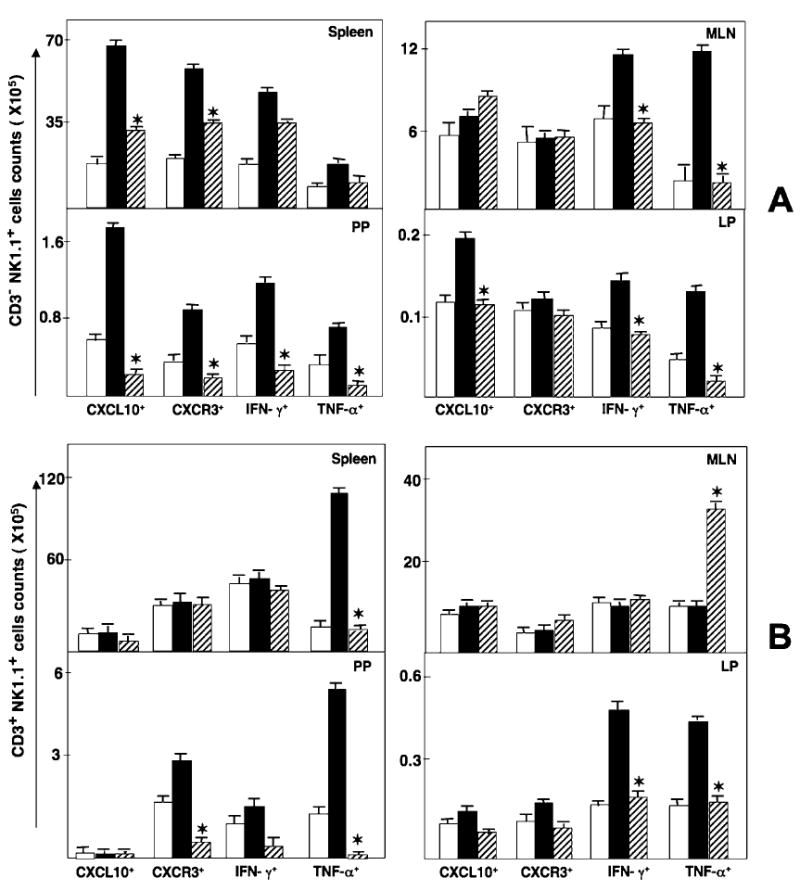
Splenic, MLN, PP, and LP lymphocytes were isolated from IL-10−/− mice before the onset of colitis (disease free, open bars) or during chronic colitis (26 weeks after the onset of symptomatic colitis) treated with either anti-CXCL10 Ab (hathed bars) or control Ab (solid bars). (A) Changes in NK cells (CD3− NK1.1+ cells) expressing IFN-γ, TNF-α, CXCL10, and CXCR3 were determined by flow cytometry and expressed as the total number of cells ± SEM. (B) Changes in NKT cells (CD3+ NK1.1+ cells) expressing IFN-γ, TNF-α, CXCL10, and CXCR3 were determined by flow cytometry and expressed as the total number of cells ± SEM. Data represent the mean of three independent experiments involving 5 mice per groups. *Statistically significant differences (p < 0.01) between anti-CXCL10-treated and control Ab-treated groups.
In contrast to NK cells, the number of splenic NKT cells that expressed CXCL10, CXCR3, or IFN-γ was unchanged during chronic colitis, but CXCL10 blockade reduced the number of splenic TNF-α+ NKT cells that were increased during this disease (Fig. 6B). NKT cells from MLN expressed TNF-α during CXCL10 blockade in IL-10−/− mice with chronic colitis. One of the most striking decreases in the number of these inflammatory cells occurred in PP-derived CXCR3+ and TNF-α+ NKT cells following CXCL10 blockade.
Our data suggested that CD4+ T cells were a source of IFN-γ in the LP during severe colitis, and the numbers of IFN-γ+-expressing as well as TNF-α+ expressing NK and NKT cells were elevated in the LP during severe colitis compared with the negative controls. CXCL10 blockade reduced the number of these cells that were isolated from this inflamed site. Although the number of CXCR3+ NK and NKT cells from the LP remained relatively unchanged, the number of CXCL10+ NK cells from the LP was significantly reduced following CXCL10 blockade. It is important to note that there were as many as 3-fold more CD4+ T cells and NKT cells than NK cells in the LP during chronic colitis. Considered together, these results indicate that CXCL10 blockade modulates the number of systemic and mucosal NKT and NK cells expressing CXCL10, CXCR3, TNF-α, and IFN-γ. Notably, NKT and CD4+ T cells appear to be sources of IFN-γ and TNF-α, whereas NK and CD4+ T cells express more CXCL10 during colitis, which is abrogated by CXCL10 inhibition.
Alteration of CXCR3, TNF-α, IFN-γ, IL-12p40, and CXCL10 expression by DCs
CXCR3 was differentially expressed by DCs from the MLN and LP during colitis (Fig. 7A). The intensity of CXCR3 expression by MLN DCs from mice with chronic colitis and anti-CXCL10 Ab-treated mice was significantly higher than that of similar cells from control Ab-treated mice with disease. In contrast, CXCR3 expression by LP DCs during chronic colitis, along with CXCL10 blockade, was significantly lower than that by similar cells from mice receiving control Ab during disease. CXCL10 blockade attenuated the number of CXCR3+ DCs in the LP and spleen of IL-10−/− mice with chronic colitis, while significantly increasing the number of CXCR3+ DCs in MLNs and PPs, back to the normal observed in controls (Fig. 7B). Mucosal DCs from IL-10−/− mice did not appear to express significant levels of CXCL10, but the numbers of CXCR3+ DCs in effector (LP) vs. inductive (MLN and PP) mucosal sites were reciprocally modulated by CXCL10 blockade.
FIG. 7.

Splenic, MLN, PP, and LP lymphocytes were isolated from IL-10−/− mice before the onset of colitis (disease free, open bars) or during colitis (26 weeks after the onset of symptomatic colitis) treated with either control Ab (solid bars) or anti-CXCL10 Ab (hathed bars). (A) Changes in the mean fluorescence intensity of CXCR3 expressed by CD11b+CD11c+ cells from MLN and LP with control Ab (dotted line) and anti-CXCL10 Ab-treated (solid line), and disease-free mice (gray line). (B) Changes in CD11b+CD11c+ cells expressing IFN-γ, TNF-α, CXCL10, IL-12p40, and CXCR3 were characterized by flow cytometry and presented by the total number of cells ± SEM. Data represent the mean of three independent experiments involving 10 mice per group. *Statistically significant differences (p < 0.01) between anti-CXCL10 Ab-treated and control Ab-treated groups.
CXCL10 modulation of costimulatory molecule expression by DCs
We have shown previously that CXCR3 ligands positively modulate CD28 expression by antigen-stimulated CD4+ T cells (Singh and others 2003b). LP and immature DCs were induced to rapid maturation and CD86 expression after entering the MLN (Huang and others 2000). To further establish the CXCL10-mediated effects on the regulation of DC costimulatory molecules, we assessed the potential of CXCL10 to modulate the expression of Ab/I-Eb, CD80, CD86, and CD30L by DCs during class II-restricted antigen stimulation of MLN leukocytes. CXCL10 significantly increased the expression of class II (Ab/I-Eb) and CD86, but not CD80, by DCs during antigen stimulation in a dose-dependent fashion (Table 2 and Fig. 8). CXCL10 also significantly downregulated the expression of CD30L by similar DCs, which supports the notion that this CXCR3 ligand modulates DC activation or maturation or both.
Table 2.
Percent Change in CD11b+ CD11cHi Cell B7, Ab/I-Eb and CD30 Ligand Expressiona
| CD11b+CD11cHi cell costimulatory molecules | Dose of CXCL10 (ng/mL)
|
|||
|---|---|---|---|---|
| 1 | 10 | 100 | 1000 | |
| CD80 | (14.2%) ± 2.1 | (10.2%) ± 1.2 | (11.82) ± 1.6 | (9.04%) ± 0.9 |
| CD86 | 29.2% ± 4.2 | 42.2%* ± 5.4 | 63.2%* ± 5.2 | 59.6%* ± 6.1 |
| CD30L | (16.5%) ± 1.9 | (15.3%) ± 2.3 | (12.5%) ± 1.1 | (1.9%)* ± 0.3 |
| Ab/I-Eb | 27.9% ± 3.3 | 35.6% ± 4.1 | 53.9%* ± 7.2 | 61.3%* ± 6.9 |
OVA-stimulated MLN CD4+ T cells and CD11b CD11cHi from DO11.10 mice were incubated with 0, 1, 10, 100, and 1000 ng/mL of CXCL10 for 48 h, at a ratio of 5:1 respectively, and prepared for FACS analysis. The percent increase (or decrease) in costimulatory molecule surface expression was calculated as the percentage of CD11b+ CD11cHi cells that were CD80+, CD86+, Ab/I-Eb+, or CD30L+ in cultures containing CXCL10 minus the percent gated double-positive cells in cultures without CXCL10 addition divided by the latter. Studies were repeated three times and presented as the mean percent increase or (decrease) in expression.
Statistically significant differences (p < 0.01) relative to culture without CXCL10.
FIG. 8.

OVA-stimulated MLN CD4+ T cells and CD11b+CD11cHi cells from DO11.10 mice were incubated with 0, 1, 10, 100, or 1000 ng/mL CXCL10 for 48 h, at a ratio of 5:1 respectively, and analyzed by flow cytometry. Changes in CD11b+CD11cHI cells expressing Ab/I-Eb, CD80, CD86, and CD30L were characterized by flow cytometry and presented as mean fluorescence intensity of cells treated with (solid line) or without CXCL10 (shaded area).
DISCUSSION
Our previous studies demonstrated that CXCL10 blockade abrogated spontaneous colitis in IL-10−/− mice (Singh and others 2003a), which is predominantly mediated by Th1-type αβ T cell receptor (TCR+) and CXCR3+ cells (Singh and others 2003b). In confirmation, it was shown recently that anti-CXCL10 Ab treatment can mitigate colitis in IL-10−/− mice by decreasing the trafficking of Th1 cells (Hyun and others 2005). We hypothesized that blocking CXCL10-CXCR3 interactions alleviates chronic colitis in IL-10−/− mice by negatively modulating Th1 cells as well as DCs, NK cells, and NKT cells. We show that anti-CXCL10 Ab treatment resolves chronic colitis, corrects weight loss, and reduces local and systemic levels of IL-1α, IL-1β, IL-6, SAA, IL-12p40, IFN-γ, and TNF-α in IL-10−/− mice with chronic colitis. Our results suggest that both systemic and mucosal CD4+ T cells, NKT cells, and NK cells differentially express IFN-γ, TNF-α, CXCL10, or CXCR3, which can be mitigated by CXCL10 blockade. We also show that although DCs do not significantly express CXCL10, IFN-γ, and TNF-α during chronic colitis, CXCR3+ DCs are elevated in the spleen as well as in LP and are diminished at mucosal inductive sites (i.e., the PP and MLN) during severe colitis. It is also important to mention that there are significant difference between WT and IL-10−/− mice. In previous studies by our laboratories and others, the numbers of CD4+ T cells were greater in IL-10−/− mice than in WT B6 mice. Indeed, anti-CXCL10 Ab did not significantly alter leukocyte populations in WT mice housed under germ-free conditions. However, IL-10−/− mice, but not WT B6 mice, housed under conventional housing undergo relatively rapid immune dysregulation in part due to CXCL10-dependent mechanisms that are susceptible to inhibition (Singh and others 2003a).
The association among CXCL10, CXCR3, and Th1-dependent immunity has been observed in several models of inflammatory diseases (Qin and others 1998). Several IBD models indicate that CD4+ T cells play a major role in the induction of IBD, and much of the intestinal damage of this disease is a result of T cell-mediated injury (Elson and others 1996). To this end, we have shown that the adoptive transfer of CXCR3+ CD4+ T cells results in colitis in TCR (β × δ)−/− mice (Singh and others 2003b). Our results confirm that the number of CD4+ T cells in the spleen, MLN, and PP represents the majority of lymphocytes that express inflammatory cytokines during colitis.
Along with TNF-α, sustained acute-phase responses are associated with both human IBD and murine colitis (Berg and others 1996). SAA is elevated during CD (De Beer and others 1982), activated macrophages increase IL-1α and IL-1β expression in patients with IBD (Casini-Raggi and others 1995), and IL-12, IL-23 (with IL-12p40 subunit), and IFN-γ play critical roles in the induction and progression of colitis (Neurath and others 1995; Parronchi and others 1997). In the present study, we demonstrate that local and systemic TNF-α, IL-1α, IL-1β, IL-12p40, and IFN-γ expression is decreased by anti-CXCL10 Ab treatment in IL-10−/− mice with chronic colitis. It seems likely that NK cell interactions might serve to initiate these factors to contribute to Th1-mediated colitis. We observed a decline in the number of NK and NKT cells in the MLNs, PP, and LP lymphocytes after CXCL10 blockade compared with similar mice treated with control Ab. In addition, the expression of CXCL10, CXCR3, TNF-α, and IFN-γ by NK cells declined after CXCL10 treatment when compared with the control Ab-treated mice. Hence, it is also possible that the Th1 imbalance in IL-10−/− mice is modulated by NK cells during colitis.
In addition to LP CD4+ T cells and NK cells, we noted that NKT cells from the LP express TNF-α. This NK cell subset represented the predominant leukocyte that expressed IFN-γ and CXCL10 during chronic colitis. However, NKT cells have been shown to contribute to the suppression of Th1-mediated colitis (Wei and others 2005; Shibolet and others 2003; Menachem and others 2005). Future studies, possibly using NK cell depletion or CD1−/− × IL-10−/− mice, will be needed to precisely and decisively demonstrate the role of NKT and NK cells in the initiation and maintenance of colitis.
Although we have shown that CXCR3+ CD4+ T, NK, and NKT cells are sources of CXCL10 and inflammatory cytokines during chronic/severe colitis, it is also likely that other cells are equally responsible for the increase in inflammatory cytokines during colitis. CXCL10 has been reported to be produced by other cell types, particularly epithelial cells (Sauty and others 1999; Spurrell and otherse 2005) and neutrophils (Singh and others 2007a). Hence, the cellular mechanisms responsible for the differentiation of naïve CD4+ T cells, in this model, are still in question, as neither mucosal inductive DCs (PP or MLN) nor effector DCs (LP) produced significant levels of IL-12. The heavily Th1-biased IL-10-deficient mouse model may not be the best to determine the exact mechanism of Th1 polarization that occurs in CD or murine colitis. Presumably, the IL-12p40+ DCs in the spleen and IL-12 and IFN-γ in the periphery aided in Th1 differentiation.
Our current findings also indicate that CXCR3+ (and often TNF-α+) DCs predominantly populate the LP during chronic colitis. CXCL10 blockade reduced colitis severity and lowered the number of CXCR3+ DCs in the spleen and LP and increased the number of DCs in MLNs and PPs to levels observed before the onset of disease. Depending on their activation state or maturation, DCs may express CCR1, CCR2, CCR5, CCR6, CXCR2, and CXCR3 (Dieu and others 1998; Sozzani and others 1998). Hence, the alteration in the number of DCs in mucosal inductive and effector sites during chronic colitis after CXCL10 blockade might be due to the upregulation of other chemokines and corresponding receptors that govern the trafficking of DCs to lymphoid organs (e.g., MLNs and PPs). For example, CCL20 and CCR6 have been shown to play a distinct role in PP DC subsets trafficking to MLNs (Iwasaki and Kelsall 1999).
Whereas immature DCs can express CCL3 and CCL5, mature DCs selectively express CCL2, CCL5, and CXCL10 upon activation (Foti and others 1999). CCL5, like CXCL10, could also participate in Th1 cell recruitment via CCR5 interactions. In the present study, however, DCs marginally expressed CXCL10 during chronic colitis. In a Mycobacterium avium paratuberculosis model of colitis, we recently observed that DCs express significant levels of CXCL11 (Singh and others 2007a), which would support CXCR3+ cell chemotaxis. The prominent increase in CXCL10+ CXCR3+ T cells, NK cells, and NKT cells in conjunction with CXCR3+ DCs suggests that these cellular mechanisms are required for the initiation and maintenance of severe/ chronic colitis.
We have shown that CXCL9, CXCL10, and CXCL11 significantly increase CD28 expression by CD3ε-stimulated CD4+ T cells (Singh and others 2003b). Other chemokines, for example, CCL3, CCL4, and CCL5, also can upregulate CD86 expression (Lillard and others 2001; Del Prete and others 1995). In this study, CXCL10 downregulated CD30L expression by DCs during OVA stimulation of DO11.10 MLN lymphocytes, which would simultaneously remove an important signal for Th2 cell differentiation (Del Prete and others 1995). Further, CXCL10 enhanced Ab/I-Eb expression by DCs in dose-dependent fashion. This coincides with reports that activated T cells could stimulate and mature APCs (Avice and others 1999; Andreae and others 2002). We present a scenario in which TCR-Ab/I-Eb and CD28-CD86 interactions are enhanced and CD30-CD30L interactions are reduced by the abundance of CXCL10 during colitis, which would activate antigen-stimulated naïve T cells to develop into Th1 cells. These findings illustrate possible mechanisms of CXCL10-mediated activation of DCs for subsequent expansion and differentiation of CD4+ T cells that propagate colitis. Considered together, our results indicate that CXCR3 and CXCL10 interactions are physiologically and pathologically important for the regulation of colitis.
Acknowledgments
The content of this paper benefited from many fruitful conversations with colleagues at the Morehouse School of Medicine, the National Institutes on Aging, the University of Alabama at Birmingham, and the University of Louisville, as well as editing by Andrew Marsh. This work was supported in part by the Crohn’s & Colitis Foundation of America, National Institutes of Health grants RR03034, GM08248, MD000525, and AI57808, the Southeast Center for Emerging Biologic Threats, and the University of Louisville.
References
- Andreae S, Piras F, Burdin N, Triebel F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223) J Immunol. 2002;168:3874–3880. doi: 10.4049/jimmunol.168.8.3874. [DOI] [PubMed] [Google Scholar]
- Avice MN, Sarfati M, Triebel F, Delespesse G, Demeure CE. Lymphocyte activation gene-3, a MHC class II ligand expressed on activated T cells, stimulates TNF-alpha and IL-12 production by monocytes and dendritic cells. J Immunol. 1999;162:2748–2753. [PubMed] [Google Scholar]
- Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) Th1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J Immunol. 1995;154:2434–2440. [PubMed] [Google Scholar]
- De Beer FC, Mallya RK, Fagan EA, Lanham JG, Hughes GR, Pepys MB. Serum amyloid-A protein concentration in inflammatory diseases and its relationship to the incidence of reactive systemic amyloidosis. Lancet. 1982;2:231–234. doi: 10.1016/s0140-6736(82)90321-x. [DOI] [PubMed] [Google Scholar]
- Del Prete G, De Carli M, Almerigogna F, Daniel CK, D’Elios MM, Zancuoghi G, Vinante F, Pizzolo G, Romagnani S. Preferential expression of CD30 by human CD4+ T cells producing Th2-type cytokines. FASEB J. 1995;9:81–86. [PubMed] [Google Scholar]
- Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Ait-Yahia S, Briere F, Zlotnik A, Lebecque S, Caux C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–386. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson CO, Beagley KW, Sharmanov AT, Fujihashi K, Kiyono H, Tennyson GS, Cong Y, Black CA, Ridwan BW, McGhee JR. Hapten-induced model of murine inflammatory bowel disease: mucosa immune responses and protection by tolerance. J Immunol. 1996;157:2174–2185. [PubMed] [Google Scholar]
- Farber JM. Mig and IP-10—CXC chemokines that target lymphocytes. J Leukoc Biol. 1997;61:246–257. [PubMed] [Google Scholar]
- Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- Foti M, Granucci F, Aggujaro D, Liboi E, Luini W, Minardi S, Mantovani A, Sozzani S, Ricciardi-Castagnoli P. Upon dendritic cell (DC) activation chemokines and chemokine receptor expression are rapidly regulated for recruitment and maintenance of DC at the inflammatory site. Int Immunol. 1999;11:979–986. doi: 10.1093/intimm/11.6.979. [DOI] [PubMed] [Google Scholar]
- Hollander GA, Simpson SJ, Mizoguchi E, Nichogiannopoulou A, She J, Gutierrez-Ramos JC, Bhan AK, Burakoff SJ, Wang B, Terhorst C. Severe colitis in mice with aberrant thymic selection. Immunity. 1995;3:27–38. doi: 10.1016/1074-7613(95)90156-6. [DOI] [PubMed] [Google Scholar]
- Huang FP, Platt N, Wykes M, Major JR, Powell TJ, Jenkins CD, MacPherson GG. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J Exp Med. 2000;191:435–444. doi: 10.1084/jem.191.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun JG, Lee G, Brown JB, Grimm GR, Tang Y, Mittal N, Dirisina R, Zhang Z, Fryer JP, Weinstock JV, Luster AD, Barrett TA. Anti-interferon-inducible chemokine, CXCL10, reduces colitis by impairing T helper-1 induction and recruitment in mice. Inflamm Bowel Dis. 2005;11:799–805. doi: 10.1097/01.mib.0000178263.34099.89. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Kelsall BL. Freshly isolated Peyer’s patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J Exp Med. 1999;190:229–239. doi: 10.1084/jem.190.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Lillard JW, Jr, Boyaka PN, Taub DD, McGhee JR. RANTES potentiates antigen-specific mucosal immune responses. J Immunol. 2001;166:162–169. doi: 10.4049/jimmunol.166.1.162. [DOI] [PubMed] [Google Scholar]
- Lillard JW, Jr, Singh UP, Boyaka PN, Singh S, Taub DD, McGhee JR. MIP-1alpha and MIP-1beta differentially mediate mucosal and systemic adaptive immunity. Blood. 2003;101:807–814. doi: 10.1182/blood-2002-07-2305. [DOI] [PubMed] [Google Scholar]
- Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10) J Exp Med. 1987;166:1084–1097. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald TT, Pettersson S. Bacterial regulation of intestinal immune responses. Inflamm Bowel Dis. 2000;6:116–122. doi: 10.1097/00054725-200005000-00008. [DOI] [PubMed] [Google Scholar]
- Menachem YT, Kolker O, Shibolet O, Alper R, Nagler A, Ilan Y. Adoptive transfer of NK 1.1+ lymphocytes in immune-mediated colitis: a pro-inflammatory or a tolerizing subgroup of cells? Microb Infect. 2005;7:825–835. doi: 10.1016/j.micinf.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Mizoguchi E, Grusby MJ, Glimcher LH, Bhan AK, Tonegawa S. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75:274–282. doi: 10.1016/0092-8674(93)80069-q. [DOI] [PubMed] [Google Scholar]
- Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, Maggi E, Pupilli C, Tonelli F, Romagnani S. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am J Pathol. 1997;150:823–832. [PMC free article] [PubMed] [Google Scholar]
- Podolsky DK. The current future understanding of inflammatory bowel disease. Best Prac Res Clin Gastroenterol. 2002;16:933–943. doi: 10.1053/bega.2002.0354. [DOI] [PubMed] [Google Scholar]
- Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnani S. Induction of Th1 and Th2 responses: a key role for the “natural” immune response? Immunol Today. 1992;13:379–381. doi: 10.1016/0167-5699(92)90083-J. [DOI] [PubMed] [Google Scholar]
- Sauty A, Dziejman M, Taha RA, Iarossi AS, Neote K, Garcia-Zepeda EA, Hamid Q, Luster AD. The T cell-specific CXC chemokines IP-10, Mig, and I-TAC are expressed by activated human bronchial epithelial cells. J Immunol. 1999;162:3549–3558. [PubMed] [Google Scholar]
- Shibolet O, Alper R, Zlotogarov L, Thalenfeld B, Engelhardt D, Rabbani E, Ilan Y. NKT and CD8 lymphocytes mediate suppression of hepatocellular carcinoma growth via tumor antigen-pulsed dendritic cells. Int J Cancer. 2003;106:236–243. doi: 10.1002/ijc.11201. [DOI] [PubMed] [Google Scholar]
- Singh UP, Singh S, Singh R, Karls RK, Quinn RD, Potter ME, Lillard JW., Jr Influence of Mycobacterium avium subsp. paratuberculosis on colitis development and specific immune responses during diseasse. Infect Immun. 2007a;75:3722–2728. doi: 10.1128/IAI.01770-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh UP, Singh S, Taub DD, Lillard JW., Jr Inhibition of IFN-gamma-inducible protein-10 abrogates colitis in IL-10(−/−) mice. J Immunol. 2003a;171:1401–1406. doi: 10.4049/jimmunol.171.3.1401. [DOI] [PubMed] [Google Scholar]
- Singh UP, Singh S, Weaver CT, Iqbal N, McGhee JR, Lillard JW., Jr IFN-α-inducible chemokines enhance adaptive immunity and acolitis. J Interferon Cytokine Res. 2003b;23:591–600. doi: 10.1089/107999003322485099. [DOI] [PubMed] [Google Scholar]
- Singh UP, Venkataraman C, Singh R, Lillard JW., Jr CXCR3 axis: role in inflammatory bowel disease and its therapeutic implication. Endocr Metab Immune Disord Drug Targets. 2007b;7:111–123. doi: 10.2174/187153007780832109. [DOI] [PubMed] [Google Scholar]
- Sozzani S, Allavena P, D’Amico G, Luini W, Bianchi G, Kataura M, Imai T, Yoshie O, Bonecchi R, Mantovani A. Differential regulation of chemokine receptors during dendritic cell maturation: a model for their trafficking properties. J Immunol. 1998;161:1083–1086. [PubMed] [Google Scholar]
- Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2005;289:L85–95. doi: 10.1152/ajplung.00397.2004. [DOI] [PubMed] [Google Scholar]
- Wei B, Velazquez P, Turovskaya O, Spricher K, Aranda R, Kronenberg M, Birnbaumer L, Braun J. Mesenteric B cells centrally inhibit CD4+ T cell colitis through interaction with regulatory T cell subsets. Proc Natl Acad Sci USA. 2005;102:2010–2015. doi: 10.1073/pnas.0409449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan YH, ten Hove T, The FO, Slors JF, van Deventer SJ, te Velde AA. Chemokine receptor CXCR3 expression in inflammatory bowel disease. Inflamm Bowel Dis. 2001;7:281–286. doi: 10.1097/00054725-200111000-00001. [DOI] [PubMed] [Google Scholar]