

SUMMARY
Hoxa13 is expressed early in the caudal mesoderm and endoderm of the developing hindgut. The tissue specific roles of Hoxa13 function have not been described. Hand-Foot-Genital syndrome, a rare dominantly inherited human malformation syndrome characterized by distal extremity and genitourinary anomalies, is caused by mutations in the Hoxa13 gene (HFGa13). We show evidence that one specific HFGa13 mutation likely acts as a dominant negative in vivo. When chick HFGa13 is overexpressed in the chick’s caudal endoderm early in development, caudal structural malformations occur. The phenotype is specific to HFGa13 expression in the posterior endoderm and includes taillessness and severe gut/genitounrinary (GGU) malformations. Finally, we show that chick HFGa13 negatively regulates expression of Hoxd13 and antagonizes functions of both endogenous Hoxa13 and Hoxd13 proteins. We suggest a fundamental role for epithelial specific expression of Hoxa13 in the epithelial-mesenchymal interaction necessary for tail growth and posterior GGU patterning.
Keywords: Abnormalities, Multiple, etiology, genetics, Animals, Body Patterning, Chick Embryo, Endoderm, physiology, Extremities, embryology, Fibroblast Growth Factor 8, Fibroblast Growth Factors, metabolism, Homeodomain Proteins, genetics, metabolism, Humans, Intestines, embryology, Limb Deformities, Congenital, etiology, genetics, Molecular Sequence Data, Mutation, Sequence Deletion, Syndrome, Tail, abnormalities, embryology, Transcription Factors, Urogenital Abnormalities, etiology, genetics, Urogenital System, embryology
Keywords: cloaca, tail, colon, hindgut, HOX, HOXA13, chick
INTRODUCTION
Vertebrate gastrulation events form the three germ layers and pattern the early anterior body region. Although disputed, the posterior region likely forms via a separate secondary event at the undifferentiated mesenchyme of the tailbud to form the tail somites, distal neural tube, notochord, and tailgut (Catala et al., 1995; Gajovic et al., 1993; Griffith et al., 1992; Holmdahl, 1925; Knezevic et al., 1998; Pasteels, 1943; Schoenwolf, 1977; Schoenwolf, 1979). A critical early event in patterning the posterior embryo is the formation of the caudal intestinal portal (CIP), which initiates the development of the hindgut, tail, and forms the cloaca (the common gut/genitourinary (GGU) chamber). The cloaca is maintained throughout life in avian and some other vertebrate species. In mammals, the cloaca exists as an embryologic structure that undergoes septation to become distinct urethral, anal, and genital orifices. Abnormal development of the cloaca causes severe congenital malformations in vertebrates including humans (Kluth et al., 1995; Martinez-Frias et al., 2000).
Tail growth is a function of a specialized region of ectoderm, the ventral ectodermal ridge (VER), which acts analogously to the apical ectodermal ridge (AER) of the limb bud. Both the AER and VER signal adjacent mesodermal tissues to maintain an undifferentiated state, facilitating growth and elongation (Goldman et al., 2000; Saunders, 1948). The VER forms before morphological tail development (St. 18 in chick (Mills and Bellairs, 1989) and E10 or 35+ somites in the mouse (Gruneberg, 1956)) from the cloacal membrane posterior to the tailbud where ecotoderm and endoderm are justaposed (Gruneberg, 1956). During the ensuing gastrulation events the tailgut develops anterior to the cloacal membrane and elongates in close association with the tail and in close proximity to its ectoderm. Apoptotic degeneration of the tailgut endoderm and VER heralds completion of tail growth (Fallon and Simandl, 1978; Miller and Briglin, 1996; Mills and Bellairs, 1989).
Despite common tail formation in early vertebrate development, tail growth ceases at different developmental time points in a species specific manner. Tail length is a characteristic phenotype amongst species yet its molecular controls are unknown. Molecular candidates include the Hox genes, homeobox containing transcription factors that function in pattern formation of many aspects of development (Krumlauf, 1994). In vertebrates the Hox genes are expressed in a characteristic spatial and temporal pattern mirroring their physiocal location in the chromosome. The most 5′ Hox genes are expressed in the posterior body region including the posterior mesodermal regions of the gut (Roberts et al., 1995; Yokouchi et al., 1995b). These play an important role in patterning the gut along the anterior-posterior axis (AP) (Roberts et al., 1995; Roberts et al., 1998; Warot et al., 1997). Hoxa13 and Hoxd13 are expressed specifically in the cloacal mesoderm and also uniquely in the hindgut and cloacal endoderm (Roberts et al., 1995; Yokouchi et al., 1995b). Their endodermal function is unknown.
Mutations in Hoxa13, both spontaneous and transgenic, exist in mice (Goodman and Scambler, 2001). The mutant phenotypes show both limb and GGU anomalies. Spontaneous murine Hoxa13 mutant, hypodactyly (hd), has a 50 base pair deletion within the first exon of Hoxa13 resulting in a mutant fusion protein (Mortlock et al., 1996). Although these mice suffer a high perinatal mortality, some homozygotes survive but are infertile due to hypoplasia of distal reproductive structures (Warot et al., 1997). As the Hoxa13 null mice are early perinatal lethal (Fromental-Ramain et al., 1996) the hd protein may act as a gain of function mutant.
Murine Hoxd13−/− male show subtle GU anomalies (Dolle et al., 1993; Kondo et al., 1996; Podlasek et al., 1997) and homozygotes have abnormalities of the lowest sacral vertebra (Dolle et al., 1993). Hoxd13−/+ embryos crossed with Hoxa13 null heterozygotes, have GGU anomalies more severe than in the Hoxd13−/− alone suggesting that cooperation and redundancy between Hoxa13 and Hoxd13 exists in their function in GGU development (Warot et al., 1997).
Missense and nonsense mutations of one allele of Hoxa13 cause hand-foot-genital (HFG) syndrome (Goodman and Scambler, 2001), a rare dominantly inherited human disease{MIM 14000}. Affected individuals have mild, fully penetrant, symmetrical, bilateral hand and foot anomalies. Affected individuals also exhibit incompletely penetrant and variably severe genitourinary tract (GU) abnormalities including hypospadias in males and Müllerian duct fusion abnormalities in females. Both sexes show abnormalities in ureter/bladder placement. The variety of mutations described includes deletions, truncations of the protein, or amino acid substitutions within the conserved homeodomain. The resulting proteins are thought to act either as a gain-of function mutation or possibly a competitor of the wild-type protein.
Mutations in human Hoxd13 cause a severe distal limb phenotype termed synpolydactyly (SPD) {MIM 186000}. This rare, dominantly inherited syndrome is caused by mutations in a polyalanine repeat within the coding region of Hoxd13, including expansions and intragenic deletions (Goodman and Scambler, 2001). Affected males often demonstrate GU abnormalities (Goodman and Scambler, 2001).
The manner in which these Hoxa13 and Hoxd13 mutations and nulls lead to these specific GGU malformations is unknown and may be due to either or both a mesodermal or endodermal effect of the mutation. Murine models to date have failed to address their possible tissue specific functions.
To study the role of Hoxa13 in GGU development we used the chick embryo model system. We studied the specific endodermal role of Hoxa13 using the avian specific retroviral expression system and in ovo electroporation to overexpress wild-type and mutant forms role of Hoxa13 in the chick hindgut endoderm in ovo. We constructed a chick Hoxa13 homolog of one of the specific mutations that causes HFG (HFGa13). We chose the originally described Hoxa13 mutation in which a small deletion of the Cterminal results in truncation of the protein (Morlock and Innis 1997). When expressed in the chick posterior endoderm a dramatic GGU and tail malformation results. We show that this effect is specific to endodermal HFGa13 expression. We show that HFGa13 likely acts by interfering with the normal function of endogenous Hoxa13 and Hoxd13.
MATERIALS AND METHODS
Embryos
Timed fertilized white leghorn eggs (SPAFAS, CT) were incubated at 38°C in a humidified incubator (Kuhl,NJ) until used experimentally. Embryos were staged according to Hamburger and Hamilton (St.) (Hamburger and Hamilton, 1951) or by embryonic day (E).
Constructs
Isolation of chicken Hoxa13 gene has been previously described (Nelson et al., 1996). Although this clone was thought to be a full-length cDNA, a recent manuscript described evolutionarily conserved 3′ sequence which was not present in our original clone (Mortlock et al., 2000). We isolated this N-terminal sequence by RT-PCR on E6 hindgut total RNA with a primer designed to amplify the conserved 3′ sequence (ATGTTCCTCTACGACAACAGC). After sequence verification and subcloning we verified the full-length chick Hoxa13 cDNA.
By PCR we mutated chick Hoxa13 to produce a truncation similar to a specific human mutation (HFGa13). Mutated reverse oligonucleotide (TCAGATTGTGACCTGTCGC) produced a premature stop codon as described by Mortlock (Mortlock and Innis, 1997).
Wild-type Hoxa13 and HFGa13 cDNAs RCAS(A) or RCAS(B) viruses and a Hoxd13 RCAS(A) virus were produced as described (Morgan and Fekete, 1996). We found no difference in any of the experimental results described below using either the short or long form Hoxa13 or HFGa13 and, therefore, we do not distinguish between them herein. Similarly, 3′ long and short forms of RCAS(A)Hoxd13 acted equivalently in a chick limb bud overexpression study (Goff and Tabin, 1997). We found no difference in the infectivity of either A or B envelope coats of RCAS, both acted equivalently.
Constructs for electroporation were prepared with wild-type Hoxa13 and HFGa13 cDNAs cloned into pCDNA3 (Invitrogen). An N-Flag epitope oligonucleotide was inserted in frame with the GAL4 DNA Binding Domain (Sadowski and Ptashne 1989). All sequences were confirmed prior to use in experiments.
Transfection studies and Western blots
Plasmids used for transfections were purified using the maxiprep reagent system (Qiagen). COS-7 at 60–80% confluence were washed twice with serum-free medium and then co-transfected with 100 ng of reporter plasmid (pG5luc containing 5 GAL4 binding site (Promega)), 10 ng of Renilla luciferase promoter vector (Promega), and 100 ng of different Hoxa13 plasmids with 3μl of LipofectAMINE (Life Technologies) in 200 μl of serum-free medium. After 30 minutes of incubation, 500 μl of medium supplemented with 10% serum was added. Cells were harvested after 36 h of culture. Luciferase assays were checked with the Dual-Luciferase™ Reporter Assay (Promega). Promoter activities were expressed as relative luciferase activity: units/Renilla units, and each values represents the mean of six separate wells.
Relative expression of the GAL4 fusion proteins was assessed by Western-blot analysis of COS-7 extracts. Transfections with wild-type Hoxa13 and HFGa13 constructs fused to GAL4 DNA Binding Domain (DBD) were done as described previously (Sadowski and Ptashne 1989). GAL4 DBD antibody (Santa Cruz Biotech, Inc.) was used as described by the manufacturer.
Cells were fixed 24 h after transfection for 30 min in 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS), permeabilized with PBS containing 0.02% Triton X-100, and incubated in 10% normal goat serum in PBS for 30 min at RT. Cells were then incubated with an anti-FLAG M5 monoclonal antibody (Kodak; 1:500) for 2 h at RT, followed by incubation with secondary antibody. Images were collected and processed on a Microphot Nikon microscope.
Viral infection
This technique has been previously described (Morgan and Fekete, 1996). Embryos at St.8–10 were used for experiments of the posterior endoderm, St. 18 for experiments in the limb. Approximately 1–5μ1 of freshly defrosted virus dyed with fast green was injected per embryo. For hindgut experiments, the virus was injected into the region lateral and posterior to the tailbud following a published fate map (Matsushita, 1999). Double injections were performed by mixing equal volumes of each viral aliquote before injection, as previously described (Bendall et al., 1999). In all cases, the HFGa13 virus was cloned in RCAS(B) and the wild-type viruses were in RCAS(A). For limb injections, the right hindlimb bud was viewed at St.18. Approximately 1–5μl of virus was injected filling the entire limb bud. Eggs were then placed at 38°C until harvest. Viral constructs injected included short and long forms (see above) of wild-type Hoxa13, HFGa13 and, as controls, wild-type Hoxd13 and GFP. More than 10 dozen embryos were injected with each construct.
Electroporation
This technique was adapted to a technique previously published (Grapin-Botton et al., 2001). E2.5 (St. 11–14) embryos were used for electroporation, at CIP invagination and tailbud formation. Plasmids were diluted to a final concentration of 2 μg/ml in PBS adding 1 μM MgCl2. To facilitate viewing of the viral aliquot and to slow diffusion, 50 μg/ml Nile Blue Sulfate, 3 mg/ml carboxymethylcellulose was added to the construct liquid. 5 to 10 μl of this solution was deposited using a microcapillary pulled micropipette positioned under embryo at the posterior most endoderm layer (just caudal to the tailbud). Quickly after injection, a cathode was positioned on the dorsal surface of the embryo at the level of the injected construct. The anode was placed parallel to the cathode under the embryo (about 3 mm deep) at the level of the injected construct. Neither electrode touched the embryo. Three square pulses of 17 Volts and 50 msec each were applied to the embryo (BTX T-280 square wave electroporator generously lent to us by the Melton Lab under supervision of Anne Grapin-Botton) to allow vector integration into the endodermal layer. Eggs were incubated at 38°C until embryonic death or harvest. Controls included solution with all components except the vector, solution with empty vector, construct containing the wild-type Hoxa13, or with vector containing GFP only. Approximately 3 dozen eggs were electroporated in experimental and control groups.
Whole embryo explants
The technique was adapted from that of Chapman et al. (Chapman et al., 2001). Dissected St. 9–13 embryos were placed ventral side up in a freshly made thin layer of albumin/agarose (Chapman et al., 2001). Using a dissecting microscope (Olympus SZH10) the posterior endoderm covering the tailbud and posterior was removed using one of these techniques: dissection alone using pulled glass needles and fine forceps, enzymatic digestion using 0.03% collagenase in PBS dropped on the embryo over the tailbud and allowed to sit for 15 minutes at 37°C and washed with 5 drop/aspirate cycles using PBS+10% chick serum followed by 5 drop/aspirate cycles using PBS + pen/strep, or a combination of both techniques. Controls had no manipulation. Transplanted embryos had caudal endoderm removed as above. Donor endoderm was removed from donor stage 17 embryos (for facility and to ensure good Hoxa13 expression by the posterior endoderm) by sharp dissection, kept oriented, and divided into thirds along the AP axis. The anterior endoderm and posterior endoderm were isolated and transplanted to host embryos (separately) by transfer pipette and forceps. Embryos were harvested immediately after manipulation, and at 24 and 48 hours. For each condition at least 12 embryos were used over five separate experiments.
Analysis
Embryos were harvested and fixed in freshly made 4% paraformaldehyde in PBS for 4–20 hours. Fixed embryos were washed in PBS and then either taken through a graded series of methanol-PBS to 100% methanol and kept at −20°C, until used for whole mount in situ hybridization studies, or were frozen in cryomount (Fisher Scientific) for cryosectioning. Whole mount in situ hybridization studies followed our published technique (Roberts et al., 1998). 10 μm cryosections on Superfrost Plus slides (Fisher Scientific) were air dried for 4–18 hours and kept at −20°C until used. Some fixed, in situ hybridized embryos were embedded in paraffin and sectioned at 5 μm for histologic analysis. Haematoxylin and eosin (H&E) staining was performed using standard techniques. Section in situ hybridizations were performed using published techniques (Smith et al., 2000). Immunohistochemical stains were performed using standard techniques and the Vectastain ABC detection system (Vector Laboratories, Inc) following the manufacturer’s directions. Hoxa13 antibody was a generous gift from A. Kuroiwa and used as previously described (Yokouchi et al 1995a). 3C2 antibody specific to RCAS GAG protein has been published before (Smith et al, 2000). Ribroprobes were transcribed using Roche ribroprobe synthesis kits per the manufacturer’s directions. All riboprobes used herein have been previously published (Nielsen et al., 2001; Roberts et al., 1995; Roberts et al., 1998; Smith et al., 2000; Vogel et al, 1996).
Genbank accession number AY030050. Gallus gallus Hoxa13.
RESULTS
Spatio-temporal expression of Hoxa13 during posterior GGU/tail development
Hoxa13 is first expressed early in the most posterior part of the embryo, adjacent to Hensen’s node in the area that will give rise to the CIP (St. 10) (Fig. 1A). Hoxa13 is expressed at St. 14 in the CIP (Fig. 1B). Later, Hoxa13 expression is restricted to the dorsal mesoderm of the tailbud, cloacal mesoderm, hindlimb bud mesoderm, and caudal endoderm (Fig. 1C,D). Hoxa13 and its product are strongly expressed in the endoderm of the hindgut and cloaca through early development of the gut (Fig. 1E,F) (Roberts et al., 1995; Yokouchi et al., 1995b).
Fig. 1.
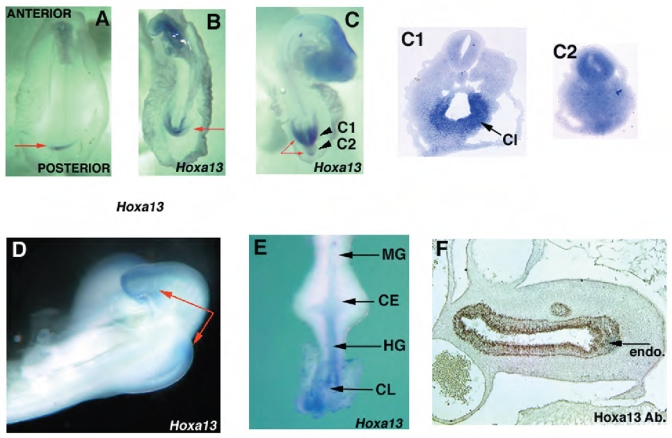
Spatio-temporal expressions of Hoxa13 (A–E) and Hoxa13 protein (F) during posterior GGU/tail development. (A) St. 10 (arrow at CIP). (B) St. 14 (arrow at CIP). (C) St. 17 (long arrows show tail tip, short arrow at hindgut and arrowhead at tailgut). Planes of cryosection are indicated by short arrows (C1, C2). Hoxa13 endodermal expression in the cloaca (CL). (D) expression in the tail and hindlimb bud at St. 22. (E) dissected E4 posterior gut (arrows at endodermal hindgut (HG) and mesodermal cloacal expression), no expression is detected in the ceca (CE) or midgut (MG). (F) protein expression at E6 section of the hindgut in the endoderm (endo) hindgut (arrow).
Human HFG and chick HFGa13 overexpressed embryos have similar phenotypes
Overexpression of Hoxa13 in the posterior embryo failed to produce a tail or gut phenotype. We did see an epithelial transformation (from midgut to hindgut) when midgut mesodermal tissues expressed Hoxa13 (see Fig. 7H for comments) as was described with ectopic Hoxd13 expression in the midgut (Roberts et al., 1998).
Fig. 7. HFGa13 interferes with the cellular functions of Hoxa13 and Hoxd13.
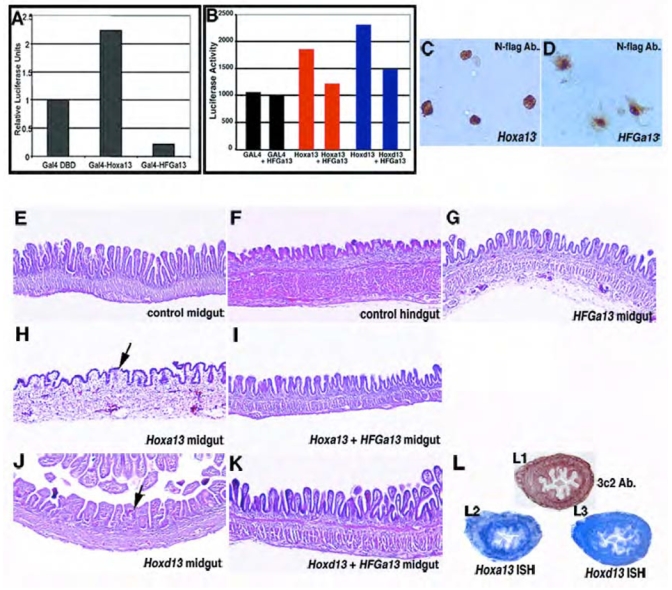
(A) Transcriptional transactivation by wild-type Hoxa13 and HFGa13 proteins in a GAL4-fusion assay in COS-7 cells. Relative luciferase activities were normalized to the empty GAL4 DNA-binding domain expression vector. Fusion protein of the GAL4 DNA-binding domain and Hoxa13 shows transcriptional activation of the synthetic reporter. In contrast, fusion protein of the GAL4 DNA-binding domain and HFGa13 fails to activate transcription of the same promoter and is able to decrease the basal activity. Luciferase assays were performed after two independent transfections, each done in triplicate (A,B). (B) Perturbation of the transcriptional transactivation of wild-type Hoxa13 and Hoxd13 by HFGa13 in a GAL4-fusion assay in COS-7 cells. In this assay, we monitored GAL4 trasncriptional activity induces by GAL4 DBD fusion proteins without or with pcDNA3-HFGa13 construct. In a same molar ratio, HFGa13 form specifically decreases Hoxa13 and Hoxd13 transcriptional activation. (C, D) Intracellular localization of Hoxa13 and HFGa13 proteins. Immunostaining of transfected N-flag tagged Hoxa13 (C) and HFGa13 (D) constructs in COS-7 cells with specific N-flag antibody show both nuclear localization. Note an additive cytoplasmic signal with the HFGa13 construct. (E–K) H&E sections of E18 control (E,F) or infected (G–K) guts. (E) Normal midgut with thin and long villi. (F) Normal hindgut with flat and short villi. (G) HFGa13 mesodermally infected midgut has wild-type midgut epithelium. Hoxa13 (H) and Hoxd13 (J) mesodermally infected midgut shows hindgut-like epithelial transformation. HFGa13 co-infected midguts with either Hoxa13 (I) or Hoxd13 (K) show rescue of the epithelial hindgut phenotype. (L) Hoxd13 and HFGa13 mesodermal midgut co-infection show presence of virus (detected by 3C2-Ab., L1), and ectopic HFGa13 (detected with Hoxa13 probe, L2) and Hoxd13 (L3) co-expressions, associated with normal epithelial phenotype.
To determine if expression of chick mutated Hoxa13 (HFGa13) results in a phenotype similar to that observed in the human HFG syndrome, we chose to construct a similar nonsense mutation as described by Mortlock and Innis, 1997. The Cterminal mutation leads to a production of a mutated Hoxa13 protein, with the last 20 last amino acids deleted. We expected that HFGa13 protein would be able to interfere with the endogenous Hoxa13.
In order to test our hypothesis, we first misexpressed HFGa13 in the hindlimb, and we were able to produce a severe morphological change reminiscent of limb defect described in HFG syndrome (Fig. 2A). The HFGa13 expressing hindlimb shows a specific malformation characterized by a substantial reduction in limb size in both the anterioposterior and dorsoventral axes compared to the uninfected contralateral control limb. HFGa13 E6 infected hindlimbs revealed a specific skeletal hypoplasia of the fibula (without changes in the tibia) and a reduction of the entire autopod area (Fig. 2A,B). This phenotype suggested a late disruption of the apical ectodermal ridge (AER). Consistent with this, Fgf8 mRNA expression was not detectable in the distal hindlimb AER of the HFGa13 hindlimbs (Fig. 2C). The distal fibular cartilage fails to properly develop and remains undifferentiated mesenchymal cells. Expression of Bapx1 was decreased only in the malformed fibula (Fig. 2D). No AER disruption or fibula maldevelopment were observed with the wild-type Hoxa13 overexpression (data not shown). These results suggest that HFGa13 interferes with the maintenance of the AER in the hindlimb and with formation of the fibula. We conclude that our construct functions to give a chick phenocopy of the human HFG syndrome.
Fig. 2.
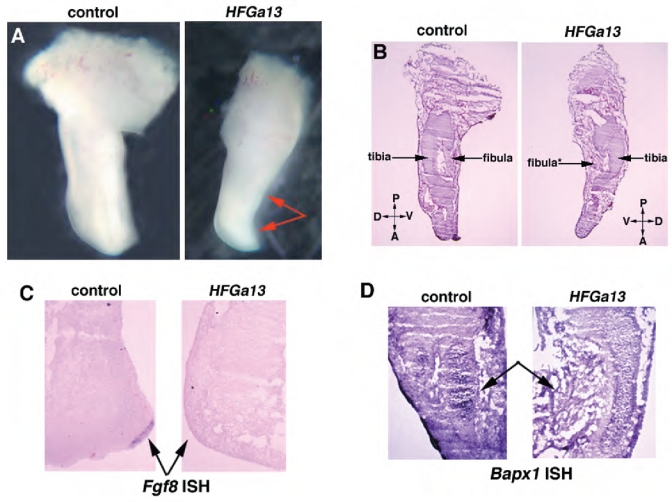
Human HFG and chick HFGa13 overexpressed embryos have similar limb phenotypes. (A–D) Control (left panel) and HFGa13-infected (right panel) hindlimbs at E6. Planar sections of E6 limbs (B), uninjected control (left) HFGa13 infected hindlimb (right, arrow shows fibula maldevelopment with undifferentiated mesenchymal cells). (C) In situ hybridization of Fgf8 in sectioned hindlimb. No Fgf8 expression is detected in the HFGa13-infected hindlimb compare to the uninfected hindlimb (arrows). (D) In situ hybridization of Bapx1 in sectioned hindlimb. Altered Bapx1 expression in the HFGa13-infected hindlimb (arrows).
Endodermally expressed HFGa13 causes abnormal hindgut and tail development
HFGa13 and control constructs were expressed in the chick in ovo by injection of virus, targeting the prospective hindgut in St. 8–10 embryos. We used this targeting technique previously to infect the midgut mesoderm (Roberts et al., 1998), but in the experiments described herein we often found strong endodermal infection in the hindgut as well (Fig. 3B).
Fig. 3.
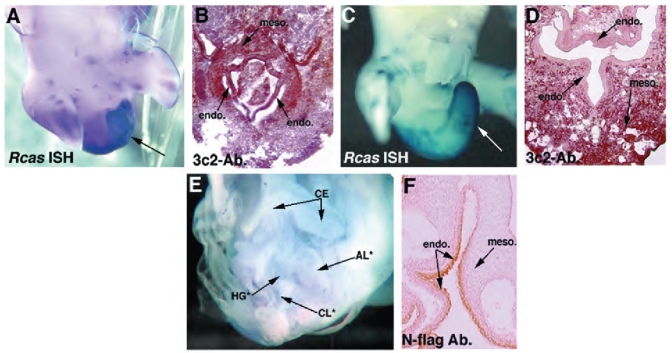
Endodermally expressed HFGa13 causes abnormal hindgut and tail development. (A) Whole-mount in situ hybridization showing RCAS expression in an E6 mutant HFGa13 infected embryo. (B) 3C2 immunohistochemistry analysis of sectioned HFGa13-infected mutant embryo shows posterior endoderm (endo.) and mesoderm (meso.) infection. (C) RCAS in situ showing absence of the mutant phenotype when HFGa13 infection is present only in the mesoderm, demonstrated in (D) with anti-gag immunohistochemistry showing no infection in the caudal endoderm (E6). (E) E6 survivors after electroporation of HFGa13 constructs in the posterior endodermal layer. The phenotype involves maldevelopment of the cloaca (CL*), hindgut (HG*), and tail. Allantoic internalization is present (AL*), ceca are unaffected (CE). (F) Anti-Nflag demonstrating expression of the tagged-HFGa13 in the endoderm of the hindgut, mesoderm is not stained. Note the electroporated endodermal cells appear undamaged and intact. Misexpression of HFGa13 and Hoxa13 constructs by electroporation show similar expression levels in the gut endoderm layer (data not shown).
Specific gross morphologic defects were obtained only with HFGa13 infections and only when the infection included the posterior endoderm (Fig. 3A,B). Hoxa13, Shh, Bmp4, and GFP constructs failed to produce this phenotype ever, even when expression was noted in the endoderm (data not shown). HFGa13 infected in the mesodermal only were phenotypically normal (Fig. 3 C,D). Our survival rate was 50–80% depending on time of incubation (survival to E3 better than to E18). Approximately 20% of HFGa13 injected embryos demonstrated the mutant phenotype. All HFGa13 embryos were analyzed for viral infection. All mutant embryos harbored posterior endodermal and mesodermal virus expression. Those without the phenotype showed no viral infection or only posterior mesodermal virus expression (no infection in the endoderm).
To determine if endodermal expression of HFGa13 is sufficient to obtain these posterior defects we used an in ovo electroporation technique (Grapin-Botton et al., 2001). Three groups of E2.5 (St. 11–16) embryos were electroporated: control group with empty vector or a GFP containing vectors, those with wild-type Hoxa13 construct; and finally a group with HFGa13. In E5 survivors, posterior short tail/hindgut atresia was present only in the HFGa13 electroporated embryos, electroporation of control constructs produced no defects (data not shown). The mutant phenotype was restricted to those embryos with hindgut epithelial expression of HFGa13 (electroporations restricted to the midgut endoderm failed to produce the posterior phenotype (data not shown)). Survival was about 80% for all groups. The posterior endodermal HFGa13 expressing embryos show similar, albeit more severe, defects as those obtained using the injection/infection technique (Fig. 3E). Confirmation of tissue integration and endodermal tissue survival were performed by tag immunostaining on all embryos (Fig. 3F). Sections of experimental embryos confirmed an intact endoderm an a histologically normal neural tube and notochord (Fig. 4). Approximately 10 embryos showed the phenotype (all the survivors and all the embryos with documented expression in the hindgut endoderm).
Fig. 4.
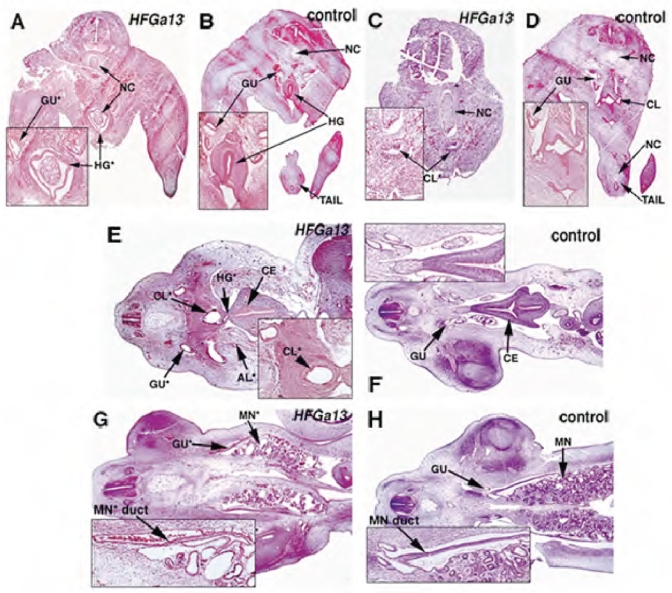
Histology analysis of mutant HFGa13-infected embryos. H&E stained transverse (A–D, E7) and longitudinal (E–H, E6) sections of mutant HFGa13-infected (A,C,E,G) and control embryos (B,D,F,H)- Ourentery is present in (A). HFGa13-infected embryos show cloacal stenosis (C), atresia of the hindgut anterior to the cloaca (E), allantoic internalization (E), and defects in the cloaca-associated viscera including more distal Müllerian duct (MN duct) atresia and cystic mesonephric (MN*) maldevelopment (G). Note the correct development of the more anterior gut structures. Ceca (CE), notochord (NC), other abbreviations as per Fig. 3.
Histological analyzes of mutant HFGa13-infected embryos were done by H&E staining. The phenotype includes malformations of the hindgut and tail (Fig. 4A,C,E and G) including maldevelopment of the tail somites and a short tail (compare Fig. 4A,C respectively with B,D). Hindgut defects include atresia of the anterior to the cloaca (compare Fig. 4E with F), malformed and malpositioned posterior cloaca (compare Fig. 4C,K respectively with D,L). Additional defects are noted in other cloaca-associated viscera including cystic mesonephric maldevelopment with normal non-atretic ureters and atresia of the distal Müllerian ducts (compare Fig. 4G with H). No associated neural tube defects or malformations are present (as shown in Fig. 4). We occasionally obtained a severe phenotype, termed ourentery (Rabaud 1900), in which the tail appears to have grown ventrally and internally to the remaining cloacal orifice, often accompanied with ventral internal malpositioning of tail structures into the hindgut and internalization of the allantois (compare Fig. 4A,C with B,D and also Fig. 3B with 3D). All these results show that HFGa13 expression, specifically in the posterior endodermal, induces maldevelopment of many of the caudal structures.
HFGa13 affects expression of Hoxd13, Fgf8 and Bapx1
Molecular analysis of HFGa13 mutant embryos were made with specific dorsoventral (DV), anteroposterior (AP), and cytodifferentiation markers. Strong down-regulation was observed with VER marker Fgf8 (ectoderm and mesoderm) and with AP marker Hoxd13 (mesoderm and endoderm) (respectively Fig. 5A and D). Bapx1 shows a diminished expression in the short tail (Fig. 5B). No change was noted in the expression of ventral mesodermal markers: Wnt5a (Fig. 5C) and Bmp4 (data not shown) and normal expression of Shh (Fig. 5E).
Fig. 5.
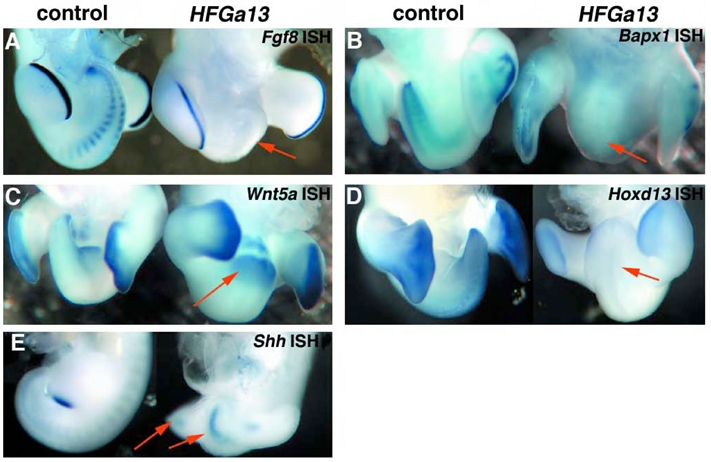
HFGa13 affects expression of Fgf8, Hoxd13 and Bapx1. (A–G) Whole-mount in situ hybridizations of control (left panel) and HFGa13-infected (right panel) embryos (arrows at malformed tail). (A) Fgf8 expression at E5, note absence of expression in the HFGa13 tail but normal expression in the hindlimb AER. (B) Bapx1 (expression) at E6. Bapx1 shows a diminished expression in the tail of the HFGa13-infected embryo, but normal expression in the non-infected hindlimbs. (C) Wnt5a gene expression shows expression in the HFGa13-infected embryo, E7. (D) Hoxd13 expression at E6 shows strong down-regulation in the tail HFGa13-infected embryo and normal expression in hindlimbs. (E) Shh expression at E4 shows is normal. Arrows at hindlimb and notochord.
Caudal gut endodermal signals are needed for normal posterior gut and tail development
To test our hypothesis that posterior gut and tail development requires specific endodermal signals we used whole embryo explant cultures in which we removed the endoderm at early (pre-CIP) stages (St. 9–11). Survival (90% of un-manipulated controls, 75% of manipulated embryos) after 2 days, to St. 20 allowed analysis of tail development. None of the control embryos had caudal defects (Fig. 6A). All embryos in which the caudal endoderm had been removed showed severe caudal defects of the tail and gut. Those embryos that survived to form hindlimb buds generally formed abnormal CIP and blunted tails similar to those produced in the HFGa13 injected or electroporated embryos (Fig. 6B). Ourentery was present in 50% of manipulated survivors (Fig. 6C). No neural tube defect was present in these embryos.
Fig. 6.
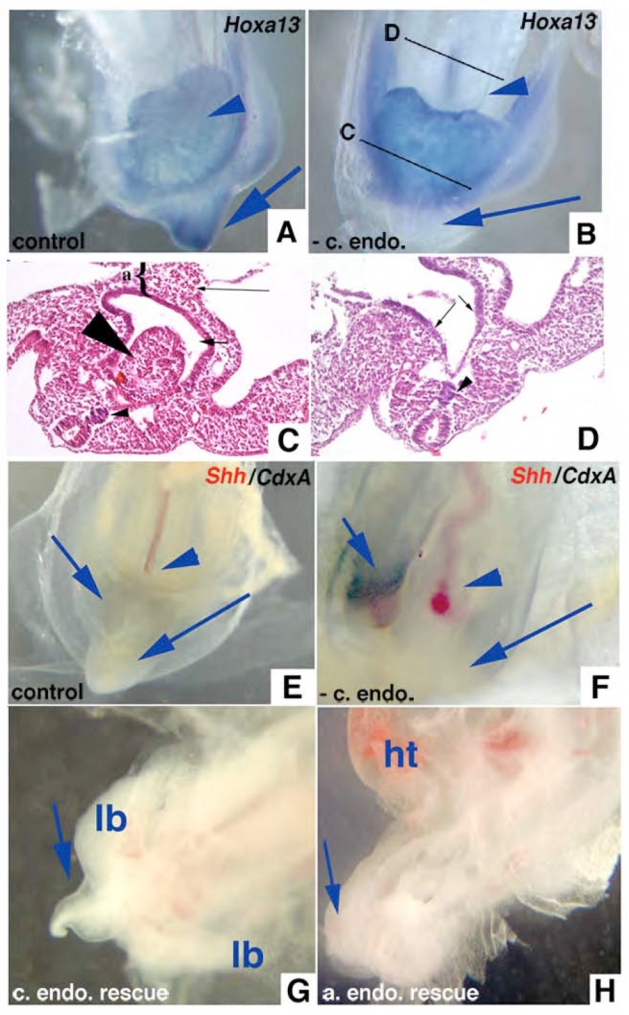
Whole embryo explants developed in culture demonstrate the physical necessity of caudal endoderm for normal tail development. (A) Whole embryo explanted at St. 10 and grown on albumen/agarose for 48 hours showing normal tail development and expression of Hoxa13 in tail (blue arrow). Arrowhead shows normal placement of allantois ventral to hindgut which expresses Hoxa13. (B) St. 10 embryo grown with caudal endoderm removed demonstrating blunted tail without Hoxa13 expression (long arrow). Short arrow marks hindgut dorsal to Hoxa13 expressing allantois. (C) H&E section through a caudal endoderm-less embryo that developed ourentery (large arrowhead) within hindgut lumen (short arrow). {shows allantois. Small arrowhead shows normal notochord. (D) Section anterior to (C) showing endoderm (arrows) and notochord (arrowhead). (E) Embryo cultured ~24 hours with caudal endoderm develops normal CIP (long arrow), expresses Shh in notochord (red stain, arrowhead, most of notochord deep to plane of photograph), and coexpresses Shh and CdxA in endoderm of CIP (short arrow, purple/black stain). (F) Embryo cultured ~24 hours without caudal endoderm fails to develop CIP or express Shh or CdxA in midline caudal endoderm (long arrow), both are coexpressed in right lateral caudal endoderm (short arrow, purple/black stain). Notochord is curved but expresses Shh (red stain, arrowhead). (G) Embryo after caudal endoderm removed and donor caudal endoderm transplanted, cultured ~48 hours showing tail growth (long arrow). lb - hindlimb buds. (H) Embryo after caudal endoderm removed and donor anterior endoderm transplanted, cultures ~48 hours showing blunted tail (long arrow), allantois (arrowhead), ht - heart.
When examined histologically, we confirmed that the midline ventral endoderm overlying the tailbud was removed (data not shown). This endoderm is absent for the first 24 hours after dissection then it appears that the adjacent endoderm occasionally re-grows over this defect (Fig. 6C). No defects are seen in the more anterior endoderm (Fig. 6D). To confirm that removal of the caudal endoderm was complete we analyzed the normal and abnormal embryos for the presence of early endodermal markers. In the caudal ventral tissues of normal embryos we found expression of Shh, Cdxa (Fig. 6E), and Hoxa13 (Fig. 6A), and Hoxd13 (data not shown). We also could not detect expression of these markers in the caudal ventral tissues of abnormal embryos at time points less than 24 hours (Fig. 6B,F). Rescue experiments included transplanting donor endoderm (either anterior or posterior) harvested from St. 17 embryos to the embryos in which the CIP endoderm had been removed. Only transplanted posterior endoderm rescued the tail and gut defect (Fig. 6G). Anterior endodermal transplants failed to rescue blunted tail phenotype (Fig. 6H). Our results show that the posterior endoderm produces signals necessary for normal tail and hindgut development.
HFGa13 interferes with the cellular functions of Hoxa13 and Hoxd13 proteins
To investigate the molecular pathway by which HFG Hoxa13 mutation functions, transactivation activities of wild-type Hoxa13 and HFGa13 were first analyzed in heterologous COS-7 cell line by luciferase assay using the synthetic GAL4 reporter system. Constructions for transfection studies were prepared using the pSG424 vector that contains the GAL4 DNA Binding Domain. Wild-type Hoxa13, HFGa13 and Hoxd13 cDNAs were subcloned into pSG424 vector in frame with the GAL4 DBD. We show that wild-type Hoxa13 protein fused to the GAL4 DNA-binding domain is able to activate transcription of this synthetic reporter (Fig. 7A). The HFGa13 construct is not able to activate the synthetic GAL4 promoter but we did notice a decrease in the basal activity using this construct. To verify expression and protein stability, we performed Western-blot analysis on whole COS-7 cell extracts of transfected cells using a specific GAL4 DBD antibody. We show that all these proteins were expressed at comparable levels as assayed by Western blotting, indicating that the inability to activate transcription is not linked to a lack of expression or instability of the HFGa13 proteins (data not shown). To determine whether a difference in intracellular localization of HFGa13 proteins could affect transcriptional activity, localization of N-flag-tagged proteins within transfected COS-7 cells was examined by immunostaining (Fig. 7C, D). Strong signals for wild type Hoxa13 and HFGa13 proteins were both observed in the nuclei of transfected COS-7 cells. However, a weak additional signal was detected in the cytoplasm with HFGa13.
Using the same synthetic GAL4 reporter system, we investigated the possible interactions between Hoxa13 and HFGa13. We used the same conditions with a plasmid containing Hoxa13 and a GAL4 DBD, but added one plasmid containing HFGa13 cDNA without GAL4 DBD (unable to bind the 5 GAL4 DBB repeats). Using this competition assay with same molar ratio we show that HFGa13 protein is able to decrease Hoxa13 transcriptional activity (Fig. 7B). Increased amounts of HFGa13 increases the strength of the repression. Interestingly, HFGa13 protein is also able to act in a dominant negative fashion with Hoxd13 by repressing Hoxd13 transcriptional activity (Fig. 7B).
In order to determine if HFGa13 acts as a dominant negative in vivo we used a competitive assay taking advantage of an epithelial phenotype alteration induced by overexpression of the wild-type Hoxa13 in the midgut. At E18, epithelium differentiation is nearly complete. Midgut epithelium is characterized by long and thin villi (Fig. 7E) whereas hindgut epithelium shows wide and flat villi (Fig. 7F). We have previously shown that ectopic Hoxd13 expression in the midgut mesoderm causes the midgut epithelium to develop with a hindgut/cloacal phenotype (Roberts et al, 1998). Herein we show that the same epithelial tranformation occurs with Hoxa13 misexpression in the midgut mesoderm (Figure 7H and J). However, infection of the midgut with HFGa13 did not transform the epithelium (Fig. 7G). If our HFGa13 acts as a dominant negative, then it should be able to repress the midgut epithelial transformation induced by ectopic Hoxa13 or Hoxd13 expressions in the midgut mesoderm. In order to test our hypothesis, we co-infected mesodermal midgut with RCAS(B)-HFGa13 and RCAS(A)-Hoxa13 or Hoxd13 retrovirus in equal titer. We used different RCAS envelope proteins to facilitate cellular co-infection as previously described (Morgan and Fekete 1996; Bendall et al., 1999). All these midgut were processed with 3c2 immunostaining to detect mesodermal virus expression (data not shown, Fig. 7L). In order to valid our co-infection experiments, we checked for viral co-expressions of both virus, and always were able to correlate viral mesodermal midgut infection (shown by 3c2 immunstaining) with ectopic co-expression of both HFGa13 and Hoxd13 (Fig. 7L). Co-infections of HFGa13 with Hoxa13 (Fig. 7I) or Hoxd13 (Fig. 7K) are sufficient to inhibit the action of Hoxa13 or Hoxd13 (compare respectively Fig. 7H with I, and Fig. 7J with K).
These experiments indicate that this HFG nonsense mutation likely functions as a dominant negative. HFGa13 may compete with the endogenous function of wild-type Hoxa13 and/or Hoxd13 proteins in vivo as a dominant-negative, like that observed with the human patient heterozygous for this mutation, probably by interfering with protein partners and/or transcriptional machinery.
DISCUSSION
It has been known for some time that there is a close association between the development of the gut and the tail or related structures (coccyx and sacral vertebrae). Human congenital malformations in one often affect the other systems. This association is seen in spontaneous and transgenic malformations in many vertebrate species (Maatman et al., 1997; Warot et al., 1997). The GGU and tail tissues derive from the tailbud mesenchyme proably via secondary body formation (Griffith et al., 1992). The factors involved in this process, if mutated, may affect multiple caudal structures. There is evidence to support this both experimentally and spontaneously produced in many vertebrate systems. When portions of the tailbud are removed early in chick development, tail truncations and cloacal anomalies are common (Schoenwolf, 1978). In some specific spontaneous murine mutants with tail truncation anomalies, GGU malformations are common, for example, Danforth’s Short Tail mutant (sd) develops anal stenosis, rectal duplications, anal atresias in association with the characteristic short tail (Dunn et al., 1940). In humans, the relationship between sacral and coccygeal vertebral defects and hindgut defects has also been documented (van der Putte, 1986) in sporadic/isolated malformations (eg. cloacal and bladder exstrophy (Loder and Dayioglu, 1990; Martinez-Frias et al., 2000)) and syndromic malformations (e.g. VATERCL syndrome that includes anal atresia and hemivertebrae (Beals and Rolfe, 1989). Another example is the limb/pelvis-hypoplasia/aplasia syndrome that includes absent fibulae, Müllerian aplasia, and sacral hypoplasia (Raas-Rothschild et al., 1988)). The elucidation of the mechanism of this association has been difficult, likely due to the complexity of the malformations as the malformations often involve anomalies of all three germ layers, deciphering the primary from the secondary effects in a transgenic model is troublesome.
Tail development is universal amongst vertebrates, although the persistence of a tail is not. Tail length is a function of the developmental time point that tailgut and VER apoptosis occurs. We suggest a functional relationship between the caudal endoderm and the VER. Gruneberg suggested that the origin of the VER is from the cloacal membrane (Gruneberg, 1956). Other experimental evidence supports the association between caudal endoderm and the VER. VER ablation causes a displacement of the tailgut dorsally due to increasing the number of cells between the VER and tailgut (Goldman et al., 2000). In murine chimeras derived from wild-type and sd mutant ES cells it was shown that the sd cells never populated the ventral hindgut or tailgut suggesting a ventral signal from the gut endoderm is absent in the sd mutant mouse (Maatman et al., 1997). It may be that the impairment of heterozygous and homozygous sd mutant cells to colonize the ventral hindgut endoderm is the earliest manifestation of the sd phenotype (Maatman et al., 1997).
The molecular controls of normal or abnormal tail/GGU are poorly understood but VER function and signals have recently been described (Goldman et al., 2000). Signals from specialized ectoderm in the limb (AER) and the tail (VER) direct elongation of their respective subjacent structures. The AER and VER do not appear to be functionally equivalent. In mice, both the AER and VER express a fibroblast growth factor and a bone morphogenic protein (Dudley and Tabin, 2000; Maatman et al., 1997). Although exogenous application of FGF or BMP to the AER rescues the limb phenotype in AER ablated embryos (Zuniga et al., 1999), these proteins when placed on VER ablated tails in vitro failed to rescue the blunted tail phenotype (Goldman et al., 2000). Transplanting the AER to VER ablated tails also fails in rescuing growth (Goldman et al., 2000). There are clearly other factors, either from the VER or other tail tissues that are important in directing tail development. We conclude that signaling between the endoderm and ectoderm at this very early stage of development is critical and independent of notochord or neural tube related inductions. We suggest that one of these factors is Hoxa13 derived from the caudal endoderm.
Clearly there are multiple factors involved in tail development. Many different model systems–genetic, mechanical, and toxic can result in the phenotype of blunted tail and cloacal anomalies. Classical anatomic literature has examples of toxic or pharmacological induction of blunted tail and ourenteric malformations in chick including exposure to insulin (Moseley, 1947), organophosphides (Wyttenbach and Thompson, 1985), and retinoic acid (Griffith and Wiley, 1989). Mechanical disruptions by transection or extirpation of the notochord, tailbud, and hindgut endoderm, shaking, or placement of a conductive glass tube all result in blunted tail and ourentery (Moseley 1947) and references therein, (Hotary and Robinson, 1992). The transgenic data in mice shows that pertubations in many different pathways result in blunted tails including FGF (Furthauer et al., 1997), BMP (Brunet et al., 1998), Wnt (Yamaguchi et al., 1999), and retinoic acid (Abu-Abed et al., 2001). What is common to these diverse “methods” of producing the combination of caudal tail/vertebrae and gut defects? We suggest one possibility may be interruption of caudal endoderm signaling needed for normal tail development.
A spontaneous genetically dominant chicken mutation resembles the phenotype of our HFGa13 embryos. Dominant Rumpless chicks develop a truncated tail, abnormal cloaca, and often show ourentery (Zwilling, 1942). It would be very interesting to determine if Hoxa13 is mutated or if abnormalities in this pathway are present in this strain. We are currently studying dominant rumpless chick embryos for Hoxa13 mutations.
In the human syndrome HFG, no sacral or coccygeal anomalies have been reported. In the murine counterpart, hd shows anal stenosis but not caudal vertebrae or tail defects (Post and Innis, 1999). Interestingly, caudal vertebrate malformations due to mutations in the paralog Hoxd13, though, have been reported both in human SPD syndrome (Akarsu et al., 1996) and in homozygous Hoxd13 knockout mice (Dolle et al., 1993). We show that our HFGa13 construct interferes with the normal expression and function of Hoxd13 (Figs. 5 and 7). Our HFGa13 phenotype may be in part an indirect phenomenon due to downregulation of Hoxd13.
It is curious that the human, chick, and murine phenotypes differ given the same genetic alteration. It may be that there are subtle vertebral defects in human individuals with HFG not described to date. Similarly, subtle murine lumbosacral or tail abnormalities may have escaped observation in the hd or Hoxa13−/− mice. Our findings in chick may relate to the particular Hoxa13 mutation we constructed or to the relative levels of wild-type and mutant proteins in the “transgenic” embryos. Or, it may be due to a different function of Hoxa13 in avian species in the posterior vertebrae compared with that of mouse and human.
A theory derived from our results suggests that the presence of a tail structure in a vertebrate species may be related to persistence of the tailgut during development. In humans and avians the tailgut undergoes apoptotic regression relatively early in development (Fallon and Simandl, 1978; Miller and Briglin, 1996), whereas in rodents the tailgut persists over a much longer relative developmental time period (Goldman et al., 2000). Although this study does not address the upstream controls of Hoxa13 expression in this caudal region, it follows that significant differences in this control would exists amongst species with different tail lengths.
Acknowledgments
We thank C. Nielsen, S. Winfield, and T. Manganaro for technical assistance. We thank C. Tabin and P. Donahoe for generous guidance, use of reagents, and expertise. We thank G. Schoenwolf, L. Pierro, and F. Goldman for helpful discussions and insights into our results. We are especially appreciative of direct guidance and sharing of equipment from D. Melton and A. Grapin-Botton. We are indebted to A. Kuroiwa and his laboratory for generous sharing of the precious Hoxa13 antibody. Thanks to S. Faure and A. de Santa Barbara for support. This work was supported by NIH grant HD34448-03 to D.J.R. to P.d.S.B. an ARC postdoctoral fellowship and an American Foundation of Urology Fellowship Grant.
References
- Abu-Abed S, Dolle P, Metzger D, Beckett B, Chambon P, Petkovich M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001;15:226–40. doi: 10.1101/gad.855001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akarsu AN, Stoilov I, Yilmaz E, Sayli BS, Sarfarazi M. Genomic structure of HOXD13 gene: a nine polyalanine duplication causes synpolydactyly in two unrelated families. Hum Mol Genet. 1996;5:945–52. doi: 10.1093/hmg/5.7.945. [DOI] [PubMed] [Google Scholar]
- Beals RK, Rolfe B. VATER association. A unifying concept of multiple anomalies. J Bone Joint Surg Am. 1989;71:948–50. [PubMed] [Google Scholar]
- Bendall AJ, Ding J, Hu G, Shen MM, Abate-Shen C. Msx1 antagonizes the myogenic activity of Pax3 in migrating limb muscle precursors. Development. 1999;126:4965–76. doi: 10.1242/dev.126.22.4965. [DOI] [PubMed] [Google Scholar]
- Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–7. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- Catala M, Teillet MA, Le Douarin NM. Organization and development of the tail bud analyzed with the quail-chick chimaera system. Mech Dev. 1995;51:51–65. doi: 10.1016/0925-4773(95)00350-a. [DOI] [PubMed] [Google Scholar]
- Chapman SC, Collignon J, Schoenwolf GC, Lumsden A. Improved method for chick whole-embryo culture using a filter paper carrier. Dev Dyn. 2001;220:284–9. doi: 10.1002/1097-0177(20010301)220:3<284::AID-DVDY1102>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Dolle P, Dierich A, LeMeur M, Schimmang T, Schuhbaur B, Chambon P, Duboule D. Disruption of the Hoxd-13 gene induces localized heterochrony leading to mice with neotenic limbs. Cell. 1993;75:431–41. doi: 10.1016/0092-8674(93)90378-4. [DOI] [PubMed] [Google Scholar]
- Dudley AT, Tabin CJ. Constructive antagonism in limb development. Curr Opin Genet Dev. 2000;10:387–92. doi: 10.1016/s0959-437x(00)00101-5. [DOI] [PubMed] [Google Scholar]
- Dunn L, Gluecksohn-Schoenheimer S, Bryson V. A new mutation in the mouse affection spinal column and urogenital system. J Hered. 1940;31:343–348. [Google Scholar]
- Fallon JF, Simandl BK. Evidence of a role for cell death in the disappearance of the embryonic human tail. Am J Anat. 1978;152:111–29. doi: 10.1002/aja.1001520108. [DOI] [PubMed] [Google Scholar]
- Fromental-Ramain C, Warot X, Messadecq N, LeMeur M, Dolle P, Chambon P. Hoxa-13 and Hoxd-13 play a crucial role in the patterning of the limb autopod. Development. 1996;122:2997–3011. doi: 10.1242/dev.122.10.2997. [DOI] [PubMed] [Google Scholar]
- Furthauer M, Thisse C, Thisse B. A role for FGF-8 in the dorsoventral patterning of the zebrafish gastrula. Development. 1997;124:4253–64. doi: 10.1242/dev.124.21.4253. [DOI] [PubMed] [Google Scholar]
- Gajovic S, Kostovic-Knezevic L, Svajger A. Morphological evidence for secondary formation of the tail gut in the rat embryo. Anat Embryol (Berl) 1993;187:291–7. doi: 10.1007/BF00195767. [DOI] [PubMed] [Google Scholar]
- Goff DJ, Tabin CJ. Analysis of Hoxd-13 and Hoxd-11 misexpression in chick limb buds reveals that Hox genes affect both bone condensation and growth. Development. 1997;124:627–36. doi: 10.1242/dev.124.3.627. [DOI] [PubMed] [Google Scholar]
- Goldman DC, Martin GR, Tarn PP. Fate and function of the ventral ectodermal ridge during mouse tail development. Development. 2000;127:2113–23. doi: 10.1242/dev.127.10.2113. [DOI] [PubMed] [Google Scholar]
- Goodman FR, Scambler PJ. Human HOX gene mutations. Clin Genet. 2001;59:1–11. doi: 10.1034/j.1399-0004.2001.590101.x. [DOI] [PubMed] [Google Scholar]
- Grapin-Botton A, Majithia AR, Melton DA. Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes Dev. 2001;15:444–54. doi: 10.1101/gad.846001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith CM, Wiley MJ. Direct effects of retinoic acid on the development of the tail bud in chick embryos. Teratology. 1989;39:261–75. doi: 10.1002/tera.1420390308. [DOI] [PubMed] [Google Scholar]
- Griffith CM, Wiley MJ, Sanders EJ. The vertebrate tail bud: three germ layers from one tissue. Anat Embryol. 1992;185:101–13. doi: 10.1007/BF00185911. [DOI] [PubMed] [Google Scholar]
- Gruneberg H. A ventral ectodermal ridge of the tail in mouse embryos. Nature. 1956;177:787–788. doi: 10.1038/177787b0. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morph. 1951;88:49–92. [PubMed] [Google Scholar]
- Holmdahl DE. Experimentelle untersuchungen uber die lage der grenze zwischen primarer and sekundarer korperentwicklung beium huhn. Anat Anz Bd. 1925;59:393–396. [Google Scholar]
- Hotary KB, Robinson KR. Evidence of a role for endogenous electrical fields in chick embryo development. Development. 1992;114:985–96. doi: 10.1242/dev.114.4.985. [DOI] [PubMed] [Google Scholar]
- Kluth D, Hillen M, Lambrecht W. The principles of normal and abnormal hindgut development. J Pediatr Surg. 1995;30:1143–7. doi: 10.1016/0022-3468(95)90007-1. [DOI] [PubMed] [Google Scholar]
- Knezevic V, De Santo R, Mackem S. Continuing organizer function during chick tail development. Development. 1998;125:1791–801. doi: 10.1242/dev.125.10.1791. [DOI] [PubMed] [Google Scholar]
- Kondo T, Dolle P, Zakany J, Duboule D. Function of Posterior HoxD genes in the morphogenesis of the anal sphincter. Development. 1996;122:2651–2659. doi: 10.1242/dev.122.9.2651. [DOI] [PubMed] [Google Scholar]
- Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- Loder RT, Dayioglu MM. Association of congenital vertebral malformations with bladder and cloacal exstrophy. J Pediatr Orthop. 1990;10:389–93. doi: 10.1097/01241398-199005000-00018. [DOI] [PubMed] [Google Scholar]
- Maatman R, Zachgo J, Gossler A. The Danforth’s short tail mutation acts cell autonomously in notochord cells and ventral hindgut endoderm. Development. 1997;124:4019–28. doi: 10.1242/dev.124.20.4019. [DOI] [PubMed] [Google Scholar]
- Martinez-Frias ML, Bermejo E, Rodriguez-Pinilla E. Anal atresia, vertebral, genital, and urinary tract anomalies: a primary polytopic developmental field defect identified through an epidemiological analysis of associations. Am J Med Genet. 2000;95:169–73. doi: 10.1002/1096-8628(20001113)95:2<169::aid-ajmg15>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Matsushita S. Fate mapping study of the endoderm in the posterior part of the 1.5-day- old chick embryo. Dev Growth Differ. 1999;41:313–9. doi: 10.1046/j.1440-169x.1999.413430.x. [DOI] [PubMed] [Google Scholar]
- Miller SA, Briglin A. Apoptosis removes chick embryo tail gut and remnant of the primitive streak. Dev Dyn. 1996;206:212–8. doi: 10.1002/(SICI)1097-0177(199606)206:2<212::AID-AJA10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Mills CL, Bellairs R. Mitosis and cell death in the tail of the chick embryo. Anat Embryol. 1989;180:301–8. doi: 10.1007/BF00315888. [DOI] [PubMed] [Google Scholar]
- Morgan BA, Fekete DM. Manipulating gene expression with replication-competent retroviruses. In: Bronner-Fraser M, editor. Methods in avian embryology. Vol. 51. San Diego: Academic Press; 1996. pp. 185–218. [DOI] [PubMed] [Google Scholar]
- Mortlock DP, Innis JW. Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet. 1997;15:179–80. doi: 10.1038/ng0297-179. [DOI] [PubMed] [Google Scholar]
- Mortlock DP, Post LC, Innis JW. The molecular basis of hypodactyly (Hd): a deletion in Hoxa 13 leads to arrest of digital arch formation. Nat Genet. 1996;13:284–9. doi: 10.1038/ng0796-284. [DOI] [PubMed] [Google Scholar]
- Mortlock DP, Sateesh P, Innis JW. Evolution of N-terminal sequences of the vertebrate HOXA13 protein. Mamm Genome. 2000;11:151–8. doi: 10.1007/s003350010029. [DOI] [PubMed] [Google Scholar]
- Moseley HR. Insulin-induced remplessness of chickens IV. Early embryology. J Exp Zool. 1947;105:279–316. doi: 10.1002/jez.1401050303. [DOI] [PubMed] [Google Scholar]
- Nelson CE, Morgan BA, Burke AC, Laufer E, DiMambro E, Murtaugh LC, Gonzales E, Tessarollo L, Parada LF, Tabin C. Analysis of Hox gene expression in the chick limb bud. Development. 1996;122:1449–66. doi: 10.1242/dev.122.5.1449. [DOI] [PubMed] [Google Scholar]
- Nielsen C, Murtaugh LC, Chyung JC, Lassar A, Roberts DJ. Gizzard formation and the role of Bapx1. Dev Biol. 2001;231:164–74. doi: 10.1006/dbio.2000.0151. [DOI] [PubMed] [Google Scholar]
- Pasteels J. Proliférations et croissance dans la gastrulation et la formation de la queue des vertébrés. Arch Biol (Liège) 1943;54:1–51. [Google Scholar]
- Podlasek CA, Duboule D, Bushman W. Male accessory sex organ morphogenesis is altered by loss of function of Hoxd-13. Dev Dyn. 1997;208:454–65. doi: 10.1002/(SICI)1097-0177(199704)208:4<454::AID-AJA2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Post LC, Innis JW. Infertility in adult hypodactyly mice is associated with hypoplasia of distal reproductive structures. Biol Reprod. 1999;61:1402–8. doi: 10.1095/biolreprod61.6.1402. [DOI] [PubMed] [Google Scholar]
- Raas-Rothschild A, Goodman RM, Meyer S, Katznelson MB, Winter ST, Gross E, Tamarkin M, Ben-Ami T, Nebel L, Mashiach S. Pathological features and prenatal diagnosis in the newly recognised limb/pelvis-hypoplasia/aplasia syndrome. J Med Genet. 1988;25:687–97. doi: 10.1136/jmg.25.10.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaud E. Etude embryologique de l’ourentérie et de la cordentérie. Journal de L’Anat et de la Physiol. 1900:619–634. [Google Scholar]
- Roberts DJ, Johnson RL, Burke AC, Nelson CE, Morgan BA, Tabin C. Sonic hedgehog is an endodermal signal inducing Bmp-4 and Hox genes during induction and regionalization of the chick hindgut. Development. 1995;121:3163–74. doi: 10.1242/dev.121.10.3163. [DOI] [PubMed] [Google Scholar]
- Roberts DJ, Smith DM, Goff DJ, Tabin CJ. Epithelial-mesenchymal signaling during the regionalization of the chick gut. Development. 1998;125:2791–801. doi: 10.1242/dev.125.15.2791. [DOI] [PubMed] [Google Scholar]
- Sadowski I, Ptashne M. A vector for expressing GAL4(1-147) fusions in mammalian cells. Nucleic Acids Res. 1989;17:7539. doi: 10.1093/nar/17.18.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders JW. The proximo-distal sequence of the origin of the parts of the chick wing and the role of the ectoderm. J Exp Zool. 1948;108:363–403. doi: 10.1002/jez.1401080304. [DOI] [PubMed] [Google Scholar]
- Schoenwolf GC. Tail (end) bud contributions to the posterior region of the chick embryo. J Exp Zool. 1977;201:227–246. [Google Scholar]
- Schoenwolf GC. Effects of complete tail bud extirpation on early development of the posterior region of the chick embryo. Anat Rec. 1978;192:289–95. doi: 10.1002/ar.1091920209. [DOI] [PubMed] [Google Scholar]
- Schoenwolf GC. Histological and ultrastructural observations of tail bud formation in the chick embryo. Anat Rec. 1979;193:131–47. doi: 10.1002/ar.1091930108. [DOI] [PubMed] [Google Scholar]
- Smith DM, Nielsen C, Tabin CJ, Roberts DJ. Roles of BMP signaling and Nkx2.5 in patterning at the chick midgut- foregut boundary. Development. 2000;127:3671–3681. doi: 10.1242/dev.127.17.3671. [DOI] [PubMed] [Google Scholar]
- van der Putte SC. Normal and abnormal development of the anorectum. J Pediatr Surg. 1986;21:434–40. doi: 10.1016/s0022-3468(86)80515-2. [DOI] [PubMed] [Google Scholar]
- Vogel A, Rodriguea C, Izpisua-Belmonte JC. Involvement of FGF-8 in initiation, outgrowth and patterning of the vertebrate limg. Development. 1996;122:1737–1750. doi: 10.1242/dev.122.6.1737. [DOI] [PubMed] [Google Scholar]
- Warot X, Fromental-Ramain C, Fraulob V, Chambon P, Dolle P. Gene dosage-dependent effects of the Hoxa-13 and Hoxd-13 mutations on morphogenesis of the terminal parts of the digestive and urogenital tracts. Development. 1997;124:4781–91. doi: 10.1242/dev.124.23.4781. [DOI] [PubMed] [Google Scholar]
- Wyttenbach CR, Thompson SC. The effects of the organophosphate insecticide malathion on very young chick embryos: malformations detected by histological examination. Am J Anat. 1985;174:187–202. doi: 10.1002/aja.1001740208. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Bradley A, McMahon AP, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–23. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- Yokouchi Y, Nakazato S, Yamamoto M, Goto Y, Kameda T, Iba H, Kuroiwa A. Misexpression of Hoxa-13 induces cartilage homeotic transformation and changes cell adhesiveness in chick limb buds. Genes Dev. 1995a;9:2509–22. doi: 10.1101/gad.9.20.2509. [DOI] [PubMed] [Google Scholar]
- Yokouchi Y, Sakiyama J, Kuroiwa A. Coordinated expression of Abd-B subfamily genes of the HoxA cluster in the developing digestive tract of chick embryo. Dev Biol. 1995b;169:76–89. doi: 10.1006/dbio.1995.1128. [DOI] [PubMed] [Google Scholar]
- Zuniga A, Haramis AP, McMahon AP, Zeller R. Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature. 1999;401:598–602. doi: 10.1038/44157. [DOI] [PubMed] [Google Scholar]
- Zwilling E. The development of dominant rumplessness in chick embryos. Genetics. 1942;27:641–656. doi: 10.1093/genetics/27.6.641. [DOI] [PMC free article] [PubMed] [Google Scholar]