

Abstract
Background
Although humans require dietary choline for methyl donation, membrane function, and neurotransmission, choline can also be derived from the de novo synthesis of phosphatidylcholine, which is up-regulated by estrogen. A recommended Adequate Intake (AI) exists for choline; however, an Estimated Average Requirement has not been set because of a lack of sufficient human data.
Objective
The objective of the study was to evaluate the dietary requirements for choline in healthy men and women and to investigate the clinical sequelae of choline deficiency.
Design
Fifty-seven adult subjects (26 men, 16 premenopausal women, 15 postmenopausal women) were fed a diet containing 550 mg choline · 70 kg−1 · d−1 for 10 d followed by <50 mg choline · 70 kg−1 · d−1 with or without a folic acid supplement (400 μg/d per randomization) for up to 42 d. Subjects who developed organ dysfunction during this diet had normal organ function restored after incremental amounts of choline were added back to the diet. Blood and urine were monitored for signs of toxicity and metabolite concentrations, and liver fat was assessed by using magnetic resonance imaging.
Results
When deprived of dietary choline, 77% of men and 80% of postmenopausal women developed fatty liver or muscle damage, whereas only 44% of premenopausal women developed such signs of organ dysfunction. Moreover, 6 men developed these signs while consuming 550 mg choline · 70 kg−1 · d−1, the AI for choline. Folic acid supplementation did not alter the subjects’ response.
Conclusion
Subject characteristics (eg, menopausal status) modulated the dietary requirement for choline, and a daily intake at the current AI was not sufficient to prevent organ dysfunction in 19 of the subjects.
Keywords: Choline deficiency, phosphatidylcholine, fatty liver, creatine phosphokinase, muscle damage
INTRODUCTION
Choline, or its metabolites, is used to form cell membranes; it is the major source of methyl groups in the diet, and it is a precursor for the neurotransmitter acetylcholine (1). We previously showed that young men had a dietary requirement for choline and that they developed fatty liver and hepatic damage when deprived of choline (2). This knowledge contributed to the recognition of choline as an essential nutrient for humans and to the establishment of a recommended Adequate Intake (AI) of 550 mg choline/d for men and of 425 mg choline/d for women; these recommendations were based on the intakes needed to prevent liver dysfunction (3). The AI is estimated to be sufficient to prevent deficiency, but adequate human data are not available to calculate the actual Estimated Average Requirement (EAR; ie, the intake that meets the dietary requirement in 50% of the population) for choline on which a Dietary Reference Intake (DRI) would be based (3). Thus, the aim of this study was to investigate the dietary choline requirement of men and women (pre- and postmenopausal) to refine these recommendations.
Choline is found in a wide variety of foods (4, 5), and the only source of choline other than diet is from the de novo biosynthesis of phosphatidylcholine catalyzed by phosphatidylethanolamine-N-methyltransferase (PEMT; EC 2.1.1.17). This enzyme methylates phosphatidylethanolamine by using S-adenosylmethionine (SAM) as a methyl donor and forms a new choline moiety (6). Studies in animal models suggest that females may have more PEMT activity than do males. Female rats are less sensitive to choline deficiency than are male rats (7), and female mice produce more phosphatidylcholine via the PEMT pathway than do male mice (8). Estrogen status is important for this increased PEMT activity (9–11). Thus, estrogen could be the mediator of increased PEMT activity in humans. If so, we would expect premenopausal women to have lower dietary choline requirements than men and postmenopausal women.
Choline, methionine, and folate metabolism are interrelated and interact at the point where homocysteine is converted to methionine (12–15). Thus, any requirement for dietary choline must be considered in relation to these other nutrients. Supplemental oral betaine can lower plasma homocysteine concentrations (16, 17), and dietary choline intake is important for lowering plasma homocysteine concentrations even when there is adequate dietary consumption of folate and other B vitamins (18, 19). Humans ingesting diets containing 100−300 mg choline/d had decreased plasma choline concentrations, and this was prevented if they also received a folic acid supplement (20, 21). This finding suggests that folic acid supplementation may be able to spare a portion of the dietary choline requirement.
We conducted a study in humans to evaluate dietary choline requirements and to identify the clinical and metabolic sequelae of choline deficiency. We found that, independent of folate status, most men and postmenopausal women developed liver or muscle dysfunction when fed a low-choline diet, whereas premenopausal women were more resistant to developing such organ dysfunction. Also, we report that the current AI for choline may not be sufficient for some men who became depleted despite this level of intake.
SUBJECTS AND METHODS
Subjects
Healthy men (n = 31) and women (n = 35; n = 20 premenopausal and n = 15 postmenopausal) were recruited for a protocol approved by the Institutional Review Board at the University of North Carolina (UNC) at Chapel Hill; informed consent was obtained from all participants. Of the initially recruited 66 subjects, 57 completed the study per protocol, 7 voluntarily dropped out or were discharged for noncompliance, and 2 completed the study but were later deemed to have violated protocol. Of these latter 2 subjects, 1 experienced steady weight loss that was substantial by the end of the study, and the other had urinary choline and betaine concentrations that indicated dietary noncompliance despite strict supervision in the metabolic ward. The primary statistical analyses were conducted on data from the remaining per-protocol study population of 57 subjects (26 men, 16 premenopausal women, and 15 postmenopausal women). The subjects ranged in age from 18 to 70 y and had body mass indexes (in kg/m2) between 19 and 33. The ethnic heritages of the 57 subjects included white American (65%), African American (25%), Asian American (5%), Native American (3%), and others (2%), which reflected the local population characteristics of the Raleigh-Durham-Chapel Hill area. Inclusion was contingent on a good state of health, a BMI of 18−34, and no history of hepatic, renal, or other chronic system disease as determined by physical examination and standard clinical laboratory tests. Subjects with an abnormal physical examination, chronic illness, anemia, or abnormal clinical laboratory values (including unacceptable hematopoietic, hepatic, and renal function) were excluded from the study. Moreover, individuals using drugs or medications known to alter liver metabolism or using choline-containing dietary supplements during the previous 3 mo were ineligible for participation in the study. Those subjects who were eating unusual diets that would interfere with the study were also excluded.
Study design
The schedule for longitudinal experimental manipulation of dietary choline is summarized in Figure 1. On entry to the study the participants were admitted to the General Clinical Research Center (GCRC) of the UNC at Chapel Hill, where they remained under the supervision of study staff for the entire duration of the study. The diets administered to the subjects—which were composed of 0.8 g/kg high biologic value protein (current DRI), 30% of energy from fat, and the remaining amount of energy from carbohydrate—were prepared in-house to protocol specifications and are described in detail elsewhere (22). Total food intake was adjusted to be isocaloric and to provide adequate intakes of macro- and micronutrients. All diets met or exceeded the Estimated Average Requirement for methionine plus cysteine and the DRI for vitamins B-12 and B-6. The baseline diet (550 mg choline · 70 kg body wt−1 · d−1 contained ≈400 dietary folate equivalents (DFE)/d, and the depletion and repletion diets contained ≈100 DFE/d—an amount at the lower end of that found in unfortified American diets (3). A multivitamin supplement (Centrum Multi-vitamin Multi-mineral liquid; Wyeth Consumer Healthcare, Madison, NJ) provided vitamins A, D, B-6, B-12, and C and thiamine, niacin, and riboflavin at or above the DRI. Mineral supplements included iron, zinc, selenium, calcium, magnesium, manganese, and chromium at or above the DRI [Caltrate 600 Plus (Wyeth Consumer Healthcare) and GNC Selenium 50 and GNC Ironchel 18 (General Nutrition Corporation, Pittsburgh, PA)].
FIGURE 1.
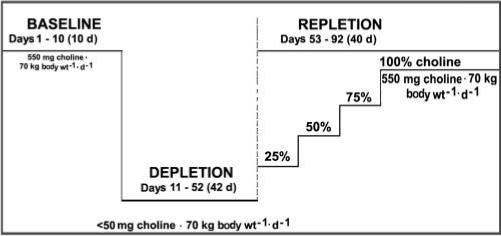
Study design. The choline content of the baseline, depletion, and repletion diets is indicated. Subjects who developed organ dysfunction when fed the low-choline depletion diet were fed graded increasing intakes of choline for 10 d each until signs of organ dysfunction resolved: 137.5 mg choline · 70 kg−1 · d−1 [25% of the Adequate Intake (AI)], 275 mg choline · 70 kg−1 · d−1 (50% of the AI), 412.5 mg choline · 70 kg−1 · d−1 (75% of the AI), and finally 550 mg choline · 70 kg−1 · d−1 (100% of the AI). The baseline diet provided 400 dietary folate equivalents (DFE)/d. The choline-depletion and -repletion diets provided 100 DFE/d. On day 11, the subjects were randomly assigned to receive a placebo or a 400-μg folic acid supplement for the remainder of the study.
Initially, all participants were fed a conventional diet of normal foods containing 550 mg choline · 70 kg body wt−1 · d−1 [the current AI (3)], 50 mg betaine · 70 kg body wt−1 · d−1, and 400 DFE/d (baseline). After 10 d of this initial diet, the subjects entered a choline-depletion phase and were assigned to 2 arms according to a randomization protocol (double-blinded) stratified by group (men, premenopausal women, and postmenopausal women—defined as women whose last spontaneous menstrual bleeding was >12 mo previously and who had a follicle-stimulating hormone concentration >30 IU/L). One arm received dietary folate only (100 DFE) for the duration of the study, whereas the other arm received a dietary supplement of 400 μg folic acid/d (total daily intake of 768 DFE) for the duration of the study. During the choline-depletion stage, both arms were fed a low-choline diet containing <50 mg choline · 70 kg body wt−1 · d−1 and 6 mg betaine · 70 kg body wt−1 · d−1, as confirmed by chemical analysis of a sample of duplicate food portions (4, 23). Periodic measurements of urinary choline and betaine concentrations (23) were made to confirm compliance with the dietary restrictions (data not shown). The subjects continued to consume the depletion diet until they developed organ dysfunction associated with choline deficiency or for 42 d if they did not. The subjects were deemed to have organ dysfunction associated with choline deficiency if they had a more than 5-fold increase in serum creatine phosphokinase (CPK) activity (24); a more than 1.5-fold increase in aspartate aminotransferase (AST), alanine aminotransferase (ALT), γ-glutamyltransferase (GGT), or lactate dehydrogenase (LD); or an increase in liver fat content of >28% during the consumption of the choline-depletion diet (see later discussion) and if these elevated measures resolved when choline was returned to the diet.
During the depletion stage, if functional markers indicated organ dysfunction associated with choline deficiency, the subjects were switched to a repletion protocol, in which choline was added back to their diet in incremental steps to a final intake of ≥550 mg choline · 70 kg−1 · d−1 for ≥3 d. The first incremental step was a diet containing 137.5 mg choline · 70 kg−1 · d−1 (25% AI; low or normal folate) for 10 d. If the functional markers indicated that deficiency status persisted, the subjects were graduated to a second step with a diet containing 275 mg choline · 70 kg−1 · d−1 (50% AI; low or normal folate) for 10 d. If markers of organ dysfunction did not indicate repletion at this time, the subjects were switched to a diet containing 412.5 mg choline · 70 kg−1 · d−1 (75% AI; low or normal folate) for 10 d. If still not replete, the subjects were fed a diet containing 550 mg choline · 70 kg−1 · d−1 (100% AI; low or normal folate) until replete. If, at any time during the depletion or repletion phases, the subjects met the stopping rules criteria (CPK ≥ 10 000 U/L, or a more than 5-fold increase above the upper limit of normal for ALT, AST, GGT, or LD), they were immediately advanced to either a 100% AI choline repletion diet or an ad libitum diet providing >100% of the AI for choline. Subjects who did not manifest signs of organ dysfunction while consuming the low-choline diet did not need to be repleted in stages and thus were only fed an ad libitum diet for 3 d before being discharged from the study.
Clinical assessment
A complete panel of laboratory tests was performed on each subject at screening, on day 1, and at the end of each dietary phase. These laboratory analyses (conducted at the Mclendon Clinical Laboratory at UNC Hospitals; Clinical Laboratory Improvement Act and College of American Pathologists accredited) included measurements of sodium, potassium, magnesium, phosphorus, chloride, fasting glucose, carbon dioxide, blood urea nitrogen (BUN), creatinine, alkaline phosphatase (AP), ALT, AST, CPK, LD, total protein, albumin, uric acid, total bilirubin, calcium, GGT, amylase, lipase, complete blood count with differential (white blood cells, red blood cells, hemoglobin, hematocrit, platelet count, mean cell volume, mean cell hemoglobin, mean cell hemoglobin adjusted for cell volume, red blood cell distribution width, neutrophils, lymphocytes, monocytes, eosinophils, and basophils), prothrombin time, partial thromboplastin time, total cholesterol, triacylglycerols, and HDL and LDL cholesterol in blood drawn by venipuncture. An abbreviated toxicity panel, which included AST, ALT, GGT, AP, LD, total bilirubin, CPK, BUN, creatinine, amylase, and uric acid, was run every 3−4 d throughout the duration of the study to monitor the depletion and repletion status of subjects. Additional blood samples were collected for the measurement of plasma choline, phosphatidylcholine, SAM, and S-adenosylhomocysteine (SAH) and serum methionine, homocysteine, methylglycine (MG), and dimethylglycine (DMG). Urinalysis was conducted at screening and at the end of each dietary phase. The urinalysis included measurements of betaine, specific gravity, pH, protein, glucose, and blood. Each subject also received a chest X-ray at screening and an electrocardiogram at screening and on the last day of the depletion and repletion diets.
Choline and methyl-group metabolites
Choline, phosphatidylcholine, and betaine were extracted from plasma by the method of Bligh and Dyer (25). Aqueous and organic compounds were separated, analyzed, and quantified directly by using liquid chromatography/electrospray ionization-isotope dilution mass spectrometry after the addition of internal standards labeled with stable isotopes to correct for recovery (23).
Methionine, homocysteine, MG, and DMG concentrations were measured in serum with the use of a capillary gas chromatography–mass spectrometry method previously described (26). Briefly, serum samples were extracted and metabolites were isolated with the use of an anion-exchange column, derivatized with N-methyl-N(tert-butyldimethylsilyl)-trifluoroacetamide, and analyzed by mass spectrometry. Deuterated standards were used to correct for recovery. SAM and SAH in plasma were measured by HPLC with fluorescence detection after conversion into their fluorescent isoindoles (27).
Fatty liver
Liver fat was measured by magnetic resonance imaging (MRI) at the beginning and end of the baseline diet (550 mg choline · 70 kg−1 · d−1), after 21 and 42 d of the low-choline diet, and at the end of each repletion period (for subjects who manifested organ dysfunction during the low-choline diet). Liver fat content was estimated by MRI with a Vision 41.5T clinical MR system (Siemens, Malvern, PA) by using a modified “In and Out of Phase” procedure (18, 28). This approach involved the use of the differences in transverse magnetization intensity after an ultrabrief time interval (FLASH; echo time = 2.2 and 4.5 ms, flip angle = 80 °, and repetition time = 140 ms). Processing of successive FLASH MRI images with software from Siemens Medical Solutions (Malvern, PA) was used to estimate fat content. Quantification of organ fat content was based on measurements across 5 images (liver slices) per subject and standardized by relating the results to the fat content of similarly measured images of spleen. (It was assumed that spleen fat remained constant during the study and could be used to normalize values over time.) In previously published reports, investigators used either a 25% or a 30% increase in liver fat as a predetermined threshold value for indicating organ dysfunction secondary to choline deficiency; for this study, we ultimately decided to use 28% as the threshold value.
Liver fat measurements were subject to being mistimed by a few days because of MRI unavailability. The evaluation at the end of baseline stage (day 10) was performed during days 8−11 for the 6 subjects who had signs of organ dysfunction while consuming the baseline diet. Of the other 51 subjects, 5 had MRI evaluations that were not normalized and hence were unusable. Of the remaining 46 subjects, the day 10 evaluation occurred during days 8−11 for 41 subjects, whereas 2 subjects were evaluated on day 13 and 3 subjects were evaluated during days 5−7. Of the 46 subjects with usable MRI data, the designated end-of-depletion stage evaluation occurred within 1 day of the last day of that stage for 17 subjects, occurred 2−3 d before the end of that stage for 28 subjects, and occurred 8 d into the repletion stage for 1 subject. These variations in timing of the MRI evaluations contributed additional variability to the liver-spleen ratio measurements but did not prevent accurate categorization of subjects as to whether they had developed fatty liver.
Statistical methods
The primary statistical analyses were performed by using the data from the 57 subjects who completed the study per protocol. In evaluating their responses to the experimental choline deprivation and repletion, the primary outcome variables were the occurrence of the choline deficiency syndrome (defined in terms of certain signs of organ dysfunction) and categorical ease of repletion (defined as level of dietary choline required to reverse those signs of organ dysfunction in subjects who responded). Plans for analyses developed before study initiation were simplified to accommodate sample size limitations and were adapted to accommodate the unexpected episodes of choline deficiency that occurred during the initial baseline stage. In addition to simple descriptive graphic and tabular statistical methods (eg, means shown with one SE unless otherwise noted), the primary inferential analyses of the incidence of choline deficiency, conditional on explanatory variables (eg, sex and age group, level of dietary choline, folic acid supplementation, changes in weight, and serum hormone concentrations), relied on likelihood-based estimation accompanied by 95% CIs (score method; 29), a Fisher's exact test procedure, logistic regression methods, and stepwise variable selection algorithms for more complete exploration of the data. Similar methods were used in the analyses of sign-specific incidence rates. The primary analyses of responses to the repletion diets focused on simple descriptive methods; the data did not support the use of time-to-event (survival) methods to the repletion data. Having established, in the primary analyses, that sex and menopausal status were associated with choline depletion and subsequent incidence rate of signs of organ dysfunction, secondary exploratory analyses of longitudinal measures of numerous biomarkers were used to generate hypotheses about underlying biological mechanisms and their differences between the 3 subgroups (men, premenopausal women, and postmenopausal women). These exploratory analyses, which were conditional on explanatory variables (eg, sex and age group, stage of study, folic acid supplementation, time in days, and changes in weight), relied on linear statistical models for longitudinal data (30). For the biomarkers known to follow distributions that are adequately approximated by log-normal distributions (eg, AST, ALT, CPK, and liver-spleen fat ratio), these analyses were performed in log10 scale. With the use of these linear models, the presented model-based analyses of changes in a given biomarker focused on the use of the last observed biomarker value in each stage of the study. P values for tests comparing mean responses at the end of the depletion stage (or end of the repletion stage) with mean responses at the end of the baseline stage were adjusted for multiple comparisons and treated in an exploratory manner for purposes of generating hypotheses about mechanisms underlying the development of organ dysfunction.
RESULTS
All diets, including the low-choline (depletion) diet, were well tolerated by the subjects. No side effects were observed, other than those associated with the removal of choline (hepatic and muscle dysfunction). The organ dysfunction associated with choline deficiency was subclinical and was not felt by the subjects; all of these signs of dysfunction resolved when choline was added back to the diet (repletion phase). At no time did the subjects feel sick or abnormal. Moreover, no study-related physical illness occurred in any subject studied.
Presentation of organ dysfunction in response to the 550-mg choline diet
Of the 57 subjects who completed the study and adhered to the protocol, 39 (68%) became choline deficient as evidenced by changes in serum CPK, AST, ALT, and LD or by hepatic steatosis detected by MRI either while consuming the baseline 550-mg choline diet (6 of 39) or while consuming the low-choline diet (33 of 39). It is reasonable to assume that the 6 subjects who developed organ dysfunction while consuming the initial 550-mg choline diet would also have become deficient had they been administered the low-choline diet first. However, the pattern of signs exhibited might have been different; therefore, we presented the results separately for these subjects who rapidly became depleted.
Of the 6 subjects (all men) who developed organ dysfunction while on the 550 mg choline baseline diet, 1 exhibited liver dysfunction alone (change in liver fat to spleen fat ratio of 60%), while an additional subject developed muscle function alone (Table 1). The remaining 4 of the 6 subjects had a combination of liver and muscle dysfunction. In this group as a whole, the mean ratio of liver fat to spleen fat did not differ between day 1 of the 550-mg choline diet (screening) and the end of the 550-mg choline diet or the end of the repletion phase (Table 2). Of these 6 subjects, increases in serum AST that met deficiency criteria were seen in 4 subjects. In the group as a whole, mean AST and ALT values at the end of the 550-mg choline diet differed from the screening values (Table 2). We also assessed other markers of liver function: albumin, bilirubin, GGT, prothrombin time, and cholesterol secreted from liver in a choline-dependent mechanism. We compared values measured at screening (or on day 1) with values measured at the end of the 550-mg choline phase and the end of the repletion phase and found that albumin concentrations were significantly decreased during the 550-mg choline diet and at repletion (Table 2). There were no substantial changes in the other measures of liver function.
TABLE 1.
Modes and frequencies of organ dysfunction associated with choline deficiency1
| Manifestation of depletion |
|||||||
|---|---|---|---|---|---|---|---|
| Depleted with 550-mg choline diet |
Depleted with low-choline diet |
||||||
| Group | Muscle dysfunction alone | Liver dysfunction alone | Liver and muscle dysfunction | Muscle dysfunction alone | Liver dysfunction alone | Liver and muscle dysfunction | Did not become depleted |
| Men (n = 26) | 1 | 1 | 4 | 1 | 8 | 5 | 6 |
| Postmenopausal women (n = 15) | 0 | 0 | 0 | 0 | 12 | 0 | 3 |
| Premenopausal women (n = 16) | 0 | 0 | 0 | 0 | 6 | 1 | 9 |
Subjects were fed a baseline diet providing 550 mg choline/70 kg body wt for 10 d as described in the text. If subjects did not manifest signs of organ dysfunction during this diet, they were advanced to a choline-depletion diet (<50 mg choline/70 kg body wt) for ≤42 d. Premenopausal women were less likely to experience manifestations of depletion (7 of 16 depleted) than were men (20 of 26 depleted; P = 0.034) and postmenopausal women (12 of 15 depleted; P = 0.046). Such a difference between men and postmenopausal women was not detected (P = 0.819).
TABLE 2.
Laboratory values in men who developed signs of organ dysfunction while consuming a diet providing 550 mg choline · 70 kg−1 · d−11
| Test and dietary phase | Value (n = 6) |
|---|---|
| Ratio of liver fat to spleen fat | |
| Screening | 1.6 ± 0.4 |
| 550 mg choline | 1.9 ± 0.7 |
| Repletion | 1.5 ± 0.2 |
| Serum AST (U/L) | |
| Screening | 26 ± 1.5 |
| 550 mg choline | 135 ± 372 |
| Repletion | 30 ± 3 |
| Serum ALT (U/L) | |
| Screening | 30 ± 4 |
| 550 mg choline | 58 ± 102 |
| Repletion | 48 ± 52 |
| Serum CPK (U/L) | |
| Screening | 115 ± 17 |
| 550 mg choline | 9081 ± 33812 |
| Repletion | 188 ± 31 |
| Serum albumin (mg/dL) | |
| Screening | 4.3 ± 0.1 |
| 550 mg choline | 4.0 ± 0.13 |
| Repletion | 4.0 ± 0.13 |
| Plasma choline (nmol/mL) | |
| Screening | 9.3 ± 0.9 |
| 550 mg choline | 9.9 ± 1.0 |
| Repletion | 10.4 ± 1.0 |
| Plasma betaine (nmol/mL) | |
| Screening | 40 ± 6 |
| 550 mg choline | 65 ± 112 |
| Repletion | 50 ± 5 |
| Plasma phosphatidylcholine (nmol/mL) | |
| Screening | 2305 ± 178 |
| 550 mg choline | 1910 ± 1743 |
| Repletion | 1949 ± 127 |
All values are x̄ ± SE. For some time points, n was smaller than indicated because of missing data (see text). Prestudy (screening) values were obtained on day 1 of the 550-mg choline diet to determine the ratio of liver fat to spleen fat; all other laboratory variables were measured during the screening visit. For all variables, measurements were also conducted at the end of the 550-mg choline diet phase and at the end of the repletion phase. AST, aspartate aminotransferase; ALT, alanine aminotransferase; CPK, creatine phosphokinase. The variables selected for inclusion in this table were identified after examination of 50 P values; ie, 42 of the 50 laboratory variables studied with the use of linear statistical models did not show clinically meaningful or significant changes at an α level of 0.05 and are not presented in the table. Statistical significance was conducted by using F tests, which were performed via a closed procedure (31) for testing multiple hypotheses, to determine differences obtained by fitting unified linear mixed-effects models to the data in appropriate scales.
2,3 Significantly different from screening:
P < 0.01
P < 0.05.
Five of these 6 early depleters exhibited muscle dysfunction as evidenced by marked elevations in serum CPK activity (Table 1). In these subjects, mean serum CPK activity increased by >7000% on day 10 of the 550-mg choline diet compared with values drawn at screening (Table 2); values in some individuals exceeded 10 000 U/L.
We also measured plasma concentrations of choline, betaine, phosphatidylcholine, homocysteine, DMG, MG, methionine, SAM, and SAH periodically throughout the study to explore the utility of these measurements as markers of choline deficiency. In subjects who developed organ dysfunction during the 550-mg choline diet, mean values of these markers at the end of baseline were compared with mean values at the end of the repletion stage and, when available, with screening values. For all of these metabolites, there were no substantial changes observed between the end of the 550-mg choline diet and the end of repletion (Table 2). However, mean plasma betaine concentrations increased and phosphatidylcholine concentration decreased when values at the end of the 550-mg diet phase were compared with screening values.
Presentation of organ dysfunction in response to the low-choline (<50 mg) diet
Liver dysfunction
For the 33 subjects who became depleted in choline while consuming the low-choline diet, 26 manifested choline deficiency with signs of liver dysfunction alone (Table 1). A combination of signs of liver dysfunction and muscle dysfunction led to detection of choline deficiency in 6 of the 33 subjects. Only 1 of these 33 subjects uniquely had signs of muscle dysfunction. Hepatic steatosis was the most common sign of choline deficiency (8 of 20 men, 12 of 15 postmenopausal women, and 6 of 16 premenopausal women).
Mean values for all liver function tests assessed are presented in Table 3 for all subjects who developed signs of organ dysfunction in response to a low-choline diet compared with those who did not. For those subjects who developed signs of organ dysfunction while consuming the low-choline diet, the ratio of liver fat to spleen fat increased from 1.9 units at baseline to 2.6 units in the depletion stage. In addition to fatty liver, some subjects, when fed the low-choline diet, developed leakage of enzymes into serum, which indicated liver cell damage. Leakage of AST into the serum occurred in a subset of the 33 subjects in whom organ dysfunction due to choline deficiency was detected. Five of the 8 subjects who exhibited AST leakage were men, 2 were premenopausal women, and 1 was a postmenopausal woman. On average, in all subjects with signs of organ dysfunction, mean serum AST, LD, and amylase activities increased at the end of the depletion phase (Table 3). Although the increase in ALT occurred less frequently in the subjects who developed organ dysfunction during the 550-mg choline or the low-choline diet (3 of 39), serum AST and ALT activity levels were correlated (r2 = 0.64). Of the other markers of liver function that were measured, mean concentrations of serum GGT, total bilirubin, and albumin and mean partial thromboplastin time exhibited negligible changes in subjects who developed organ dysfunction. Moreover, serum total cholesterol, LDL-cholesterol, and HDL-cholesterol concentrations did not change in either group of subjects in response to the low-choline diet. AP activity (a marker for liver and bone damage) increased in all subjects in response to the low-choline diet regardless of whether they manifested organ dysfunction (Table 3).
TABLE 3.
Clinical measures in humans fed diets with various choline contents1
| Test and dietary phase | Subjects with signs of organ dysfunction (n = 33) | Subjects with no signs of organ dysfunction (n = 18) |
|---|---|---|
| Ratio of liver fat to spleen fat | ||
| 550 mg choline | 1.9 ± 0.1 | 1.4 ± 0.1 |
| <50 mg choline | 2.6 ± 0.22 | 1.4 ± 0.1 |
| Repletion | 2.1 ± 0.2 | NA |
| Serum AST (U/L) | ||
| 550 mg choline | 29 ± 3 | 22 ± 2 |
| <50 mg choline | 43 ± 63 | 20 ± 1 |
| Repletion | 26 ± 1 | NA |
| Serum ALT (U/L) | ||
| 550 mg choline | 34 ± 4 | 25 ± 2 |
| <50 mg choline | 35 ± 3 | 22 ± 2 |
| Repletion | 33 ± 3 | NA |
| Serum GGT (U/L) | ||
| 550 mg choline | 23 ± 1.2 | 22 ± 1.3 |
| <50 mg choline | 24 ± 1.9 | 22 ± 1.6 |
| Repletion | 24 ± 1.7 | NA |
| Serum total bilirubin (mg/dL) | ||
| 550 mg choline | 0.5 ± 0.04 | 0.5 ± 0.07 |
| <50 mg choline | 0.5 ± 0.04 | 0.6 ± 0.07 |
| Repletion | 0.6 ± 0.05 | NA |
| Serum LD (U/L) | ||
| 550 mg choline | 444 ± 14 | 414 ± 23 |
| <50 mg choline | 485 ± 183 | 395 ± 22 |
| Repletion | 433 ± 17 | NA |
| Serum albumin (mg/dL) | ||
| 550 mg choline | 3.9 ± 0.09 | 3.9 ± 0.11 |
| <50 mg choline | 3.8 ± 0.09 | 3.9 ± 0.09 |
| Repletion | 3.8 ± 0.07 | NA |
| Serum amylase (U/L) | ||
| 550 mg choline | 56 ± 3.8 | 72 ± 6.0 |
| <50 mg choline | 51 ± 3.74 | 67 ± 5.8 |
| Repletion | 57 ± 5.4 | NA |
| Serum AP (U/L) | ||
| 550 mg choline | 76 ± 2.7 | 63 ± 2.8 |
| <50 mg choline | 81 ± 2.75 | 67 ± 3.13 |
| Repletion | 81 ± 3.1 | NA |
| PTT (s) | ||
| 550 mg choline | 28 ± 0.5 | 31 ± 0.9 |
| <50 mg choline | 28 ± 0.5 | 31 ± 0.9 |
| Repletion | 28 ± 0.6 | NA |
| Serum total cholesterol (mg/dL) | ||
| 550 mg choline | 174 ± 7 | 159 ± 10 |
| <50 mg choline | 175 ± 8 | 159 ± 10 |
| Repletion | 180 ± 9 | NA |
| Serum LDL (mg/dL) | ||
| 550 mg choline | 90 ± 5 | 94 ± 10 |
| <50 mg choline | 93 ± 6 | 91 ± 8 |
| Repletion | 101 ± 7 | NA |
| Serum HDL (mg/dL) | ||
| 550 mg choline | 52 ± 1.7 | 51 ± 2.9 |
| <50 mg choline | 52 ± 2.0 | 48 ± 2.53 |
| Repletion | 51 ± 2.0 | NA |
| Serum CPK (U/L) | ||
| 550 mg choline | 187 ± 60 | 132 ± 36 |
| <50 mg choline | 803 ± 302 | 89 ± 12 |
| Repletion | 98 ± 11 | NA |
| Serum uric acid (mg/dL) | ||
| 550 mg choline | 4.5 ± 0.15 | 4.1 ± 0.23 |
| <50 mg choline | 5.4 ± 0.202 | 5.0 ± 0.262 |
| Repletion | 5.4 ± 0.184 | NA |
| Serum blood urea nitrogen (mg/dL) | ||
| 550 mg choline | 9.7 ± 0.48 | 7.8 ± 0.49 |
| <50 mg choline | 8.9 ± 0.48 | 7.7 ± 0.35 |
| Repletion | 10 ± 0.66 | NA |
| Serum creatinine (mg/dL) | ||
| 550 mg choline | 0.9 ± 0.03 | 0.8 ± 0.03 |
| <50 mg choline | 0.8 ± 0.03 | 0.8 ± 0.04 |
| Repletion | 0.8 ± 0.03 | NA |
All values are x̄ ± SE. The subjects were fed a baseline diet providing 550 mg choline · 70 kg body wt−1 · d−1 for 10 d. If the subjects did not manifest organ dysfunction during this diet, they were advanced to a choline-depletion diet (<50 mg choline · 70 kg body wt−1 · d−1) until they developed signs of organ dysfunction or for ≤42 d. The subjects who developed organ dysfunction were fed choline-repletion diets until signs of organ dysfunction were reversed. Data from subjects (n = 51) who were offered the low-choline diet are included; data from 6 subjects who were depleted during the 550-mg choline baseline diet are shown in Table 2. Laboratory measures were performed at the end of each dietary phase. For some time points, n was smaller than indicated because of missing data (see text). Statistical significance was conducted by using F tests to determine differences obtained by fitting unified linear mixed-effects models to the data (n = 51) in appropriate scales. For all biomarkers except HDL, the interactions of diet and depletion with subgroup (men, premenopausal women, and postmenopausal women) were not statistically significant at the α = 0.05 level. For HDL, with the use of a closed testing procedure (31) for testing multiple hypotheses, one interaction was detected; we rejected the null hypothesis that pre- and postmenopausal women are not different with respect to the difference between those who experience choline deficiency—related organ dysfunction and those who do not (P ≤ 0.05). CPK, creatine phosphokinase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, γ-glutamyl transferase; AP, alkaline phosphatase; LD, lactate dehydrogenase; PTT, partial thromboplastin time; NA, not available.
2−5 Significantly different from 550 mg choline:
P < 0.0001
P < 0.05
P < 0.01
P < 0.001.
Muscle dysfunction
In the 33 subjects who developed organ dysfunction while consuming the low-choline diet, mean serum CPK activity increased from 187 U/L at baseline to 803 U/L during the low-choline diet (Table 3). However, for the subgroup of 6 men and 1 premenopausal woman who developed organ dysfunction, which included muscle dysfunction, the mean during the low-choline diet was 3993 U/L and the maximum was 6849 U/L. In all subjects who developed muscle dysfunction, CPK activities returned to screening or baseline levels after the choline-repletion diet.
We previously established (24) that the elevations in CPK activities seen in our subjects resulted because of damage to skeletal muscle rather than to cardiac muscle. The current subjects had no muscle pain or weakness, and electrodiagnostic studies in 3 of the subjects and muscle biopsy specimens from 2 of the subjects (at the time that CPK values were elevated) were normal.
Other signs of organ dysfunction associated with a low-choline diet
Renal function as assessed on the basis of BUN and serum creatinine did not change in subjects in response to a low-choline diet (Table 3). Serum uric acid concentrations (a measure of cell death) increased in all subjects fed the low-choline diet, irrespective of whether they manifested organ dysfunction. Moreover, uric acid concentrations remained significantly different from baseline concentrations at the end of the choline-repletion diet (note that repletion data were not collected for subjects who did not develop signs of organ dysfunction). The following laboratory variables were measured but are not presented because they did not exhibit clinically important changes related to the low-choline diet: total protein, hematocrit, red blood cell count, mean cell volume, mean cell hemoglobin, mean cell hemoglobin adjusted for cell volume, neutrophil count, platelet count, absolute lymphocyte count, ratio of BUN to creatinine, chloride, carbon dioxide, sodium, potassium, calcium, anion gap, glucose, lipase, magnesium, phosphorus, prothrombin time, red cell distribution width, and urine nitrogen.
Sex differences in susceptibility to developing organ dysfunction during a low-choline diet
Of all the men studied, 20 of 26 (77%) were classified as having developed organ dysfunction associated with choline deficiency (Table 1). Of the premenopausal women studied, 7 of 16 (44%) were classified as having developed organ dysfunction associated with choline deficiency (Table 1). One of these 7 women who developed organ dysfunction was subsequently determined to be perimenopausal; she had not had a menstrual cycle for >6 mo. Of the postmenopausal women, 12 of 15 (80%) were classified as choline deficient when they experienced hepatic steatosis (1 with elevations in serum AST and ALT activities; Table 1). Two of 15 postmenopausal women were receiving hormone replacement therapy; 1 developed organ dysfunction and was classified as choline deficient, whereas the other did not.
Premenopausal women were less likely to develop choline-deficiency associated organ dysfunction than were men (P = 0.034) and postmenopausal women (P = 0.046). Such a difference between men and postmenopausal women was not detected (P = 0.819). These hypothesis test results were obtained via an exact test procedure in the context of a logistic regression model for depletion conditional on demographic group (men, premenopausal women, or postmenopausal women.) The results were the same when folate treatment was taken into account. Men were more likely than premenopausal women and postmenopausal women to manifest depletion via an elevation in serum CPK activity (P = 0.001). In fact, only one woman (premenopausal) in the study manifested an elevation in serum CPK activity with choline deficiency. All postmenopausal women who manifested organ dysfunction in response to a low-choline diet developed fatty liver. Of these, one postmenopausal woman also developed elevations in serum enzymes (AST or ALT), which indicated liver damage and fatty liver.
Effects of folic acid supplementation on the likelihood of developing organ dysfunction associated with a low-choline diet
All subjects received 400 DFE during the baseline phase, including those who developed organ dysfunction while consuming this diet. Of the 51 subjects who were fed the depletion diet, half were assigned by a stratified randomization procedure to receive a dietary supplement containing 400 μg folic acid/d during the depletion and repletion phases and the other half to receive a placebo dietary supplement. The foods in the depletion and repletion diets provided only 100 DFE/d. We detected no effect of folic acid supplementation on the likelihood of manifesting organ dysfunction in response to a low-choline diet (P = 0.784; Fisher's exact test). Of the subjects who developed organ dysfunction, 16 were randomly assigned to receive the folic acid supplement, whereas 17 received a placebo. Of the 18 subjects who did not develop organ dysfunction, 10 received folic acid and 8 received placebo.
Choline and biochemically related metabolites in plasma
Of the 51 subjects fed the low-choline diet, we measured plasma concentrations of metabolites of choline and methyl metabolism (Table 4). Of note, mean plasma choline concentrations decreased by 28−33% in all subjects, independent of whether they developed organ dysfunction and irrespective of folate status. Choline concentrations at the end of the repletion stage remained statistically significantly different from baseline concentrations in subjects with signs of organ dysfunction. Similarly, mean plasma betaine concentrations decreased by ≈50% in all subjects who consumed a low-choline diet. Plasma betaine concentrations remained statistically significantly depressed at the end of the repletion phase in subjects with signs of organ dysfunction. Mean plasma phosphatidylcholine concentrations decreased only in subjects with signs of organ dysfunction during the low-choline diet; concentrations measured at the end of the repletion phase did not differ from baseline concentrations in those who developed organ dysfunction. Repletion data were not available for subjects who did not manifest signs of organ dysfunction. Mean serum concentrations of DMG and MG, both end products of choline metabolism, decreased at the end of the depletion period in all subjects, regardless of whether organ dysfunction occurred and irrespective of folate status. Concentrations of these metabolites at the end of the repletion phase in subjects who manifested organ dysfunction remained significantly different from baseline concentrations.
TABLE 4.
Plasma choline and related methyl-group metabolite concentrations1
| Metabolite and dietary phase | Subjects with signs of organ dysfunction (n = 33) | Subjects with no signs of organ dysfunction (n = 18) |
|---|---|---|
| nmol/mL | ||
| Plasma choline | ||
| 550 mg choline | 10 ± 0.3 | 9.5 ± 0.5 |
| <50 mg choline | 6.7 ± 0.22 | 6.8 ± 0.32 |
| Repletion | 9.4 ± 0.43 | NA |
| Plasma betaine | ||
| 550 mg choline | 60 ± 4 | 60 ± 4 |
| <50 mg choline | 27 ± 22 | 30 ± 22 |
| Repletion | 42 ± 32 | NA |
| Plasma phosphatidylcholine | ||
| 550 mg choline | 1925 ± 58 | 1667 ± 54 |
| <50 mg choline | 1700 ± 512 | 1604 ± 67 |
| Repletion | 1889 ± 69 | NA |
| Serum dimethylglycine | ||
| 550 mg choline | 6.0 ± 0.4 | 5.1 ± 0.4 |
| <50 mg choline | 3.1 ± 0.22 | 3.0 ± 0.32 |
| Repletion | 4.8 ± 0.44 | NA |
| Serum methylglycine | ||
| 550 mg choline | 1.9 ± 0.1 | 1.5 ± 0.09 |
| <50 mg choline | 0.9 ± 0.052 | 0.9 ± 0.062 |
| Repletion | 1.6 ± 0.13 | NA |
| Serum homocysteine | ||
| 550 mg choline | 6.9 ± 0.3 | 5.5 ± 0.3 |
| <50 mg choline | 8.0 ± 0.42 | 6.7 ± 0.42 |
| Repletion | 7.8 ± 0.45 | NA |
| Serum methionine | ||
| 550 mg choline | 27 ± 0.7 | 25 ± 1.2 |
| <50 mg choline | 24 ± 0.75 | 25 ± 1.0 |
| Repletion | 25 ± 0.93 | NA |
| Plasma SAM | ||
| 550 mg choline | 85 ± 3.6 | 75 ± 3.2 |
| <50 mg choline | 80 ± 3.1 | 76 ± 3.8 |
| Repletion | 85 ± 4.2 | NA |
| Plasma SAH | ||
| 550 mg choline | 24 ± 2.4 | 18 ± 2.6 |
| <50 mg choline | 22 ± 1.6 | 20 ± 2.7 |
| Repletion | 24 ± 2.1 | NA |
All values are x̄ ± SE. The subjects were treated as described in Table 3. Data from subjects (n = 51) who were offered the low-choline diet are included; data from 6 subjects who were depleted during the 550-mg baseline diet are shown in Table 2. Laboratory measures were performed at the end of each dietary phase. For some time points, n was smaller than indicated because of missing data (see text). Statistical significance was conducted by using F tests to determine differences obtained by fitting linear mixed-effects models to the data in appropriate scales. For all biomarkers except methionine, the interactions of diet and depletion with subgroup (men, premenopausal women, and postmenopausal women) were not statistically significant at the α = 0.05 level. For methionine, all of the tests for individual interaction terms [performed via a closed procedures for testing multiple hypotheses (53)] yielded P values > 0.05 except for methionine: P = 0.031 for the test of the null hypothesis that pre- and postmenopausal women are not different with respect to the difference between those who experience choline deficiency syndrome and those who do not and P = 0.012 for the test of the null hypothesis that pre- and postmenopausal women are not different with respect to the difference between the baseline diet and the depletion diet. SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; NA, not available.
2−5 Significantly different from 550 mg choline:
P < 0.0001
P < 0.01
P < 0.05
P < 0.001.
Mean plasma concentrations of homocysteine increased at the end of the depletion phase in all subjects, regardless of whether organ dysfunction occurred and irrespective of folate status (Table 4), and remained elevated at repletion in subjects who manifested signs of choline deficiency. As expected, at the end of the depletion period, participants who did not receive a folate supplement had higher plasma homocysteine concentrations than did those who did (P = 0.008; data not shown). Mean plasma concentrations of methionine in subjects who developed signs of organ dysfunction were reduced at the end of the depletion and repletion diets relative to the baseline diet. This difference was not seen in subjects without signs of organ dysfunction. Concentrations of SAM and SAH did not differ with diet in either group of subjects.
Variability in choline requirements
Twenty of the 26 men who completed the protocol developed organ dysfunction associated with choline deficiency. Of these 20 subjects, 6 manifested signs of choline depletion in response to the baseline diet (550 mg choline). One such subject who rapidly became depleted became replete on a diet containing 825 mg choline · 70 kg−1 · d−1. Specific repletion data are missing for the remaining 5 rapid depleters because they presented with greatly elevated laboratory values that met our stopping criteria. Thus, per protocol design, they were immediately switched to a diet providing 550 mg choline · 70 kg−1 · d−1 or an ad libitum diet, and their markers of deficiency returned to baseline values. Of the remaining 14 male subjects who developed organ dysfunction in response to a low-choline diet, 6 subjects became replete (organ function returned to normal) during the 25%-choline diet (137.5 mg · 70 kg−1 · d−1), 2 subjects became replete during the 50%-choline diet (275 mg · 70 kg−1 · d−1), and 3 subjects required 75% of the choline AI (412.5 · 70 kg−1 · d−1) to reverse signs of deficiency (Figure 2). Choline concentrations in one additional subject, who developed fatty liver when choline deficient, did not return to baseline after completion of the 550 mg choline · 70 kg−1 · d−1 choline-repletion diet. This subject then consumed an ad libitum diet for 2 wk, which was successful in fully reversing signs of deficiency. Finally, repletion data were missing for 2 subjects because they were erroneously classified as not being choline deficient. These errors were due to improper calculations during the initial weeks of the study.
FIGURE 2.
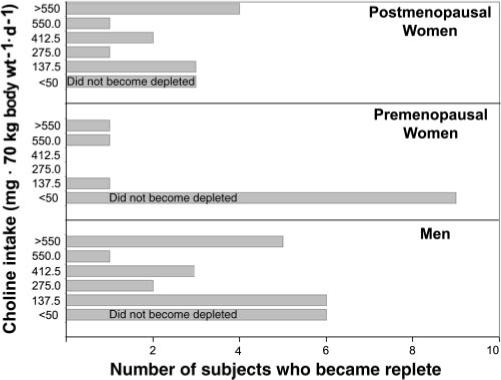
The number of subjects whose choline status returned to normal at each dietary choline intake. The subjects were treated as described in the legend for Figure 1. There was considerable variation in dietary choline requirements between the subjects. Those who manifested signs of organ dysfunction in response to the baseline or low-choline diet (depletion phase) became replete after graded increases in choline intakes were fed and signs of organ dysfunction resolved, if stopping rules did not dictate immediate advancement to a 100% or ad libitum repletion diet.
Of the 7 premenopausal women who became choline deficient, the oldest (49 y) woman did not recover fully after consuming the 550 mg choline · 70 kg−1 · d−1 diet, but did so when placed on an ad libitum diet. A second premenopausal woman became replete during the 137.5 mg choline · 70 kg−1 · d−1 diet. One woman was directly advanced to a 550 mg choline · 70 kg−1 · d−1 diet, which successfully returned markers of deficiency to baseline concentrations. Repletion data were missing for 3 subjects because initial values were not properly calculated as indicating deficiency. Repletion data are missing for one additional woman because she voluntarily left the study before completing the repletion phase.
Of the 12 postmenopausal women who manifested organ dysfunction related to choline deficiency, 3 became replete with the 137.5 mg choline · 70 kg−1 · d−1 diet, 1 became replete with the 275 mg choline · 70 kg−1 · d−1 diet, and 2 became replete with the 412.5 mg choline · 70 kg−1 · d−1 repletion diet. Liver fat values trended downward in 4 subjects after choline was reintroduced into their diets; however, they did not become fully replete with the 550 mg choline · 70 kg−1 · d−1 diet. Again, 550 mg choline · 70 kg−1 · d−1 may have been insufficient for these particular women, who eventually became replete after ≥2 wk of an ad libitum diet. Complete repletion data were missing for one woman because she elected to leave the study after completing the 50% repletion phase. Although this subject was not fully replete at this time, organ function had returned to normal when she returned after consuming an ad libitum diet for 2 wk. Repletion data were missing for one additional subject because initial values were not properly calculated as indicating deficiency. Model-based analyses of time-to-repletion (days), conditional on subgroup, did not detect an association between subgroup membership and time to repletion.
DISCUSSION
This is the first study of choline requirements that included women. We found that most men and postmenopausal women developed organ dysfunction when deprived of choline, whereas most premenopausal women did not. Susceptibility to choline deficiency was not altered by supplementation with folic acid.
We identified a group of rapid depleters who developed organ dysfunction when fed the AI of choline (550 mg choline · 70 kg−1 · d−1). We presume that these men, when free-living, consumed diets that had a higher choline content; this would explain the decreased mean plasma phosphatidylcholine concentrations that we observed after 10 d of the 550-mg choline diet. Similarly, our experimental diet was likely higher in betaine than was the subjects’ ad libitum diets, perhaps explaining the increased mean plasma betaine concentration observed. These rapid depleters developed fatty liver, muscle damage, or both that reversed when they consumed a high-choline diet (825 mg · 70 kg−1 · d−1 diet) or an ad libitum diet. Five of 6 subjects developed especially high CPK activities in plasma. These men are worthy of further study; it is possible that these men have a defect in CTL1, the mammalian homolog of the yeast choline transporter gene responsible for choline uptake into muscle (32). Because the ad libitum diet differed from the repletion diet in more nutrients than just choline, we cannot be sure that the reversal of organ dysfunction observed was solely due to choline restoration. For this reason, we segregated the analysis of data from these subjects and did not include them in the analyses of the other 51 subjects studied.
Liver and muscle dysfunction occurred in response to a low-choline diet in both men and women. Fatty liver (hepatosteatosis) is common in human and animal models of choline deficiency (2, 33) because a specific lack of phosphatidylcholine limits the export of excess triacylglycerol from the liver (34, 35). Leakage of enzymes (eg, AST, ALT, AP, LD, and CPK) from tissues of liver and muscle into blood likely occurs because choline deficiency induces apoptotic pathways in these cells (24, 36, 37). Because choline deficiency is associated with DNA damage and apoptosis (37–39), it is not surprising that choline-depleted humans have hyperuricemia because uric acid, a product of purine metabolism, is released into blood after tissue lysis (40).
The glomerulus of the kidney uses 2 metabolites of choline (betaine and glycerophosphocholine) as osmolytes (41, 42), and choline-deficient rodents have renal dysfunction (1, 43). We did not observe dietary choline-related changes in urinalysis or urine-specific gravity in our subjects (data not shown).
Choline is derived not only from the diet but from de novo synthesis of phosphatidylcholine catalyzed by PEMT. As discussed earlier, PEMT activity is increased by estrogen in animal models. We hypothesize that this is the reason why premenopausal women were more resistant to developing signs of organ dysfunction when fed a low-choline diet. We are currently examining whether the human PEMT gene has an estrogen response element in its promoter. Only 2 postmenopausal women in our study were treated with hormone replacement therapy; 1 became deplete and 1 did not during the low-choline diet. The subject who became deplete was using a transdermal patch and was receiving a dose lower than that of the other subject who took hormone replacement therapy orally. Further studies are needed before we can determine whether the choline requirement in postmenopausal women is decreased by estrogen therapy.
The requirement for choline in the diet is quite variable. A portion of the men and women we studied required more than the recommended AI for choline, whereas others required <50 mg choline · 70 kg−1 · d−1. Some subjects became deplete quickly and some took almost 7 wk to develop organ dysfunction when fed a low-choline diet. Estrogen status accounts for much of this variability. In addition, we recently reported that genetic polymorphisms may account for the rest of the variability in dietary choline requirements. We identified a single nucleotide polymorphism in the promoter region of the PEMT gene (rs12325817), for which 18 of the 23 female carriers of the variant C allele (78%) developed organ dysfunction when fed a low-choline diet (odds ratio: 25; P = 0.002); the variant C allele was relatively common: 18% of subjects were CC, 56% were GC, and 26% were GG genotype (44). In addition, premenopausal women who were carriers of the common 5,10-methylenetetrahydrofolate dehydrogenase-1958A (MTHFD1) gene allele were >15 times as likely as were noncarriers to develop signs of choline deficiency (P < 0.0001) during the low-choline diet unless they were also treated with a folic acid supplement (45).
We previously published that choline metabolism is interrelated to homocysteine metabolism (18). It is interesting that premenopausal women, whatever the response group, had a lower plasma homocysteine concentration at baseline (with signs: 5.7 ± 0.4 nmol/mL; without signs: 4.6 ± 0.2 nmol/mL) than did men (with signs: 7.4 ± 0.3 nmol/mL; without signs: 6.9 ± 0.6 nmol/mL); however, when subjects were fed the low-choline diet, homocysteine concentrations uniformly increased 20% in men (with signs: 8.8 ± 0.6 nmol/mL; without signs: 8.6 ± 0.7 nmol/mL), premenopausal women (with signs: 6.9 ± 0.6 nmol/mL; without signs: 5.8 ± 0.2 nmol/mL), and postmenopausal women (data not shown). Also, we report that plasma concentrations of methylated end products of choline and methionine metabolism changed in predicted directions (Table 4).
We observed no effect of folic acid supplementation on susceptibility or on mode of presentation. Previously published studies suggesting that folic acid supplementation might decrease requirements for choline used diets much higher in choline (150−300 mg choline/d) than ours (< 50 mg/d) (20, 21). Perhaps the effects of folate become apparent only when marginally adequate amounts of choline are supplied and not when diets are almost devoid of choline.
Plasma concentrations of choline, betaine, and phosphatidylcholine decreased when subjects were fed a low-choline diet, but the decrease was not highly correlated with susceptibility to developing organ dysfunction while this diet was being consumed. Thus, decreased plasma concentrations of choline metabolites are a necessary, but not sufficient, criterion for predicting choline deficiency–associated organ dysfunction. Plasma concentrations likely do not fully reflect intracellular concentrations of these metabolites.
This study, in combination with our previous work, establishes a panel of measurements that can be used to define individuals who are sufficiently deplete of choline to develop liver and muscle dysfunction. Factors that we identified that increase susceptibility to developing organ dysfunction in humans fed low-choline diets, such as menopausal status and genetic polymorphisms, are likely to be of clinical importance. Humans fed intravenously with solutions low in choline (total parenteral nutrition) often develop liver dysfunction that sometimes resolves when a choline source is added to their feeding solution (46); it may be that susceptible individuals can be identified based on the factors we identified and treatment appropriately modified. Poor dietary intake of choline contributes to adverse outcomes during pregnancy, a time when choline demand is high (47, 48). Deficient maternal dietary intake of choline during pregnancy in humans is associated with a 4-fold increased risk of having a baby with a neural tube defect (47); it is possible that susceptible mothers can be identified through examination of the genetic polymorphisms we describe. In rodent models, maternal dietary choline intake influences brain development. Rodents fed choline-deficient diets during late pregnancy have offspring with diminished progenitor cell proliferation and increased apoptosis in fetal hippocampus (49, 50), insensitivity to long-term potentiation when they were adult animals (51), and decremented visuospatial and auditory memory (52). For these reasons, understanding sources of variability in human choline requirements is important.
Of the 57 subjects fed the low-choline or baseline diets, the current AI for choline was sufficient to prevent or reverse organ dysfunction associated with choline deficiency in 46 individuals (81%); the remainder needed 825 mg choline · 70kg−1 · d−1 or the amount of choline in an ad libitum diet (>550 mg · 70 kg−1 · d−1 (53). This data should help inform the Institute of Medicine as they refine estimates for DRIs for choline.
Acknowledgments
We thank Marjorie Busby, Beth MacIntosh, and the nutrition research staff at the UNC GCRC for designing and preparing the research diets used in this study; Conrad Wagner and his laboratory staff for the SAM and SAH analyses; and Mei-Heng Mar and Jay Patel for their assistance with the choline analyses.
The authors’ responsibilities were as follows——LMF: supervised the human study; KAdC: collected samples and supervised the choline and metabolite analyses; LK: supervised the MRI analyses; PWS and T-SL: conducted the statistical analyses; SPS and RHA: conducted the methionine metabolite analyses; T-SL: conducted the statistical computations for data analysis and research database management; PWS: planned and designed the study, developed the plans and strategies for the data analysis, led in the construction of the database for analysis, and led in the data analysis; SHZ: conducted the statistical analyses, interpreted the data, provided major input in the writing of the manuscript, and conceptualized, implemented, and designed the human study. None of the authors had a financial conflict of interest in relation to this study. SHZ received grant support from Mead Johnson Nutritionals and the Egg Nutrition Research Center for studies other than those described in this article.
Footnotes
Supported by a grant from the National Institutes of Health (DK55865) and by grants from the National Institutes of Health to the University of North Carolina Clinical Nutrition Research Unit (DK56350), Center for Environmental Health and Susceptibility (ES10126), and General Clinical Research Center (RR00046). The Solae Company provided the lecithin used to formulate the diets.
REFERENCES
- 1.Zeisel SH, Blusztajn JK. Choline and human nutrition. Annu Rev Nutr. 1994;14:269–96. doi: 10.1146/annurev.nu.14.070194.001413. [DOI] [PubMed] [Google Scholar]
- 2.Zeisel SH, daCosta K-A, Franklin PD, et al. Choline, an essential nutrient for humans. FASEB J. 1991;5:2093–8. [PubMed] [Google Scholar]
- 3.Institute of Medicine, National Academy of Sciences USA . Dietary reference intakes for folate, thiamin, riboflavin, niacin, vitamin B12, panthothenic acid, biotin, and choline. National Academy Press; Washington, DC: 1998. Choline. pp. 390–422. [PubMed] [Google Scholar]
- 4.Zeisel SH, Mar MH, Howe JC, Holden JM. Concentrations of choline-containing compounds and betaine in common foods. J Nutr. 2003;133:1302–7. doi: 10.1093/jn/133.5.1302. [DOI] [PubMed] [Google Scholar]
- 5.Zeisel SH, Mar M-H, Howe JC, Holden JM. Concentrations of choline-containing compounds and betaine in common foods. J Nutr. 2003;133:1302–07. doi: 10.1093/jn/133.5.1302. (Erratum in J Nutr 2003;133:2918.) [DOI] [PubMed] [Google Scholar]
- 6.Blusztajn JK, Zeisel SH, Wurtman RJ. Developmental changes in the activity of phosphatidylethanolamine N-methyltransferases in rat brain. Biochem J. 1985;232:505–11. doi: 10.1042/bj2320505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tessitore L, Sesca E, Greco M, Pani P, Dianzani M. Sexually differentiated response to choline in choline deficiency and ethionine intoxication. Int J Exp Pathol. 1995;76:125–9. [PMC free article] [PubMed] [Google Scholar]
- 8.Noga AA, Vance DE. A gender-specific role for phosphatidylethanolamine N-methyltransferase-derived phosphatidylcholine in the regulation of plasma high density and very low density lipoproteins in mice. J Biol Chem. 2003;278:21851–9. doi: 10.1074/jbc.M301982200. [DOI] [PubMed] [Google Scholar]
- 9.Drouva SV, LaPlante E, Leblanc P, Bechet JJ, Clauser H, Kordon C. Estradiol activates methylating enzyme(s) involved in the conversion of phosphatidylethanolamine to phosphatidylcholine in rat pituitary membranes. Endocrinology. 1986;119:2611–22. doi: 10.1210/endo-119-6-2611. [DOI] [PubMed] [Google Scholar]
- 10.Young DL. Estradiol- and testosterone-induced alterations in phosphatidylcholine and triglyceride synthesis in hepatic endoplasmic reticulum. J Lipid Res. 1971;12:590–5. [PubMed] [Google Scholar]
- 11.Vigo C, Vance DE. Effect of diethylstilboestrol on phosphatidylcholine biosynthesis in the liver of roosters. Biochem Soc Trans. 1981;9:98–9. doi: 10.1042/bst0090098. [DOI] [PubMed] [Google Scholar]
- 12.Weisberg IS, Jacques PF, Selhub J, et al. The 1298A→C polymorphism in methylenetetrahydrofolate reductase (MTHFR): in vitro expression and association with homocysteine. Atherosclerosis. 2001;156:409–15. doi: 10.1016/s0021-9150(00)00671-7. [DOI] [PubMed] [Google Scholar]
- 13.Finkelstein JD. Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Haemost. 2000;26:219–25. doi: 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- 14.Sunden S, Renduchintala M, Park E, Miklasz S, Garrow T. Betainehomocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch Biochem Biophys. 1997;345:171–4. doi: 10.1006/abbi.1997.0246. [DOI] [PubMed] [Google Scholar]
- 15.Mudd SH, Ebert MH, Scriver CR. Labile methyl group balances in the human: the role of sarcosine. Metabolism. 1980;29:707–20. doi: 10.1016/0026-0495(80)90192-4. [DOI] [PubMed] [Google Scholar]
- 16.Steenge GR, Verhoef P, Katan MB. Betaine supplementation lowers plasma homocysteine in healthy men and women. J Nutr. 2003;133:1291–5. doi: 10.1093/jn/133.5.1291. [DOI] [PubMed] [Google Scholar]
- 17.Wendel U, Bremer H. Betaine in the treatment of homocystinuria due to 5,10-methylenetetrahydrofolate reductase deficiency. Eur J Pediatr. 1984;142:147–50. doi: 10.1007/BF00445602. [DOI] [PubMed] [Google Scholar]
- 18.da Costa KA, Gaffney CE, Fischer LM, Zeisel SH. Choline deficiency in mice and humans is associated with increased plasma homocysteine concentration after a methionine load. Am J Clin Nutr. 2005;81:440–4. doi: 10.1093/ajcn.81.2.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olthof MR, Brink EJ, Katan MB, Verhoef P. Choline supplemented as phosphatidylcholine decreases fasting and postmethionine-loading plasma homocysteine concentrations in healthy men. Am J Clin Nutr. 2005;82:111–7. doi: 10.1093/ajcn.82.1.111. [DOI] [PubMed] [Google Scholar]
- 20.Jacob RA, Pianalto FS, Henning SM, Zhang JZ, Swendseid ME. In vivo methylation capacity is not impaired in healthy men during short-term dietary folate and methyl group restriction. J Nutr. 1995;125:1495–502. doi: 10.1093/jn/125.6.1495. [DOI] [PubMed] [Google Scholar]
- 21.Jacob RA, Jenden DJ, Allman-Farinelli MA, Swendseid ME. Folate nutriture alters choline status of women and men fed low choline diets. J Nutr. 1999;129:712–7. doi: 10.1093/jn/129.3.712. [DOI] [PubMed] [Google Scholar]
- 22.Busby MG, Fischer L, Da Costa KA, Thompson D, Mar MH, Zeisel SH. Choline- and betaine-defined diets for use in clinical research and for the management of trimethylaminuria. J Am Diet Assoc. 2004;104:1836–45. doi: 10.1016/j.jada.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 23.Koc H, Mar MH, Ranasinghe A, Swenberg JA, Zeisel SH. Quantitation of choline and its metabolites in tissues and foods by liquid chromatography/electrospray ionization-isotope dilution mass spectrometry. Anal Chem. 2002;74:4734–40. doi: 10.1021/ac025624x. [DOI] [PubMed] [Google Scholar]
- 24.da Costa KA, Badea M, Fischer LM, Zeisel SH. Elevated serum creatine phosphokinase in choline-deficient humans: mechanistic studies in C2C12 mouse myoblasts. Am J Clin Nutr. 2004;80:163–70. doi: 10.1093/ajcn/80.1.163. [DOI] [PubMed] [Google Scholar]
- 25.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 26.Allen RH, Stabler SP, Lindenbaum J. Serum betaine, N,N-dimethylglycine and N-methylglycine levels in patients with cobalamin and folate deficiency and related inborn errors of metabolism. Metabolism. 1993;42:1448–60. doi: 10.1016/0026-0495(93)90198-w. [DOI] [PubMed] [Google Scholar]
- 27.Davis SR, Quinlivan EP, Shelnutt KP, et al. The methylenetetrahydrofolate reductase 677C→T polymorphism and dietary folate restriction affect plasma one-carbon metabolites and red blood cell folate concentrations and distribution in women. J Nutr. 2005;135:1040–4. doi: 10.1093/jn/135.5.1040. [DOI] [PubMed] [Google Scholar]
- 28.Fishbein M, Gardner K, Potter C, Schmalbrock P, Smith M. Introduction of fast MR imaging in the assessment of hepatic steatosis. Magn Reson Imaging. 1997;15:287–93. doi: 10.1016/s0730-725x(96)00224-x. [DOI] [PubMed] [Google Scholar]
- 29.Wilson EB. Probable inference, the law of succession, and statistical inference. J Am Stat Assoc. 1927;22:209–12. [Google Scholar]
- 30.Muller K, Stewart P. Linear model theory: univariate, multivariate, and mixed models. Wiley; New York, NY: 2006. [Google Scholar]
- 31.Marcus R, Peritz E, Gabriel K. On closed testing procedures with special reference to ordered analysis of variance. Biometrika. 1976;63:655–60. [Google Scholar]
- 32.Yuan Z, Wagner L, Poloumienko A, Bakovic M. Identification and expression of a mouse muscle-specific CTL1 gene. Gene. 2004;341:305–12. doi: 10.1016/j.gene.2004.07.042. [DOI] [PubMed] [Google Scholar]
- 33.Buchman A, Dubin M, Moukarzel A, et al. Choline deficiency: a cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatology. 1995;22:1399–403. [PubMed] [Google Scholar]
- 34.Yao ZM, Vance DE. Head group specificity in the requirement of phosphatidylcholine biosynthesis for very low density lipoprotein secretion from cultured hepatocytes. J Biol Chem. 1989;264:11373–80. [PubMed] [Google Scholar]
- 35.Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem. 1988;263:2998–3004. [PubMed] [Google Scholar]
- 36.Albright CD, da Costa KA, Craciunescu CN, Klem E, Mar MH, Zeisel SH. Regulation of choline deficiency apoptosis by epidermal growth factor in CWSV-1 rat hepatocytes. Cell Physiol Biochem. 2005;15:59–68. doi: 10.1159/000083653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albright CD, Lui R, Bethea TC, da Costa K-A, Salganik RI, Zeisel SH. Choline deficiency induces apoptosis in SV40-immortalized CWSV-1 rat hepatocytes in culture. FASEB J. 1996;10:510–6. doi: 10.1096/fasebj.10.4.8647350. [DOI] [PubMed] [Google Scholar]
- 38.da Costa K, Niculescu M, Craciunescu C, LM F, Zeisel S. Choline deficiency increases lymphocyte apoptosis and DNA damage in humans. Am J Clin Nutr. 2006;84:88–94. doi: 10.1093/ajcn/84.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.James S, Miller B, Basnakian A, Pogribny I, Pogribna M, Muskhelishvili L. Apoptosis and proliferation under conditions of deoxynucleotide pool imbalance in liver of folate/methyl deficient rats. Carcinogenesis. 1997;18:287–93. doi: 10.1093/carcin/18.2.287. [DOI] [PubMed] [Google Scholar]
- 40.Tsimberidou AM, Keating MJ. Hyperuricemic syndromes in cancer patients. Contrib Nephrol. 2005;147:47–60. doi: 10.1159/000082541. [DOI] [PubMed] [Google Scholar]
- 41.Nakanishi T, Burg MB. Osmoregulation of glycerophosphorylcholine content of mammalian renal cells. Am J Physiol. 1989;257:C795–801. doi: 10.1152/ajpcell.1989.257.4.C795. [DOI] [PubMed] [Google Scholar]
- 42.Kempson SA, Montrose MH. Osmotic regulation of renal betaine transport: transcription and beyond. Pflugers Arch. 2004;449:227–34. doi: 10.1007/s00424-004-1338-6. [DOI] [PubMed] [Google Scholar]
- 43.Baxter JH. A study of hemorrhagic-kidney syndrome of choline deficiency. J Nutr. 1947;34:333. doi: 10.1093/jn/34.3.333. [DOI] [PubMed] [Google Scholar]
- 44.da Costa K, Kozyreva OG, Song J, Galanko JA, Fischer LM, Zeisel SH. Common genetic polymorphisms have major effects on the human requirement for the nutrient choline. FASEB J. 2006;20:1336–44. doi: 10.1096/fj.06-5734com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohlmeier M, da Costa KA, Fischer LM, Zeisel SH. Genetic variation of folate-mediated one-carbon transfer pathway predicts susceptibility to choline deficiency in humans. Proc Natl Acad SciUSA. 2005;102:16025–30. doi: 10.1073/pnas.0504285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchman AL, Ament ME, Sohel M, et al. Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: proof of a human choline requirement: a placebo-controlled trial. JPEN J Parenter Enteral Nutr. 2001;25:260–8. doi: 10.1177/0148607101025005260. [DOI] [PubMed] [Google Scholar]
- 47.Shaw GM, Carmichael SL, Yang W, Selvin S, Schaffer DM. Periconceptional dietary intake of choline and betaine and neural tube defects in offspring. Am J Epidemiol. 2004;160:102–9. doi: 10.1093/aje/kwh187. [DOI] [PubMed] [Google Scholar]
- 48.Zeisel SH, Mar M-H, Zhou Z-W, da Costa K-A. Pregnancy and lactation are associated with diminished concentrations of choline and its metabolites in rat liver. J Nutr. 1995;125:3049–54. doi: 10.1093/jn/125.12.3049. [DOI] [PubMed] [Google Scholar]
- 49.Albright CD, Tsai AY, Friedrich CB, Mar MH, Zeisel SH. Choline availability alters embryonic development of the hippocampus and septum in the rat. Brain Res Dev Brain Res. 1999;113:13–20. doi: 10.1016/s0165-3806(98)00183-7. [DOI] [PubMed] [Google Scholar]
- 50.Albright CD, Friedrich CB, Brown EC, Mar MH, Zeisel SH. Maternal dietary choline availability alters mitosis, apoptosis and the localization of TOAD-64 protein in the developing fetal rat septum. Brain Res Dev Brain Res. 1999;115:123–9. doi: 10.1016/s0165-3806(99)00057-7. [DOI] [PubMed] [Google Scholar]
- 51.Jones JP, Meck W, Williams CL, Wilson WA, Swartzwelder HS. Choline availability to the developing rat fetus alters adult hippocampal long-term potentiation. Brain Res Dev Brain Res. 1999;118:159–67. doi: 10.1016/s0165-3806(99)00103-0. [DOI] [PubMed] [Google Scholar]
- 52.Meck WH, Williams CL. Choline supplementation during prenatal development reduces proactive interference in spatial memory. Brain Res Dev Brain Res. 1999;118:51–9. doi: 10.1016/s0165-3806(99)00105-4. [DOI] [PubMed] [Google Scholar]
- 53.Fischer LM, Scearce JA, Mar MH, et al. Ad libitum choline intake in healthy individuals meets or exceeds the proposed adequate intake level. J Nutr. 2005;135:826–9. doi: 10.1093/jn/135.4.826. [DOI] [PMC free article] [PubMed] [Google Scholar]