

Abstract
Poly(ortho ester) (POE) microspheres have been previously shown to possess certain advantages for the in vivo delivery of DNA vaccines. In particular, timing of DNA release from POE microspheres in response to acidic phagosomal pH was shown to be an important factor in determining immunogenicity, which was hypothesized to be linked to the natural progression of antigen presenting cell uptake, transfection, maturation, and antigen presentation. Here we report in vitro characterization of the enhanced the efficacy of POE microspheres by blending poly(ethylenimine) (PEI), a well-characterized cationic transfection agent, into the POE matrix. Blending of a tiny amount of PEI (approximately 0.04 wt%) with POE caused large alterations in POE microsphere properties. PEI provided greater control over the rate of pH-triggered DNA release by doubling the total release time of plasmid DNA and enhanced gene transfection efficiency of the microspheres up to 50-fold without any significant cytotoxicity. Confocal microscopy with labeled PEI and DNA plasmids revealed that PEI caused a surface-localizing distribution of DNA and PEI within the POE microsphere as well as focal co-localization of PEI with DNA. We provide evidence that upon degradation, the microspheres of POE-PEI blends released electrostatic complexes of DNA and PEI, which are responsible for the enhanced gene transfection. Furthermore, blending PEI into the POE microsphere induced 50% to 60% greater phenotypic maturation and activation of bone marrow-derived dendritic cells in vitro, judged by up-regulation of co-stimulatory markers on the cell surface. Physically blending PEI with POE is a simple approach for modulating the properties of biodegradable microspheres in terms of gene transfection efficiency and DNA release kinetics. Combined with the ability to induce maturation of antigen-presenting cells, POE-PEI blended microspheres may be excellent carriers for DNA vaccines.
Keywords: Polyorthoester, Microsphere, Immune response, Controlled drug release, Gene therapy
1. Introduction
Whereas traditional vaccination strategies entail direct administration of an antigen, DNA vaccines are composed of a plasmid carrying a gene for the desired antigen. Plasmid-based vaccination can safely activate both the humoral and cell-mediated immune response pathways leading to antibody and cytotoxic T-lymphocyte (CTL) responses as well as strong memory induction [1]. Additionally, DNA vaccines allow for both prophylactic and therapeutic vaccination strategies and have been targeted against infectious agents, various cancers, and other diseases related to immune dysfunction [2]. An amazing variety of materials, physical formulations, and vector design strategies exist for the development of non-viral delivery systems for nucleic acid-based therapeutics including DNA vaccines. Non-viral strategies for plasmid transfection have included electroporation and physical methods such as ballistic “gene gun” [3], microparticles such as PLGA microspheres [4], nanoparticle formulations in liposomes [5] or polymer nanoparticles [6, 7], and recently release from polyelectrolyte multilayered materials [8, 9]. However, very few non-viral vectors have progressed to the stage of clinical trials [10]. The poor in vivo performance of non-viral delivery systems has been generally attributed to poor transfection efficiency leading to massive dosing requirements, lack of targeting, toxicity, and non-specific effects [6, 10, 11].
Polymer microspheres, mostly based on poly(lactic-co-glycolic acid) (PLGA), have been developed to protect plasmids from degradation, control DNA release, and target phagocytic antigen-presenting cells (APCs) on the basis of micrometer (1–10 μm) size [12–14]. However, these formulations often suffer from low gene transfection efficiency [4]. Efforts to increase the transfection efficiency of PLGA have mainly focused on introducing modifications with cationic surfactants such as CTAB (cetyl-trimethyl-ammonium-bromide) [15] and cationic polymers such as PEI (polyethylenimine) [16–18], and PBAE (poly(beta-aminoesters)) [19, 20]. In these reports, incorporation of the cationic molecules increased transfection efficiency and boosted immune responses. Yet the use of PLGA microspheres for gene delivery is still not optimal due to low DNA loading [12], undesirably slow release rates [21, 22], and DNA-damaging acidic degradation by-products [23, 24].
Poly(ortho ester)s (POE) are a class of pH-sensitive polymeric materials originally developed in the 1970’s to be biodegradable and non-toxic alternatives to poly(lactic acid) and poly(glycolic acid) for polymer-mediated drug delivery [25]. We previously reported the development of two 4th-generation POE polymer specifically designed for DNA delivery [26], herein referred to as P1 and P2. Both polymers have similar backbone components to achieve similar polymer physical properties (rigidity, hydrophilicity, latent acid content), but in addition, P2 incorporates tertiary amines in the backbone. These polymers functioned as effective DNA vaccine carriers in vivo. Antibodies to the model antigen β-galactosidase were induced in a gene-specific manner. Furthermore, activation of cytotoxic T lymphocytes (CTL) was gene-specific, induced memory responses, and resulted in tumor regression when mice were vaccinated against the model SIY antigen and then challenged with an SIY-bearing syngenic tumor. The potential of these POE microspheres to stimulate the immune system was indeed polymer-specific. The P1 polymer, which did not contain tertiary amine groups, did not achieve immune responses as robust as the amine-containing P2 polymer. We hypothesized that this disparate immune function was correlated to the timing of DNA release. The addition of a tertiary amine in the polymer backbone, which would be protonated and charged at the acidic pH of the late endosome, slowed the release of DNA from the degrading matrix. We proposed a model that immune responses will be enhanced if DNA release and gene expression are more closely matched to the natural progression of APC migration to the draining lymph nodes, APC maturation, and antigen presentation.
Here we hypothesized that the blending of a cationic polymer into the POE microsphere matrix could further enhance DNA vaccine delivery by POE microspheres. PEI is a well-characterized, commercially-available, cationic transfection reagent that forms stable polyplexes via electrostatic interaction with DNA and exhibits very high transfection efficiency [27, 28]. In this paper, we hypothesized that blending limited amounts of PEI into the POE matrix would increase DNA loading, increase transfection efficiency, and modulate release kinetics to be better tuned with the natural progression of immune responses. Furthermore, we investigated the ability of POE and POE-PEI blends to induce APC maturation.
2. Materials / Methods
2.1 Materials
POE was provided as a gift by AP Pharma (Redwood, CA) as previously characterized [26]. Poly(lactic-co-glycolic acid) acid (PLGA) (Resomer RG503) was from Boehringer Ingelheim Chemicals with a 50:50 ratio of lactide:glycolide and molecular weight of ~35 kD. Branched PEI 25kD was purchased from Sigma. Poly(vinyl alcohol) (PVA) ~25 kD was purchased from Polysciences, Inc. Heparin sodium was purchased from Celsus Laboratories. Endotoxin-free plasmid DNA was expanded and purified by Elim Biopharmaceuticals. Two plasmids encoding model antigens, pCMV-Luc (expressing luciferase) and pCI-Neo (expressing the SIY antigen), were used as previously reported [26].
2.2 Microsphere Preparation
A modified double-emulsion (w:o:w) method for encapsulation of DNA plasmids into a PEI/POE blend was developed to incorporate PEI into the organic phase, use 100-mg batches of polymer, and reproducibly generate 5-μm microspheres. Briefly, PEI was co-dissolved at 0.1875 mg/mL with 100 mg of POE in 2 mL of CH2Cl2. DNA plasmid was dissolved at 3 mg/mL in TE-lactose buffer (20 mM Tris, 1 mMEDTA, 300 mM lactose, pH 8.0). The aqueous phase (with DNA or TE-lactose alone for preparing blank microspheres) was added (125 uL) to 2 mL of the PEI/POE/CH2Cl2 solution. The aqueous/organic mixture was dispersed by sonication (power 3, continuous, for 12 seconds) and added to 25 mL of 5 wt% PVA in TE-lactose. The w:o:w emulsion was immediately homogenized at 4700 RPM with a 7/10” bit for 35 seconds and finally added to 50 mL of 1 wt% PVA in TE-lactose. The final mixture was stirred for 2 to 3 hours to allow solvent evaporation. Microspheres were recovered by centrifugation at 450 RCF for 10 min, washed twice in water (pH 8.0), and lyophilized. Microparticle size was determined using a Beckman Coulter Multisizer III equipped with a 30 μm pore. Zeta-potential was determined with a Brookhaven Instruments Zeta-PALS. Microspheres were analyzed for levels of endotoxin using the QCL-1000 LAL endotoxin detection kit (Lonza Walkersville).
2.3 DNA Extraction and Encapsulation Efficiency
We developed an organic to aqueous solvent extraction protocol utilizing the ability of heparin to break apart PEI-DNA complexes for DNA quantification [29]. DNA was extracted from microspheres by dissolving 5 mg of microsphere in 500 μL of a 50:48:2 mixture of phenol/chloroform/isoamyl alcohol (Invitrogen) and extracted to 300 μL TE buffer containing 12.5 mg/mL of heparin by shaking vigorously at 37°C for 1 hour, then centrifuging to separate the organic and aqueous layers at 18,000 RCF for 5 min. Supernatant samples were diluted in TE-heparin and complexed with Picogreen reagent (Invitrogen) following manufacturer’s directions for 5 minutes in a 96-well black plate; fluorescence intensity was determined at 485 nm (excitation)/535 nm (emission). Serial dilutions of DNA in TE-heparin buffer were used as standards.
2.4 Cytotoxicity and Transfection
The murine macrophage cell line P388D1 was obtained from ATCC and cultured in RPMI 1640 medium supplemented with 10% FCS, 1mM MEM sodium pyruvate, 10 mM HEPES, and 100 U/mL penicillin/streptomycin (all from Invitrogen). P388D1 cells were plated at 10,000 cells per well in clear 96-well tissue culture plates (Corning) in 200 μL of media. After incubation for 24 hours, 100 μL of culture media was removed from each well. Microspheres containing plasmid DNA, diluted in culture media, were then added to wells to achieve a final concentration of 4, 2, 1, 0.5, and 0.25 mg microsphere per well (n=5). PEI-DNA complexes (N/P 7.7:1) at 15 μg/mL PEI were also incubated with P388D1 cells for 48 hours as a control. To assess cytotoxicity, total metabolic activity was assayed after 48 hours of incubation with microspheres by MTT-formazan (ATCC) production according to the manufacturer’s protocols. Wells were not washed prior to adding MTT reagent to each well. After 4 hours of MTT incubation, cells were lysed and MTT-formazan concentration was determined by optical density at 570 nm. OD570 readings were normalized to untreated cells for comparison.
To assess transfection efficiency, P388D1 cells were plated at 10,000 cells per well in white 96-well tissue culture plates (Corning) in 200 μL of media. Microspheres containing pCMV-Luc plasmid were diluted in culture media at concentrations normalized to DNA content as determined by encapsulation efficiency to achieve the same DNA concentrations across all samples. After incubating for 24 hours, culture media in each well was replaced with media containing microspheres (n=5) to achieve a final dose of 1 μg, 0.2 μg, or 0.04 μg DNA per well in 200 μL media. Microspheres formulated without DNA (just TE-lactose) were also incubated with cells at a concentration matched per mg of microsphere to the 1 μg DNA per well dose for each polymer type. At days 3 and 6 after adding microspheres, cell culture media was completely removed and cells were lysed in the presence the Bright-Glo luciferase assay reagent for 2 minutes according to manufacturers protocols (Promega). Luminescence was assessed for a collection time of 0.4 seconds per well on a Mithras luminometer (Berthold Technologies). Background luminescence as measured with untreated cells was subtracted from all measurements. Analysis of variance (ANOVA) followed by Student’s t-test was used to analyze the differences between spheres containing PEI and spheres without PEI.
2.5 Release Kinetics
To determine timing of DNA release, 5 mg samples of microspheres (n=6) were incubated in 1.5 mL of phosphate buffered saline (PBS), pH 7.4, in a microcentrifuge tube constantly rotating at 37°C. After 24 hours half of the samples (n=3) were switched to a 25 mM sodium acetate buffer at pH 5 to simulate acidification of the phagosome. All buffers included 12.5 mg/mL heparin. Samples were taken periodically. Tubes were centrifuged at 2,000 RCF for 4 min to pellet microspheres, 1.25 mL supernatant samples were removed and replaced with fresh buffer, and microspheres were resuspended by vigorous pipetting. Sampling was repeated until microspheres had completely degraded. Samples were saved at −20° C until analysis by complexing with Picogreen. Blank (TE-lactose only encapsulated) microspheres were also analyzed. Background signal as measured from TE-releasing microspheres was subtracted and quantitative DNA release was normalized to amount of initial DNA.
2.6 Confocal Microscopy
Fluorescently labeled components were used to formulate microspheres of P2 polymer to investigate distribution of PEI and DNA within the POE microsphere matrix. Fluorescein isothiocyanate (FITC) (Sigma) was dissolved to 1 mg/mL in a pH 8 bicarbonate buffer, and PEI was dissolved at 5 mg/mL in in bicarbonate buffer. FITC was reacted with PEI at a mass ratio of 1:20 by mixing 8 mL of PEI solution with 2 mL of FITC solution for 2 hours at room temperature. The reaction mixture was incubated with 1 g of DOWEX 1-X8, 200–400 mesh, (-OH) anion exchange resin (Sigma) for 15 minutes, filtered, and dialyzed at 3500 MWCO (Pierce) against 2 L of double-distilled water to remove free FITC. PEI was lyophilized prior to formulation. Rhodamine-labeled DNA was purchased from Genlantis, lyophilized, and reconstituted in TE-lactose. Rhod-DNA was diluted 1:6 in unlabelled plasmid DNA prior to encapsulation. Plasmid DNA was also labeled with a Cy5 fluorophore using the Mirus Label-IT Cy5 Labeling Kit per manufacturer’s protocols (Mirus Bio). Microspheres were formulated with FITC-PEI and Rhod-DNA as described above, resuspended in PBS, and immediately dried onto glass slides. Prolong Gold anti-fade reagent (Invitrogen) was added onto dried microspheres prior to sealing with a glass cover slip. Images were taken at 0.2 to 0.3 um slices and vertical stacks projected into single flattened images for print.
2.7 BMDC Isolation, Maturation, and FACS Analysis
All animals were cared for in compliance with the guidelines of the Massachusetts Institute of Technology. Bone marrow was extracted from healthy C57BL/6 mice (Taconic Farms) and cultured in the presence of GM-CSF and IL-4 in RPMI-1640 supplemented with 10% FCS, 50 uM 2-ME, 2mM glutamine, 0.1 mM non-essential amino acids, 1 mM MEM sodium pyruvate, 10 mM HEPES, and 100 U/mL penicillin/streptomycin. Half of the media was replaced once every 2 days. Primary bone marrow-derived dendritic cells (BMDCs) were purified from culture by magnetic-assisted cell sorting for CD11c+ cells per manufacturer’s protocol (Miltenyi Biotech).
Sorted cells were seeded at 10,000 cells per well in 96-well plates and incubated with increasing amounts of microspheres at 0.01, 0.1, and 1 mg/mL. As controls LPS was added at 10 ug/mL (~50,000 EU/mL), naked DNA was added at 10 ug/mL, and some cells were left untreated. After 24 hours of incubation, cells were incubated with purified anti-mCD16/32 (BioLegend) to block non-specific binding of FcGamma receptors, then stained with propidium iodide (Invitrogen) and fluorescently-tagged antibodies: mCD11c-APC (BioLegend), mMHCII-Iab-PE (BD-Biosciences), and one of three FITC-conjugated antibodies (BD-Biosciences) to mCD40, mCD86, or mF4/80. Cells were analyzed for fluorescent staining on a Becton Dickinson LSR II FACS machine with a standard laser set. Cells were gated for forward scatter, side scatter, live-dead, and fluorescent levels using FlowJo analysis software (Treestar).
3. Results
3.1 Microsphere formulation and physical properties
Physical properties of representative batches of microspheres are summarized in Table 1. PLGA microspheres were formulated for comparison to POE microspheres. Blending PEI into the POE or PLGA matrix achieved higher (more positive) zeta-potentials indicating the presence of PEI at the surface of the microsphere. The surface charge was further increased by the inclusion of DNA in the microsphere. Total yields of DNA were typically between 60% to 70%. All POE microspheres exhibited similar surface features on SEM imaging (Fig. 1). There was little noticeable change in POE-PEI microsphere morphology suggesting that PEI blending does not change the structural properties of microspheres. PEI/DNA clusters were not visible on the microsphere surface. All microspheres contained less than 10 EU of endotoxin per mg of sphere.
Table 1.
Physical Properties of Representative Batches of Poly(ortho ester) Microspheres
| Sphere | Size
(μm) |
Zeta Potential
(mV) |
DNA Loading
(ng DNA/mg sphere) |
|---|---|---|---|
| P1-DNA | 5.08 | −22.66 | 2780 |
| P1-TE | 4.83 | −5.75 | |
| P1-PEI-DNA | 6.54 | 17.83 | 730 |
| P1-PEI-TE | 4.96 | 13.72 | |
| P2-DNA | 4.94 | 34.14 | 3510 |
| P2-TE | 5.33 | 45.53 | |
| P2-PEI-DNA | 5.70 | 65.97 | 4040 |
| P2-PEI-TE | 4.96 | 54.81 | |
| PLGA-DNA | 4.63 | −38.42 | 2610 |
| PLGA-TE | 5.22 | −63.60 | |
| PLGA-PEI-DNA | 5.34 | −12.50 | 702 |
| PLGA-PEI-TE | 4.77 | 15.72 |
Figure 1.

Electron micrographs at 5,000X of (a) P1-PEI (b) P2-PEI (c) P1 (d) P2 microspheres
Measuring the encapsulation of DNA in PEI-containing microspheres was challenging due to binding of PEI to DNA. UV absorption assays following base-digestion of microspheres was unreliable (data not shown), likely due to PEI-DNA complexation and interference from POE degradation products. A modified extraction and quantification method was developed to be compatible with the presence of PEI. Because PEI extracts to the aqueous phase and strongly binds to DNA (preventing Picogreen binding), heparin sodium was added to break PEI-DNA complexes [29]. PEI-DNA binding in the presence of various concentrations of heparin was assayed as DNA retardation on gel electrophoresis. An appropriate heparin concentration for reproducibly disrupting PEI-DNA complexes was determined to be 12.5 mg/mL. Heparin does not prevent DNA binding to the Picogreen reagent ([29] and data not shown).
Without PEI, DNA encapsulation efficiency into P2, which contains tertiary amines, is greater than DNA encapsulation in P1 (Table 1). This difference was further amplified by PEI blending, which increased DNA loading in the P2 polymer but decreased loading in the P1 polymer. A similar decrease in loading was observed with blending PEI into PLGA.
3.2 Cytotoxicty
We used an MTT-based assay to measure cellular activity following incubation of the phagocytosing P388D1 murine macrophages with PEI-containing POE microspheres. After 48 hours of incubation, POE-PEI microspheres showed low levels of toxicity across a large range of microsphere concentrations from 0.25 mg/mL to 2 mg/mL (Fig. 2). At 4 mg/mL, cells incubated with POE-PEI microspheres exhibited decreased viability particularly for the P1 polymer. At this concentration POE-PEI microspheres contain approximately 15 μg/mL PEI. For comparison, PEI-DNA complexes (N/P 7.7:1) at 15 μg/mL of PEI were also incubated with P388D1 cells for 48 hours and showed marked cytotoxicity at this high concentration.
Figure 2.

Cytotoxicity of increasing concentrations of POE-PEI blended microspheres in the P388D1 murine macrophage cell line; PEI nanoparticles at N/P 7.7. Viability expressed relative to untreated control cells, n=5.
3.3 Transfection Efficiency
The phagocytic P388D1 murine macrophage cell line was used to investigate gene expression following incubation with POE-containing microspheres. Transfection with and without blended PEI was compared between the two POE polymer types, P1 and the amine-containing P2, as well as PLGA. Microsphere concentration was normalized to DNA content, and P388D1 cells were incubated with 1 μg DNA or 0.2 μg DNA per well in 96-well plates. Luciferase activity was measured 3 days (Fig. 3a,3c,3e) and 6 days (Fig. 3b,3d,3f) later. Transfection with PEI-DNA nanoparticles was orders of magnitude higher than microsphere-based transfection (103 to >104 RLU, data not shown) and was omitted for scale. Background luminescence as controlled for by blank TE-containing microspheres (no DNA) was low to undetectable for all polymer types; naked plasmid DNA was also at background levels (data not shown).
Figure 3.

Luciferase activity expressed as relative light units following transfection of the P388D1 murine macrophage cell line shown at day 3 (a,c,e) and day 6 (b,d,f) for P1 polymer (a,b), P2 polymer (c,d), and PLGA (e,f) microspheres in red, or PEI-blended microspheres in blue (n=5). Blank spheres (TE-loaded) shown for comparison. Activity expressed as light units relative to untreated cells. Comparison between PEI-containing and non-PEI microspheres by ANOVA and student t-test: * indicates p < 0.05; † indicates p < 0.07. Note y-axis scale change for P1 polymer (a,b).
The blending of PEI into the microsphere matrix resulted in increased transfection for both types of POE microspheres (P1 and P2) at 3 and 6 days (Fig. 3a–d). Transfection levels of P1 and P1-PEI microspheres were greater than P2 and P2-PEI microspheres (note scale change between Fig. 3a–b and Fig. 3c–f). Highest transfection occurred early for P1-PEI microspheres at 3 days compared to P2-PEI microspheres at 6 days. PEI increased PLGA transfection efficiency to above background levels only at the highest dosage of DNA at the longer incubation time, day 6. Transfection levels for P1-PEI microspheres were approximately 80-fold greater on average than PEI-PLGA microspheres; P2-PEI microspheres had transfection levels approximately 7.4-fold greater than PLGA-PEI microspheres.
3.4 Release Kinetics
The timing of microsphere degradation and DNA release appears to have a significant modulating effect on the natural progression of antigen presenting cell (APC) activation, migration to lymph nodes, gene expression, and antigen presentation to stimulate immune responses [26]. We hypothesized that addition of PEI to the polymer matrix would retard DNA release. We performed initial trials to estimate DNA release rates using Picogreen to quantify free DNA in solution from PEI-containing POE microspheres. Picogreen binds to double-stranded DNA and then fluoresces similarly to FITC. We observed that this binding interaction can be prevented by complexing agents such as PEI or detergents such as sodium dodecyl sulfate (SDS). The electrostatic interaction of PEI with DNA, which prevents Picogreen binding, can be disrupted using poly-anionic materials such as heparin to compete DNA off of PEI [29]. Picogreen binding was not inhibited by the presence of heparin at working concentration. Interestingly, we were unable to quantify DNA release from microspheres containing PEI without adding heparin to the release media indicating the presence of PEI-DNA complexes in the release media.
We compared the kinetics of pH-triggered release of DNA from POE microspheres containing PEI to those without PEI in the presence of 12.5 mg/mL heparin sodium (fig 4a–b). To simulate acidification in the phagosome of an APC following phagocytosis, microspheres were incubated in a pH 7.4 solution for 24 hours and then switched to a pH 5.0 solution; a second batch of microspheres was maintained at pH 7.4 continuously. While PEI has high capacity to buffer an acidic solution, a property crucial for its gene delivery function [30], pH sensitive release was preserved because the amount of PEI present was too low to have a significant buffering effect.
Figure 4.

Cumulative release of DNA from P1 (a) and P2 (b) microspheres into heparin-containing media at 37°C (n=3). POE microspheres shown in red, and POE-PEI blended microspheres shown in blue, were incubated in a PBS-buffered solution at pH 7.4 for 24 hours. To visualize pH-dependence of DNA release, microsphere were either kept in PBS (dashed lines) or switched to a sodium acetate-buffered solution at pH 5 (solid lines). DNA release was quantified by Picogreen assay, and DNA release relative to total DNA encapsulation was determined after correcting for background signal with blank (TE-loaded) microspheres.
An initial burst release of DNA was observed for all types of microspheres with relatively slow release of DNA at pH 7.4. Burst release was the greatest for P1-PEI microspheres. After changing to pH 5.0 the rate of DNA release drastically increased for all sets of spheres. However, PEI-containing microspheres lagged behind non-PEI containing microspheres. The effect of PEI on DNA release at pH 5.0 was more pronounced with the P2 polymer (Fig. 4b) than with the P1 polymer (Fig. 4a).
3.5 Confocal Microscopy
Release kinetics data and physical properties indicated a large effect of PEI on the P2 polymer microsphere. Fluorescently labeled components were used to formulate microspheres of P2 polymer to investigate distribution of PEI and DNA within the microsphere matrix. PEI can be seen distributing preferentially to the surface of the microspheres (Fig. 5a) and in smaller focal clusters distributed throughout the polymer matrix. The plasmid DNA also seems to follow a similar distribution preferentially in the periphery and in small focal clusters (Fig. 5b). Encapsulation of FITC-PEI and Rhod-DNA together allowed for simultaneous visualization of DNA and PEI (Fig. 5c). We observed evidence of co-localization of PEI and DNA as seen as yellow signal as well as a broader distribution of free PEI, seen as green signal, at the surface. To visualize plasmid DNA distribution without PEI in the POE microsphere, plasmid DNA was labeled with a Cy5 dye and incorporated into P2 microspheres (Fig. 5d). DNA was observed in focal clusters preferentially in the center of the microsphere with little clustering at the periphery.
Figure 5.

Confocal micrographs of P2 microspheres at 100x optical magnification with (a) FITC-labeled PEI (b) Rhodamine-labeled plasmid DNA and unlabelled PEI (c) Both FITC-PEI and Rhodamine-DNA (d) Cy5-labelled DNA (without PEI). Scale bar indicates 5 um.
3.6 BMDC Maturation
Dendritic cells are a very important type of APC for generating immune responses [31]. Upon activation, these cells undergo phenotypic changes into a mature state by expressing activation markers on the cell surface. These activation markers include receptors and ligands necessary for inducing strong immune responses such as CD40, CD80 (B7.1), and CD86 (B7.2). Dendritic cells can be activated by a variety of immunologic ligands such as lipopolysaccharide (LPS), a gram negative bacterial cell wall component, through specific receptors such as Toll-like receptor 4 (TLR4). Our microspheres contained very low but detectable levels of endotoxin (< 10 EU/mg). Other groups have reported similar levels in PLGA microspheres [21]. To control for endotoxin effects on DC maturation, we compared microsphere-mediated activation to activation induced by large quantities of LPS.
To investigate the potential of POE microspheres to activate APCs, primary bone marrow-derived dendritic cells (BMDCs) were generated from healthy C57BL/6 mice. These cells were purified by magnetic separation and incubated with various types of microspheres. FACS analysis was used to quantify expression of co-stimulatory markers. The surface antigens MHCII, CD40, CD80, and CD86 are all critical components in the process of antigen presentation for immune stimulation. These antigens are upregulated upon APC maturation. The antigen F4/80 is a macrophage lineage marker and its surface expression is downregulated in dendritic cells upon maturation. The change in expression of maturation markers was expressed as a ratio to compare activation of BMDCs incubated with PEI containing microspheres to those incubated with regular POE microspheres (n=3). MHCII, which is generally highly expressed on APCs such as BMDCs, is upregulated as dendritic cells become activated to multiple stimuli including phagocytosis. CD40 and CD86 levels are highly expressed only upon BMDC maturation. Since F4/80 is downregulated upon activation in dendritic cells, it serves as a control for non-specific binding of antibodies to cells or microspheres themselves.
All types of POE microspheres, with or without PEI, exhibited dose-dependent activation of dendritic cells as exhibited by increased MHCII, CD80, and CD86 expression (data not shown). For the P1microspheres, the addition of PEI caused a 66% increase in CD40, 36% increase in CD86, and a 52% decrease in F4/80 on average. For the P2 microspheres, the addition of PEI caused a 55% increase in CD40, 23% increase in CD86, and a 43% decrease in F4/80 on average. It should be noted that while PEI caused an increase in MHCII expression, non-PEI containing microspheres had high levels MHCII expression.
At a dose of 0.1 mg/mL of microspheres, BMDCs showed an activated phenotype with increased expression of both CD40 and CD86 and MHCII (Fig. 6d–g) compared to untreated controls (Fig. 6b) and LPS (Fig. 6c). The addition of PEI also enhanced the activated phenotype (Fig. 6e, 6g). Naked plasmid DNA did not cause activation. Microspheres contained less than 10 EU of endotoxin per mg of sphere. At 0.1 and 1 mg/mL microsphere doses (<1 EU/ml and <10 EU/mL), activation of dendritic cells greatly exceeded levels achieved with LPS at 10 μg/mL (approximately 50,000 EU/mL).
Figure 6.

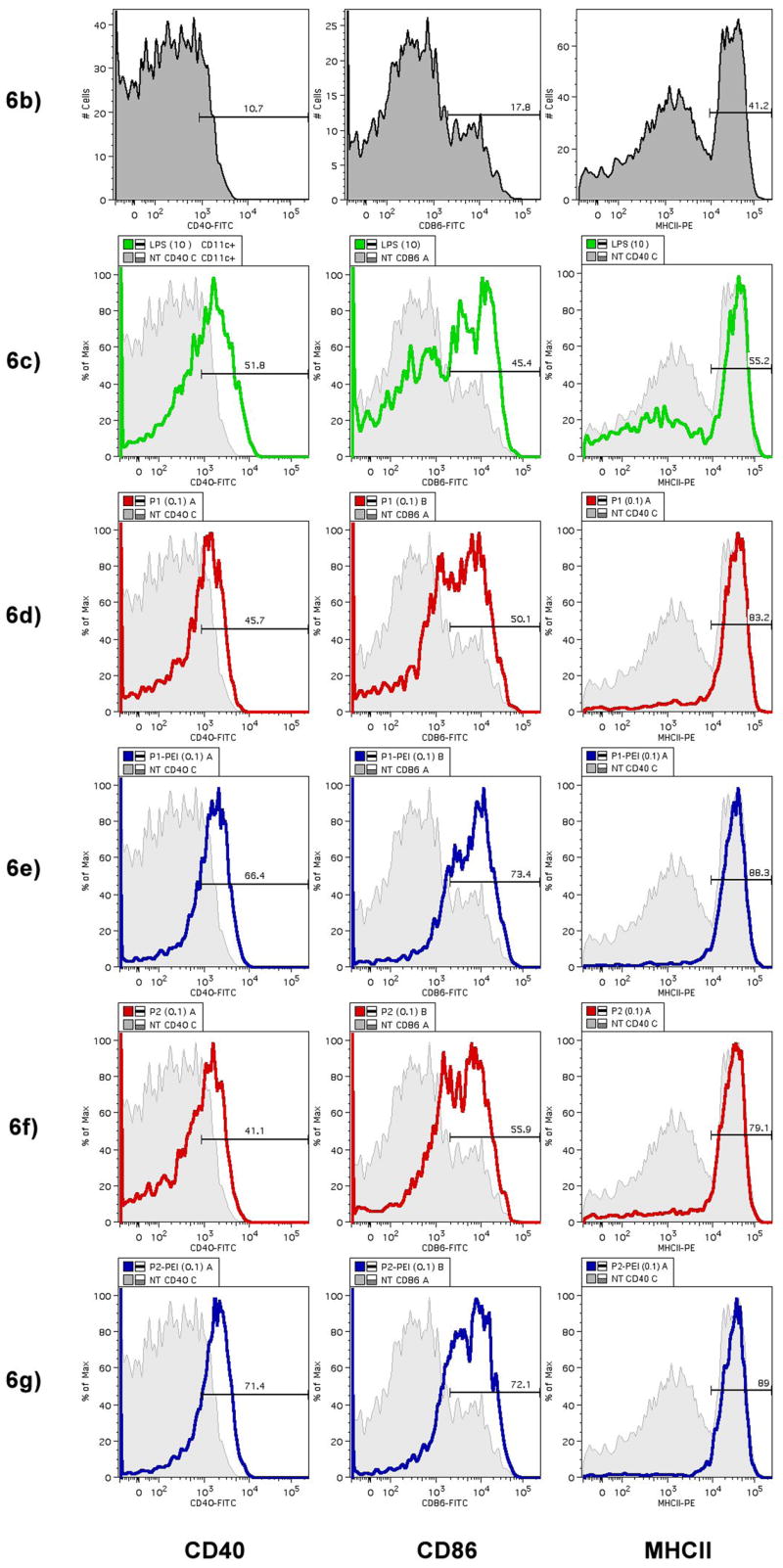
POE microspheres containing plasmid DNA were incubated with primary CD11c+ B6 mouse bone marrow-derived dendritic cells (BMDCs). After 24 hours, BMDCs were stained for expression of activation markers CD40, CD86, and F4/80, which is down-regulated upon activation, and MHCII. Activation marker expression was analyzed by FACS. (a) Relative levels of expression of maturation and co-stimulatory markers after incubation with PEI-containing microspheres compared to incubation with non-PEI microspheres (n=3) expressed as a ratio of POE-PEI blended activation to unblended POE control activation. Values greater than 1 indicate increased expression of the surface marker with PEI blendding; less than 1 indicates decreased expression levels. (b–g) Histogram plots of expression of CD40, CD86, and MHCII expression after incubation with 0.1 mg/mL of POE microsphere. Untreated controls are shown in grey (b) and as background for comparison to incubation with LPS at 10 μg/mL (c), P1 (d), P1-PEI (e), P2 (f), and P2-PEI (g).
4. Discussion
The POE polymer is acid-labile at the orthoester linkage, and the inclusion of latent acid monomers in the POE chain provides a self-catalyzing degradation meachanism [25]. This pH-sensitive character of POEs is advantageous for gene delivery to phagocytic APCs such as macrophages and dendritic cells, where upon acidification in the late phagosome POE particles should rapidly degrade and release their cargo. Our previous work indicated a role for timing of DNA release in the overall immunogenicity of POE microspheres for DNA vaccine delivery. The slower-releasing P2 polymer had increased immune responses compared to the rapidly releasing P1 polymer. However, we find in this study that the transfection efficiency of P1 is greater than that of P2, further emphasizing the important role of timed DNA release in the generation of effective immune responses. While P1 exhibited higher transfection efficiency than P2 (Fig. 3), the timing of DNA release from P2 microspheres might be better tuned to immune responses and therefore could account for the higher immunogenicity demonstrated previously [26].
It has been reported that encapsulating pre-formed DNA-PEI nanoparticles into PLGA microspheres can increase transfection efficiency of PLGA microspheres [18]. Other groups have shown increased transfection rates with PLGA microspheres surface-modified with PEI [17]. However, these methods were all based on PLGA, a polymer which was not designed for optimal DNA encapsulation and release. Additionally, large amounts of PEI were necessary to achieve enhanced transfection efficiencies. The POE polymers used in this study were designed for gene delivery and were previously demonstrated to be superior to PLGA for microsphere-mediated DNA vaccination [26]. We hypothesized that inclusion of PEI into the POE polymer matrix would enhance function as a carrier for DNA vaccines by changing release kinetics as well as increasing transfection efficiency.
Modified POE microspheres were made by blending PEI into the POE matrix during encapsulation by co-dissolving PEI in the organic phase with POE. Conditions were optimized to produce microspheres approximately 5 μm in diameter. PEI was added to achieve a 1:1 w/w ratio with DNA, or approximately 7.7 N/P ratio, resulting in an overall 0.375 wt% of PEI in the microsphere formulation. Considering the relatively low amount of PEI added to the microspheres, PEI had a profound effect on both transfection efficiency and release kinetics.
The addition of PEI to the POE matrix increased transfection efficiencies of both P1 and P2 (Fig. 3). P1-PEI microspheres exhibited 10- to 50-fold more luciferase gene expression at day 3 and 2- to 7-fold more expression at day 6 (Figs. 3a,3b). The increase in luciferase activity was up to 3 orders of magnitude over baseline and was significant for all doses of DNA investigated at both 3 and 6 days. At 3 days, transfection efficiency of P2 microspheres was overall low (Fig. 3c) due to the delayed release of DNA (Fig. 4), but at day 6 P2 microsphere DNA transfection was also increased 10- to 50-fold by the incorporation of PEI (Fig. 3d). However, P2-PEI microspheres were still 10 times less efficient for transfection than P1-PEI microspheres perhaps due to more rapid quantitative release of DNA from P1 microspheres. PEI did increase PLGA microsphere transfection efficiency at the highest DNA dose but these levels were still many folds lower than levels achieved with POE microspheres (Fig. 3f). Very slow DNA release from PLGA (on the order of weeks) [18, 21] may contribute to the low transfection efficiencies observed with PLGA microspheres even after adding PEI. Additionally, acidic products of PLGA hydrolysis may bind to PEI preventing complexation with DNA or preventing PEI release.
The addition of PEI had a greater effect of slowing the release rate of DNA on P2 compared to P1, likely due to rapid and complete degradation of the P1 polymer matrix within 1 day (Fig. 4). As a hydrophilic molecule at the surface of the POE microsphere, PEI may facilitate degradation of the microsphere by increasing hydration and facilitating hydrolysis at the ortho-ester bond. However, the cationic nature of PEI may electrostatically retard DNA release. In the P2 polymer, the addition of PEI delayed pH-mediated release of DNA from P2 plasmids between 1 to 2 days. While complete release of DNA from P2 microspheres was approached approximately 2 days after switching from pH 7.4 to pH 5, PEI-blended P2 microspheres released DNA for almost 5 days (over 100% timed delay) (Fig. 4b). It is unlikely that heparin in the release media caused fundamental differences in DNA release from POE as we observed a similar characteristic release profile from non-PEI containing POE microspheres without heparin in the past [26].
During microsphere formulation, PEI was dissolved in the organic (methylene chloride) phase together with the POE polymer. Surprisingly, PEI decreased DNA loading efficiency in P1 microspheres, although this effect was more than compensated for by increased transfection efficiency. We also observed a broader distribution in particle sizes for PEI-containing P1 spheres than non-PEI containing P1 microspheres (data not shown). These observations indicate that PEI destabilizes the oil-in-water emulsion of P1 microspheres. One possible explanation for the preferential destabilization of P1 emulsions by PEI is lower co-solubility of PEI with P1 than with P2. Whereas the P1 polymer backbone is mostly hydrophobic, the P2 polymer is synthesized with 17.5 mol% of its monomers with tertiary amines that may lead to increased co-solubility with PEI due the hydrophilic nature of PEI [26]. Because PEI is hydrophilic, it likely partitions to the water/organic interface during the process of DNA encapsulation and microsphere formation. Confocal microscopy evidence (Fig. 5) coupled with increased zeta-potentials (Table 1) indicate a diffuse surface distribution of PEI within the POE microsphere matrix with little PEI in the core. Surface segregation of PEI during the DNA encapsulation process may also facilitate DNA segregation to the surface as well, which was observed in P2 microspheres with PEI (Fig. 5b) whereas microspheres without PEI exhibited DNA cluster preferentially distributed in the core (Fig. 5d). DNA brought closer to the surface by PEI would also increase burst release effects as evidenced in P1 microspheres (Fig. 4a).
Dense clusters of co-localized PEI and DNA within the microsphere matrix were also observed (Fig. 5). It is possible that these clusters are indeed preformed PEI-DNA complexes. Comparison of TE-containing and DNA-containing microspheres indicates that the encapsulation of plasmid DNA, a large poly-anion, paradoxically increases the zeta-potential of both P1-PEI and P2-PEI microspheres. The increased surface charge can only be explained by increased loading of PEI into microspheres in the presence of DNA and is further evidence of PEI-DNA interactions during microsphere formation.
Nanoparticles of PEI and DNA are known to be highly efficient transfection agents. The clinical use of PEI as free polyplexes, however, is limited due to potential toxicity when used at doses necessary for therapeutic effects [6, 7, 32]. Large amounts of surface PEI is not readily labile as PEI-containing microspheres did not exhibit the highly cytotoxic profile expected of free PEI (Fig. 2) indicating a slow release of PEI at sub-toxic levels. PEI-DNA complexes have been shown to increase endosomal escape of plasmid DNA through the proton sponge mechanism [28] as well as facilitate active transport of PEI-DNA complexes towards the nuclei [33]. We cannot distinguish between these two mechanisms of action in the POE microsphere system with the data presented. Based on results with PLGA and the difference between transfection at days 3 and 6 with P2 microspheres, it appears that the increase in transfection from blended PEI is due to release of both PEI and DNA. We have evidence that free DNA is not released in large amounts from POE-PEI microspheres since quantification of DNA release over time (Fig. 4) by binding to Picogreen was only possible in the presence of heparin sodium, which prevents the PEI-DNA interaction. These results along with the significantly increased transfection efficiencies of PEI-containing particles extending over multiple days (Fig. 3) are evidence of simultaneous controlled release of PEI-DNA complexes that could either be pre-formed in the encapsulation process or formed immediately after independent release.
In addition to transfection and antigen presentation, dendritic cell activation is a key step in DNA vaccination. Microscale and nanoscale materials have been shown to directly activate dendritic cells, though this mechanism of activation is not entirely clear [34, 35]. We observed that POE-based microspheres are potent inducers of dendritic cell activation (Fig. 6) exceeding the levels of activation induced by free LPS even without PEI. This observation is consistent with our previous findings that POE microspheres adjuvant DNA vaccine responses [26]. This activation is dependent on dosage of microspheres. MHCII expression was upregulated upon phagocytosis of all microspheres, indicating that a similar percentage of BMDCs encountered or phagocytosed microparticles regardless of PEI content. However, compared to levels of MHCII+ BMDCs, a disproportionately larger number of BMDCs expressed the co-stimulatory markers CD40 and CD86 after incubation with PEI-containing microspheres. Therefore, the addition of PEI to the POE microsphere greatly increased the activating potential of POE microspheres that cannot be explained simply by increased interaction with or uptake of microparticles. Our results are consistent with a surface distribution of PEI in the POE microparticles that increases DC activation over that of POE microspheres alone. Similar results have been achieved by incorporating cationic materials into PLGA microparticles [19, 21], though the magnitude of increases were either small [21] or required up to 25 wt% cationic material [19]. In one of these studies it was determined that surface characteristics were correlated to increased adjuvant activity [21]. Since P2 particles have a positive zeta potential even without PEI, the specific maturation of BMDCs following POE-PEI microspheres is not only a result of a more positive surface. DNA at the surface of microparticles may increase BMDC activation interaction with pattern recognition receptors that recognize foreign DNA like TLR9 [36]. It has also been recently appreciated that a cytosolic intracellular receptor of DNA, TBK-1, may be critical for dendritic cell activation and DNA vaccine responses [37]. As demonstrated by increased transfection efficiencies, PEI likely increases intracellular concentrations of DNA, at least transiently, that could facilitate engagement of the TBK1 receptor.
5. Conclusion
POE microspheres have already been shown to be a promising means for the purpose of delivering DNA vaccines in a mouse model [26]. The methods described herein, for blending PEI with a POE matrix can serve multiple purposes in the goal of increasing the immunogenicity of POE-based microspheres for DNA vaccines. PEI slows the release rate of DNA, which could synchronize gene transfection with the natural progression of immune responses. PEI increases POE microsphere gene transfection efficiency up to 50-fold without sacrificing biocompatibility, and primary dendritic cells are more activated by POE-PEI microspheres. These results were achieved with very low amount of PEI (less than 0.4 wt%) indicating the robustness of this strategy for modulating the POE microsphere system. Our in vitro investigation as described indicates that tiny amounts of PEI can significantly alter POE microsphere functions at many levels. Further in vivo evaluation is underway for potential modulation of POE-mediated DNA vaccine responses. These POE microspheres that release blended PEI and DNA in a controlled fashion could provide further insight into the effects of release kinetics, transfection efficiency, and dendritic cell stimulation on immune responses in vivo.
Acknowledgments
The authors would like to acknowledge support from NIH Grant # EB000244, the NSF Graduate Research Fellowship, and the Whitaker Foundation Graduate Research Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pachuk CJ, McCallus DE, Weiner DB, Satishchandran C. DNA vaccines--challenges in delivery. Curr Opin Mol Ther. 2000;2(2):188–198. [PubMed] [Google Scholar]
- 2.Donnelly JJ, Wahren B, Liu MA. DNA vaccines: progress and challenges. J Immunol. 2005;175(2):633–639. doi: 10.4049/jimmunol.175.2.633. [DOI] [PubMed] [Google Scholar]
- 3.Wells DJ. Gene therapy progress and prospects: electroporation and other physical methods. Gene Ther. 2004;11(18):1363–1369. doi: 10.1038/sj.gt.3302337. [DOI] [PubMed] [Google Scholar]
- 4.Jilek S, Merkle HP, Walter E. DNA-loaded biodegradable microparticles as vaccine delivery systems and their interaction with dendritic cells. Adv Drug Deliv Rev. 2005;57(3):377–390. doi: 10.1016/j.addr.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release. 2006;116(2):255–264. doi: 10.1016/j.jconrel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 6.Putnam D. Polymers for gene delivery across length scales. Nat Mater. 2006;5(6):439–451. doi: 10.1038/nmat1645. [DOI] [PubMed] [Google Scholar]
- 7.Pack DW, Hoffman AS, Pun S, Stayton PS. Design and Development of Polymers for Gene Delivery. Nature Reviews Drug Discovery. 2005;4(7):581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Montanez SI, Jewell CM, Lynn DM. Multilayered Films Fabricated from Plasmid DNA and a Side-Chain Functionalized Poly(β-amino Ester): Surface-Type Erosion and Sequential Release of Multiple Plasmid Constructs from Surfaces. Langmuir. 2007;23(22):11139–11146. doi: 10.1021/la702021s. [DOI] [PubMed] [Google Scholar]
- 9.Uta Reibetanz CCETJHED. Defoliation and Plasmid Delivery with Layer-by-Layer Coated Colloids. Macromolecular Bioscience. 2006;6(2):153–160. doi: 10.1002/mabi.200500163. [DOI] [PubMed] [Google Scholar]
- 10.Liu MA, Ulmer JB, Jeffrey C. Human Clinical Trials of Plasmid DNA Vaccines. In: Hall JCDTF, Veronica van H, editors. Advances in Genetics. Academic Press; 2005. pp. 25–40. [DOI] [PubMed] [Google Scholar]
- 11.Glover DJ, Lipps HJ, Jans DA. Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet. 2005;6(4):299–310. doi: 10.1038/nrg1577. [DOI] [PubMed] [Google Scholar]
- 12.O’Hagan DT, Singh M, Ulmer JB. Microparticles for the delivery of DNA vaccines. Immunological Reviews. 2004;199(1):191–200. doi: 10.1111/j.0105-2896.2004.00153.x. [DOI] [PubMed] [Google Scholar]
- 13.Hedley ML, Curley J, Urban R. Microspheres containing plasmid-encoded antigens elicit cytotoxic T-cell responses. Nat Med. 1998;4(3):365–368. doi: 10.1038/nm0398-365. [DOI] [PubMed] [Google Scholar]
- 14.Jones DH, Corris S, McDonald S, Clegg JC, Farrar GH. Poly(DL-lactide-co-glycolide)-encapsulated plasmid DNA elicits systemic and mucosal antibody responses to encoded protein after oral administration. Vaccine. 1997;15(8):814–817. doi: 10.1016/s0264-410x(96)00266-6. [DOI] [PubMed] [Google Scholar]
- 15.Denis-Mize KS, Dupuis M, MacKichan ML, Singh M, Doe B, O’Hagan D, et al. Plasmid DNA adsorbed onto cationic microparticles mediates target gene expression and antigen presentation by dendritic cells. Gene Ther. 2000;7(24):2105–2112. doi: 10.1038/sj.gt.3301347. [DOI] [PubMed] [Google Scholar]
- 16.Oster CG, Kim N, Grode L, Barbu-Tudoran L, Schaper AK, Kaufmann SH, et al. Cationic microparticles consisting of poly(lactide-co-glycolide) and polyethylenimine as carriers systems for parental DNA vaccination. J Control Release. 2005;104(2):359–377. doi: 10.1016/j.jconrel.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Kasturi SP, Sachaphibulkij K, Roy K. Covalent conjugation of polyethyleneimine on biodegradable microparticles for delivery of plasmid DNA vaccines. Biomaterials. 2005;26(32):6375–6385. doi: 10.1016/j.biomaterials.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 18.De Rosa G, Quaglia F, Bochot A, Ungaro F, Fattal E. Long-term release and improved intracellular penetration of oligonucleotide-polyethylenimine complexes entrapped in biodegradable microspheres. Biomacromolecules. 2003;4(3):529–536. doi: 10.1021/bm025684c. [DOI] [PubMed] [Google Scholar]
- 19.Little SR, Lynn DM, Ge Q, Anderson DG, Puram SV, Chen J, et al. Poly-beta amino ester-containing microparticles enhance the activity of nonviral genetic vaccines. Proc Natl Acad Sci U S A. 2004;101(26):9534–9539. doi: 10.1073/pnas.0403549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Little SR, Lynn DM, Puram SV, Langer R. Formulation and characterization of poly (beta amino ester) microparticles for genetic vaccine delivery. J Control Release. 2005;107(3):449–462. doi: 10.1016/j.jconrel.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 21.Jilek S, Ulrich M, Merkle HP, Walter E. Composition and surface charge of DNA-loaded microparticles determine maturation and cytokine secretion in human dendritic cells. Pharm Res. 2004;21(7):1240–1247. doi: 10.1023/b:pham.0000033012.16152.5d. [DOI] [PubMed] [Google Scholar]
- 22.Pack DW. Timing is everything. Nat Mater. 2004;3(3):133–134. doi: 10.1038/nmat1084. [DOI] [PubMed] [Google Scholar]
- 23.Fu K, Pack DW, Klibanov AM, Langer R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm Res. 2000;17(1):100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 24.Walter E, Moelling K, Pavlovic J, Merkle HP. Microencapsulation of DNA using poly(DL-lactide-co-glycolide): stability issues and release characteristics. J Control Release. 1999;61(3):361–374. doi: 10.1016/s0168-3659(99)00151-0. [DOI] [PubMed] [Google Scholar]
- 25.Heller J, Barr J. Poly(ortho esters)--from concept to reality. Biomacromolecules. 2004;5(5):1625–1632. doi: 10.1021/bm040049n. [DOI] [PubMed] [Google Scholar]
- 26.Wang C, Ge Q, Ting D, Nguyen D, Shen HR, Chen J, et al. Molecularly engineered poly(ortho ester) microspheres for enhanced delivery of DNA vaccines. Nat Mater. 2004;3(3):190–196. doi: 10.1038/nmat1075. [DOI] [PubMed] [Google Scholar]
- 27.Neu M, Fischer D, Kissel T. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J Gene Med. 2005;7(8):992–1009. doi: 10.1002/jgm.773. [DOI] [PubMed] [Google Scholar]
- 28.Akinc A, Thomas M, Klibanov AM, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J Gene Med. 2005;7(5):657–663. doi: 10.1002/jgm.696. [DOI] [PubMed] [Google Scholar]
- 29.Moret I, Esteban Peris J, Guillem VM, Benet M, Revert F, Dasi F, et al. Stability of PEI-DNA and DOTAP-DNA complexes: effect of alkaline pH, heparin and serum. J Control Release. 2001;76(1–2):169–181. doi: 10.1016/s0168-3659(01)00415-1. [DOI] [PubMed] [Google Scholar]
- 30.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A. 1995;92(16):7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiele L, Merkle HP, Walter E. Phagocytosis of synthetic particulate vaccine delivery systems to program dendritic cells. Expert Rev Vaccines. 2002;1(2):215–226. doi: 10.1586/14760584.1.2.215. [DOI] [PubMed] [Google Scholar]
- 32.Park TG, Jeong JH, Kim SW. Current status of polymeric gene delivery systems. Advanced Drug Delivery Reviews. 2006;58(4):467–486. doi: 10.1016/j.addr.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 33.David M, Drake DWP. Biochemical investigation of active intracellular transport of polymeric gene-delivery vectors. Journal of Pharmaceutical Sciences. 2008;97(4):1399–1413. doi: 10.1002/jps.21106. [DOI] [PubMed] [Google Scholar]
- 34.Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat Nano. 2007;2(8):469–478. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 35.Reddy ST, Swartz MA, Hubbell JA. Targeting dendritic cells with biomaterials: developing the next generation of vaccines. Trends Immunol. 2006;27(12):573–579. doi: 10.1016/j.it.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451(7179):725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]