

Abstract
During pancreatic development insulin+ cells co-express the transcription factors MafB and Pax6, and transition from a MafA− to MafA+ state. To examine the role of Pax6 and MafB in the development of β cells, we analyzed embryonic pancreata from Pax6- and MafB- deficient mice. Pax6 deficiency, as manifest in the Pax6Sey-Neu allele, reduced not only the number of cells expressing insulin or glucagon, but also the number of MafB, PDX-1 and MafA expressing cells. We show that MafB can directly activate expression of insulin and glucagon, and a MafB protein engineered to contain N248S mutation in the MafB (krENU) results in significantly reduced activation. Furthermore, pancreata from MafB deficient (krENU/krENU) mice exhibited reduced number of cells expressing insulin, glucagon, PDX-1 and MafA, with only a minor reduction in MafB expressing cells. MafB deficiency does not affect endocrine specification but does affect the lineage commitment of the endocrine cells and their maturation. Similar to Pax6 deficient mice, MafB deficient mice showed reductions both in insulin and glucagon expressing cells and in the ability of MafB and PDX-1 expressing cells to activate expression of these hormones. However, MafB deficient mice exhibited no effect on Pax6 expression. These results suggest that MafB may function as a downstream mediator of Pax6 in regulating the specification of insulin and glucagon expressing cells. Interestingly, the remaining insulin+ cells in these knockouts preferentially express Hb9, suggesting the existence of an alternate pathway for the generation of insulin expressing cells, even in the absence of Pax6 and MafB function. Thus, Pax6 acts upstream of MafB, which in turn may trigger the expression of insulin and regulate the PDX-1 and MafA expression required for β-cell maturation.
Keywords: MafB, MafA, Pax6, Sey, Kreisler, insulin gene transcription factor, pancreatic development, endocrine differentiation, pancreatic islets
Introduction
To develop reliable sources of glucose-responsive β-cells for the treatment of diabetes, it is essential to understand how β-cells form during development. The pancreas develops as a dorsal and ventral evagination from the developing foregut that expresses the homeodomain transcription factor PDX-1 (Edlund, 2002; Leonard et al., 1993; Miller et al., 1994; Ohlsson et al., 1993). PDX-1 is essential for the normal proliferation and differentiation of embryonic pancreatic precursors (Edlund, 2002; Jonsson et al., 1994; Offield et al., 1996). Another transcription factor, Ngn3, regulates the differentiation of endocrine cells (Gradwohl et al., 2000). In addition, several other transcription factors play critical roles in both pancreatic development and endocrine differentiation (Collombat et al., 2006; Grapin-Botton and Melton, 2000; Jensen, 2004; Kim and MacDonald, 2002; Murtaugh, 2007). For example, transcription factors Arx and Pax4 play critical roles in regulating specification of endocrine progenitors towards α- or β- and δ-cell fates (Collombat et al., 2005; Collombat et al., 2006; Collombat et al., 2007). Pax6 is required for maintaining these hormone-expressing cells and for regulating specification of ghrelin-expressing ε-cells (Ashery-Padan et al., 2004; Heller et al., 2005; Wang et al., 2004). In addition, members of the Nkx and Maf families regulate the differentiation of hormone positive cells.
The identification of MafA as a β-cell specific basic leucine zipper (bZIP) transcription factor has initiated the systematic characterization of Maf factors in pancreatic development and in β–cell function (Olbrot et al., 2002; Kataoka et al., 2002; Matsuoka et al., 2003). MafA (formerly known as RIPE3b1) is a glucose-responsive insulin gene transcription factor that binds to the insulin Maf responsive element (MARE) and activates insulin gene expression (Sharma and Stein, 1994; Olbrot et al., 2002; Kataoka et al., 2002; Matsuoka et al., 2003). Another Maf factor, MafB [also known as Kreisler (Krml1/MafB)], is expressed in pancreatic endocrine cells and regulates differentiation of several cell types (Eichmann et al., 1997; Manzanares et al., 1997; Moriguchi et al., 2006; Sieweke et al., 1996). The gene was first identified as the locus mutated in the Kreisler (kr) mouse, and a second ethylnitrosourea (ENU) induced mutation [krENU (also known as MafBENU)] in this gene has been described (Cordes and Barsh, 1994). MafB, expressed only in α-cells in adults, is expressed in the insulin positive cells during pancreatic development (Artner et al., 2006; Nishimura et al., 2006). We proposed that endocrine progenitors after committing to the insulin positive lineage continue a further differentiation/maturation process. Early insulin+ cells express MafB, and following the induction of PDX-1, mature into insulin+ and MafA+ cells (Nishimura et al., 2006). MafA deficient Mice have normal pancreatic islets at birth, but the ratio of β to α cells gradually reduce after birth, resulting in glucose intolerance by 8–12 weeks (Zhang et al., 2005). This phenotype suggests a critical role of MafA in the maturation step required for the function and survival of β cells. The importance of this maturation step is highlighted by a recent publication on the differentiation of human ES cells into insulin+ cells (D’Amour et al., 2006). Although these insulin+ cells expressed MafB, they did not maintain the expression of PDX-1 and did not secrete insulin in response to glucose (D’Amour et al., 2006). We hypothesize that these cells may not have switched from MafB+MafA− to MafB−MafA+ state. It is likely that the lack of MafA expression prevents early insulin+ cells from expressing the downstream targets of MafA that are required for the insulin synthesis and secretion (Kato et al., 2006; Wang et al., 2007). Thus, elucidation of the mechanisms that regulate the conversion of insulin+ cells into mature functional β-cells should facilitate our ability to generate glucose-responsive, insulin-producing cells for cell-based diabetes therapy.
In the present study, we characterized the roles of Pax6 and MafB in the differentiation of insulin expressing cells. Loss of either Pax6 or MafB function results in reduced numbers of cells that express insulin, glucagon, PDX-1 or MafA, and decreases the ability of MafB+ and PDX-1+ cells to express insulin. Our results suggest that MafB may function as a downstream mediator of Pax6 in regulating the formation of insulin and glucagon positive cells. We present data supporting the initiation of insulin expression in the endocrine progenitors by at least two pathways: the formation of insulin-expressing cells from one pathway requires the function of Pax6 and MafB, while specification of the remaining insulin-expressing cells may occurs via a Pax4-Hb9 dependent pathway. These results suggest that MafB can regulate the initiation of insulin expression in some, but not all, endocrine progenitors destined to express insulin.
Material and methods
Construction of expression vectors
Various expression vectors were constructed by conventional molecular biology techniques. Insulin promoter luciferase reporter constructs −238 WT LUC and its derivative mutant 121–22m LUC have been described previously (Harrington and Sharma, 2001). The glucagon promoter luciferase reporter GLU LUC was kindly provided by Dr. Dan Drucker (Toronto, Canada); the PDX-1 promoter luciferase reporter (Eto et al., 2007), by Dr. Melissa Thomas (Massachusetts General Hospital, Boston, MA). The expression vectors pcDNA MafA, pCMVSport6 MafB and pcDNA cMaf have been described previously (Nishimura et al., 2006). pCMVSport6 krENU expression vector was generated by PCR amplification of homozygous krENU mouse genomic DNA with oligonucleotide primers (5′-GACCCGCCAGGACTCACAGAAA-3′and 5′-CCGCGCAACAGCTACCCACTA-3′) for 35 cycles of 30 sec at 94.0C, 40 sec at 62.3C and 1 min at 72C. PCR product was digested with AflII and PvuII and was used to replace the corresponding fragment from pCMVSport6 MafB to give full-length krENU cDNA containing a point mutation at base pair 743 in the DNA binding region of MafB. Constructs were confirmed by sequencing.
Luciferase Assays
HeLa cells were transfected with 1μg of reporter constructs of −238 WT LUC, 121–22m LUC, GLU LUC or PDX-1 promoter, 1 μg of pSVβ-gal plasmid (Promega, Madison, WI) as an internal control and 1 μg of indicated expression vector. Whole cell extracts were prepared and luciferase activity was measured as previously described (Nishimura et al., 2005).
Animals
Mice with mutation in kreisler/MafB generated by ethylnitrosourea (ENU) induced chemical mutagenesis have been reported earlier (Cordes and Barsh, 1994). Mutagenesis results in a point mutation in the DNA-binding region of MafB that changes amino acid 248 from asparagine to serine (krENU or MafBENU) leading to compromised DNA binding affinity of krENU to Maf Response Element (MARE)(Sadl et al., 2002). Heterozygous mice with the ENU-induced krENU allele were received from Dr. Greg Barsh (Stanford, CA), were maintained on C57Bl/6J background for more than 10 generation and used in this study. Small eye mutant (Pax6Sey-Neu) mice (Glaser et al., 1994; Hill et al., 1991; Sander et al., 1997), also generated by chemical mutagenesis, have a point mutation in a splice donor site of 3′ end of the homeobox that results in incorrect splicing and truncated Pax6 protein lacking the carboxy-terminal 115 amino acids corresponding to the transcriptional activation domain. The day of vaginal-plug discovery was designated as embryonic day E 0.5.
Immunohistochemistry and quantification
Immunohistochemical and immunofluorescent analyses were performed as described previously (Nishimura et al., 2006). The primary antibodies used were: guinea pig anti-insulin (Linco, Billerica, MA); guinea pig anti-glucagon (Linco); rabbit anti-somatostatin (Santa Cruz Biotechnology, CA); rabbit anti-pancreatic polypeptide (Linco); rabbit anti-Ghrelin (Phoenix Pharmaceuticals, Belmont, CA); rabbit Hb9 antibody (provided by Dr. S. Pfaff); rabbit anti-MafB (Bethyl Laboratories, Montgomery, TX); mouse Nkx2.2 (Developmental Studies Hybridoma Bank, Iowa City, IA); rabbit anti-Nkx6.1 (provided by Dr. P. Serup); mouse Neurogenin3 (provided by Drs. Serup and Madsen, Beta Cell Biology Consortium); goat anti-PDX-1 (provided by Dr. C. Wright); rabbit anti-Pax6 (Covance, Princeton, NJ). Rabbit anti-MafA antibody was described previously (Nishimura et al., 2006). For amplification, biotinylated anti-rabbit, anti-mouse or anti-goat antibodies (Jackson ImmunoResearch, West Grove, PA) were used at a 1:400 dilution followed by streptavidin-conjugated Texas red (1:400) or streptavidin-conjugated Alexa fluor 488 (1:400) (Molecular Probes, Eugene, OR). The secondary antibodies were: FITC- or Texas red-conjugated anti-rabbit, anti-mouse or anti-guinea pig (Jackson ImmunoResearch). Nuclear counterstaining was performed by DAPI mounting medium (Vector, Burlingame, CA). For the double staining using rabbit anti-MafB and rabbit anti-Ghrelin antibodies, pancreatic sections were first stained with rabbit anti-MafB antibody using secondary biotinylated anti-rabbit antibody followed by streptavidin-conjugated Alexa flour 488, and images of areas expressing MafB were taken. The same imaged sections were next stained with rabbit anti-Ghrelin antibody and Texas red conjugated anti-rabbit secondary antibody and images of the same areas were taken again to examine co-expression of MafB and Ghrelin. The results of immunohistochemistry and quantification were derived from several replicates of at least three embryonic pancreases for each genotype. Every 10th section of embryonic pancreas was analyzed for immunofluorescent hormone-positive areas as a proportion of epithelial area. For total number of transcription factor+ cells (for Figure 2A), every 10th section was stained with DAB and counted. For number of transcription factor+ insulin+ cells per total number of indicated transcription factor+ cells or total number of insulin+ cells, cells were counted from at least three different pancreatic blocks. Immunofluorescence data were collected as confocal images on Zeiss LSM410 (Zeiss, Thornwood, NY); all DAB-stained images were analyzed on an Olympus BH2 bright field microscope. Images were quantitated using NIH Image J software.
Figure 2. Pax6-deficiency reduces the proportion of MafB+ and PDX-1+ cells that express insulin.

Quantification of immunostained E15.5 embryonic pancreatic sections from wild type and SeyNeu/SeyNeu embryos (n = 6 each). (A) Total number of Nkx6.1+, MafB+ and MafA+ cells in wild type and SeyNeu/SeyNeu embryonic pancreas were determined along with the number of insulin+ cells in the same sections. In SeyNeu/SeyNeu embryonic pancreas, the numbers of MafB+, MafA+ and insulin+ cells were significantly reduced (p = 0.03, 0.01 and 0.01 respectively), while the number of Nkx6.1+ cells remained unchanged (p = 0.46). (B) The ability of transcription factor expressing cells to express insulin was determined by quantifying the proportion of transcription factor+ insulin+ cells per total number of transcription factor+ cells, as represented by cells in green circle in adjacent Venn diagram. Proportions of MafB+Ins+ cells / total MafB+ cells and PDX-1+Ins+ cells / total PDX-1+ cells in SeyNeu/SeyNeu pancreas were significantly less than the wild-type (p = 0.005 and 0.01 respectively), while all of the MafA+ cells express insulin. (C) To determine the ability of insulin+ cells to undergo further maturation in the absence of Pax6 function, the proportion of insulin+ cells expressing these transcription factors was quantified, as represented by cells in red circle in adjacent Venn diagram. The proportion of MafB+ Ins+, PDX-1+Ins+ and MafA+Ins+ cells to the total Ins+ cells was not significantly different between wild type and SeyNeu/SeyNeu mice.
Western blot analysis
Cell lysates used in luciferase assays were subjected to 10% SDS-PAGE and gels were transferred to polyvinyldenedifluoride membrane filters (BioRad Laboratories, Hercules, CA). Western bolt analyses were performed using MafB and β-actin antibody (Santa Cruz Biotechnology, CA) and detected with horseradish peroxidase using ECL reagents (Amersham).
Results
Pax6 deficiency results in reduced numbers of MafA, MafB and PDX-1 expressing cells
Previously, we showed that Pax6+ insulin+ cells transition from a MafB+ MafA− stage to a mature MafA+ MafB− phenotype (Nishimura et al., 2006). An association between Pax6 and Maf factors has been reported in different systems (Reza et al., 2002; Reza and Yasuda, 2004). Pax6 deficiency results in the inability of endocrine cells to express markers of terminal differentiation and maturation (Ashery-Padan et al., 2004). Hence, we examined the role of Pax6 in regulating the expression and function of Maf factors during pancreatic development. E15.5 Pax6Sey-Neu/Sey-Neu (subsequently referred as SeyNeu/SeyNeu) pancreata completely lack Pax6 expressing cells (antibody recognizes C-terminal domain of Pax6), while wild type littermates have significant numbers of Pax6+ cells, some of which co-expressed insulin (Suppl. Figure 1). As reported earlier (Sander et al., 1997; Wang et al., 2004), loss of Pax6 results in a significant reduction in insulin and glucagon-expressing cells but does not affect the pancreatic expression of Nkx transcription factors at E15.5. Nkx6.1 expression in the pancreas marks the area of endocrine differentiation and, as shown in Suppl. Figure 1, was unaffected by loss of Pax6 function. Similarly, expression of the pro-endocrine gene Ngn3 was not affected in Pax6 deficient mice (Suppl. Figure 1), consistent with the earlier observation (Ashery-Padan et al., 2004; Wang et al., 2004) that Pax6 is not required for the initiation of endocrine differentiation.
We next examined whether the loss of Pax6 affected the expression of Maf factors. The number of cells in Pax6-deficient (SeyNeu/SeyNeu) E15.5 pancreas expressing MafB, PDX-1 (induced PDX-1 expression during secondary transition or PDX-1high) and MafA (Figures 1B–D, F–H) were reduced. Some cells still expressed these transcription factors demonstrating the presence of a Pax6 independent mechanism for their expression. The number of insulin+ cells expressing these three transcription factors was significantly reduced in SeyNeu/SeyNeu mice (Figures 1B–D, F–H). Thus, the loss of Pax6 function not only reduces the expression of terminal hormone markers but also reduces the expression of the transcription factors implicated in the terminal differentiation process.
Figure 1. SeyNeu/SeyNeu embryos have reduced expression MafB, PDX-1 and MafA.
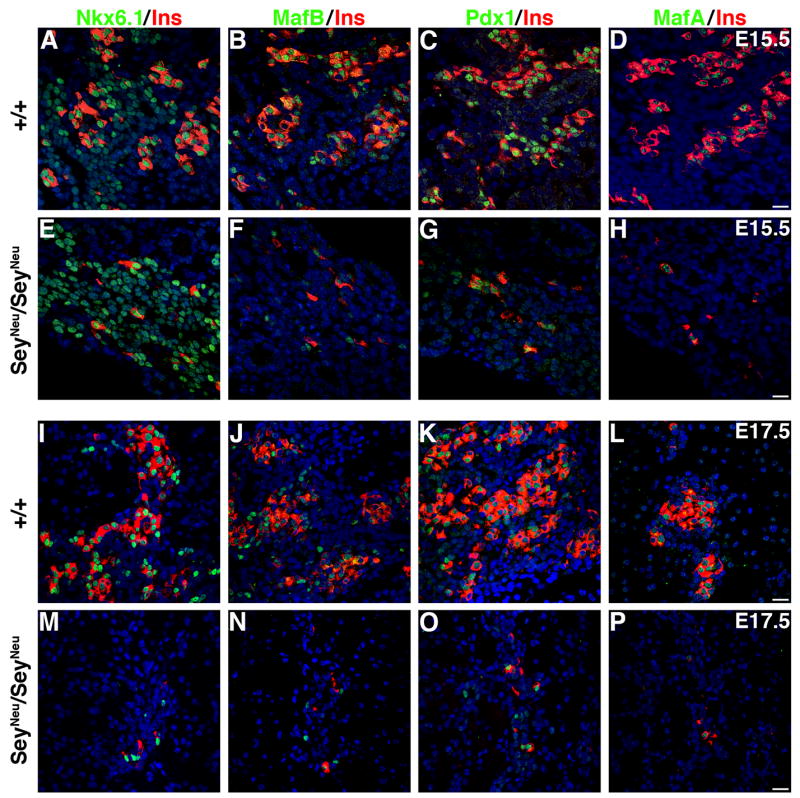
Adjacent sections from E15.5 wild type (A–D) and SeyNeu/SeyNeu (E–H) embryonic pancreata (n = 6 each) were stained to detect the expression of Nkx6.1, MafB, PDX-1 and MafA in green and insulin in red. In wild type pancreas adjacent sections show cells expressing Nkx6.1, MafB, PDX-1 and MafA, but in SeyNeu/SeyNeu embryos MafB, PDX-1 and MafA expression was significantly reduced. E17.5 pancreata from wild type (I–L) and SeyNeu/SeyNeu (M–P) embryos (n = 3 each) were stained for Nkx6.1, MafB, PDX-1 and MafA in green and insulin in red. At this stage loss of Pax6 function results in significant reduction in the number of insulin+ cells, and the majority of Nkx6.1+, MafB+, PDX-1high and MafA+ cells are restricted to these insulin+ cells. Bars: 20μm.
We next examined whether expression of MafB, PDX-1high and MafA were maintained in insulin+ cells in the absence of Pax6 function (Figures 1J–L, N–P). As reported previously (Sander et al., 1997), at E17.5 the number of insulin+ cells remained low in the SeyNeu/SeyNeu embryos. Additionally, MafA and PDX-1 expression was restricted to insulin+ cells, and the number of cells expressing these transcription factors was reduced (Figures 1K, L, O, P); most Nkx6.1+ cells expressed insulin, with only occasional Nkx6.1+ insulin− cells (Figures 1I, M). We also observed a greater reduction in insulin+ and MafB+ cells in the SeyNeu/SeyNeu embryos at E17.5 (Figure 1N) than at E15.5 (Figure 1F).
Pax6-deficiency reduces the proportion of MafB+ and PDX-1 high cells expressing insulin
Our observation that MafB+ pancreatic cells are present in the SeyNeu/SeyNeu embryos (Figures 1F, N) is consistent with the conclusion of Artner and colleagues (Artner et al., 2006) that Pax6 and Pax4 are not essential for MafB expression. To determine whether Pax6 deficiency reduces only the number of cells expressing MafB, PDX-1 and MafA or has additional effects on their function, we quantified stained E15.5 pancreatic sections (Figure 2). The number of MafB+ cells was significantly reduced in SeyNeu/SeyNeu pancreas (Figures 1F, 2A). While the number of Nkx6.1+ cells did not change, those of insulin+, MafB+ and MafA+ cells were reduced by about 75% (Figures 2A), clearly demonstrating that loss of Pax6 function significantly affects MafB+ and MafA+ cells.
Since Pax6 is not required for the specification of endocrine cells but it regulates their ability to acquire final differentiation markers (Suppl. Figure 1, Ashery-Padan et al., 2004; Heller et al., 2005; Wang et al., 2004), we analyzed in E15.5 pancreas whether the loss of Pax6 expression affected 1) the ability of MafB+, PDX-1high and MafA+ cells to express insulin and 2) the ability of remaining insulin+ cells to express normal levels of these factors. MafA was expressed only in insulin+ cells, while several MafB+ and PDX-1high cells did not express insulin. Similarly, several insulin+ cells did not express these transcription factors (Figures 1). Quantification was performed to determine the proportion of transcription factor+ cells expressing insulin (Figure 2B) and the proportion of insulin+ cells expressing different transcription factors (Figure 2C), as depicted in the Venn diagrams. Nearly 65–70% of MafB+ cells (390 MafB+, 273 Ins+ and 259 MafB+Ins+ cells; 259 out of 390), 75–80% of PDX-1high cells (346 PDX-1high, 324 Ins+ and 276 PDX1highIns+ cells) and all MafA+ cells (179 MafA+, 268 Ins+ and 179 MafA+Ins+ cells) expressed insulin in wild type pancreas whereas loss of Pax6 function significantly reduced the ability of MafB+ (~35%)(125 MafB+, 58 Ins+ and 49 MafB+Ins+ cells; 49 out of 125) and PDX-1high (~45%)(82 PDX-1high, 58 Ins+ and 49 PDX1highIns+ cells) cells to co-express insulin. Importantly, while the number of insulin+ cells was reduced in SeyNeu/SeyNeu pancreas, there was no change in the proportion of MafA+ cells expressing insulin (100%)(50 MafA+, 81 Ins+ and 50 MafA+Ins+ cells), which suggests that MafA is expressed only after insulin+ cells have reached a latter maturation stage. Next, we quantified the proportion of insulin+ cells expressing these transcription factors (Figure 2C). Unlike the reduction in the proportion of MafB+ and PDX-1high cells expressing insulin in the absence of Pax6, the proportion of insulin+ cells expressing these different transcription factors in wild type and SeyNeu/SeyNeu mice remained unchanged. Nearly 95%, 75% and 65% of insulin+ cells were MafB+, PDX-1high and MafA+, respectively, in both wild type and Pax6 deficient mice (Figure 2C). This observation suggests that the remaining insulin+ cells derived from a Pax6 independent pathway appear to undergo normal maturation.
Pax6 deficiency reduces the proportion of MafB+ cells expressing glucagon
Since Pax6 deficiency reduced the number of MafB+ cells that co-express insulin, we also used SeyNeu/SeyNeu mice to examine whether the MafB+ cells that did not express insulin instead expressed glucagon. At E12.5, Nkx6.1 and PDX-1, two transcription factors that mark the developing pancreatic epithelium at this stage, are not expressed in the majority of glucagon+ cells in either wild type or SeyNeu/SeyNeu pancreas (Figures 3A, B, E, F). It is important to note that at E12.5, PDX-1 expression was not affected by Pax6 loss of function. Thus, the reduced PDX-1 in older SeyNeu/SeyNeu embryos (Figures 1, 2) may reflect the ability of Pax6 to selectively regulate the induction of PDX-1 expression (PDX-1high) in insulin+ cells during secondary transition.
Figure 3. Pax6-deficiency reduces the proportion of MafB+ cells expressing glucagon.

Adjacent sections from E12.5 wild type (A–C) and SeyNeu/SeyNeu (E–G) (n = 5 wild type, 4 mutant) pancreata were stained to detect Nkx6.1, PDX-1 and MafB expression in green and glucagon in red. In addition, sections from E15.5 embryonic pancreata (D, H) were stained for MafB in green and glucagon in red. In E12.5 SeyNeu/SeyNeu pancreata Nkx6.1 and PDX-1 expression was normal, while the expression of MafB and glucagon expression was reduced at both E12.5 and E15.5. Arrow denotes MafB+glucagon+ cells in SeyNeu/SeyNeu pancreas at E12.5. Bars: 20μm. (I) Proportion of MafB+glucagon+ cells to total number of MafB+ or glucagon+ cells were quantified from wild type and SeyNeu/SeyNeu pancreatic sections from E12.5 and E15.5. Absence of Pax6 function reduces the proportion of glucagon+ cells that express MafB at both E12.5 and E15.5, while the proportion of MafB+ cells expressing glucagon is reduced only in E15.5 pancreata.
The quantification of glucagon+ and MafB+ cells at E12.5 and 15.5 provides important insights into the roles of Pax6 and MafB in the induction of hormone expression and in the differentiation of early endocrine cells. At E12.5 in wild type pancreas, the majority of MafB+ cells co-express glucagon, and conversely the majority of glucagon+ cells are MafB+ (260 MafB+, 251 Glu+ and 239 MafB+Glu+ cells). The number of MafB+ cells at E12.5 in SeyNeu/SeyNeu pancreata was extremely low, but all MafB+ cells co-expressed glucagon (7 MafB+, 37 Glu+ and 7 MafB+Glu+ cells), and a number of glucagon+ cells did not express MafB (Figure 3G). At E15.5, with the induction of insulin expression, nearly 40% MafB+ cells expressed glucagon in wild type (500 MafB+, 204 Glu+ and 198 MafB+Glu+ cells), and this number decreased to less than 10% in SeyNeu/SeyNeu pancreata (216 MafB+, 48 Glu+ and 15 MafB+Glu+ cells) (Figure 3I). Nearly 65% of MafB+ cells at E15.5 express insulin in wild type pancreas (Figure 2B), suggesting that most of the MafB+ cells at E15.5 in wild type either express insulin or glucagon. Together, these results (Figures 2, 3) suggest increased numbers of MafB+ cells that do not express either insulin or glucagon in SeyNeu/SeyNeu pancreas at E15.5. Deficiency of Pax6 (Heller et al 2005) and Nkx2.2 (possibly via reducing Pax6 expression) (Prado et al 2004) changes cell-fate of endocrine precursors to ghrelin expressing ε-cells at the expense of the other endocrine cell types. We observed a similar increase in ghrelin only expressing ε-cells in Pax6 deficient mice, but the ghrelin-expressing ε-cells did not express MafB (Suppl. Figure 2).
MafB can activate insulin and glucagon gene expression
To determine whether MafB itself plays an important role in the differentiation of endocrine cells, we first examined the ability of ENU induced N248S mutation in the MafB coding region (krENU) to activate the insulin and glucagon promoters. Two plasmid constructs were made from wild-type MafB plasmid (pCMV Sport MafB), one carrying the ENU induced N248S mutation (krENU) and another lacking the C-terminal DNA binding and bZIP domain (ΔCMafB) (Figure 4A). All three plasmids transfected into HeLa cells expressed a protein recognized by anti- MafB antibody in Western blots (Figure 4B). Quantification with normalization to β-actin (Figure 4C) indicated that krENU is expressed at a level slightly higher than the wild-type MafB protein. Co-transfection of these plasmids with insulin (Figure 4D) or glucagon (Figure 4E) promoter luciferase reporter constructs demonstrated that wild type MafB activated both insulin and glucagon promoters, as previously reported (Nishimura et al., 2006; Zhao et al., 2005). ΔCMafB was ineffective in inducing expression of these genes, and krENU showed significant impairment in its ability to activate insulin and glucagon gene expression (Figures 4D, E). Co-transfecting these expression plasmids with the insulin reporter construct -122.121m Luc (Harrington and Sharma, 2001) containing a mutated insulin MARE significantly reduced the MafB and krENU mediated activation of luciferase, suggesting that krENU retains some residual activity to regulate insulin expression via its cognate binding site. Thus, in principal, krENU mice should represent a model of MafB deficiency, but not absence, of MafB function.
Figure 4. The krENU allele encodes a reduced ability to activate insulin and glucagon gene expression.

(A) Schematics of wild type MafB and its krENU mutant and deletion derivatives showing activation and DNA binding domains. (B, C) Western blot analyses to detect MafB protein expression from various MafB constructs. Lysates used in transient transfection assays in (D) and (E) were subjected to Western blot to detect expression of MafB (B) and β-actin (C). (D) Insulin promoter:Luciferase reporter constructs, wild type (−238 WT LUC, red) and insulin promoter with mutation in −121−122bp (−121−122m LUC, pink), and (E) glucagon promoter:Luciferase reporter constructs (GLU LUC, green) were transfected into HeLa cells with the indicated expression plasmids and pSVβ-gal as an internal control. Luciferase and β-gal activities were determined. Results are presented relative to activity of wild-type luciferase construct ± S.E. (n = 4)
MafB deficiency results in reduction of insulin+ and glucagon+ cells
In adult mouse pancreas MafB expression is restricted to α-cells, but during embryonic development MafB expression is also seen in insulin+ cells (Nishimura et al., 2006). Since our results indicate a correlation between reduced MafB expression and the number of insulin+ and glucagon+ cells in SeyNeu/SeyNeu mice (Figures 1–3), we hypothesized that MafB might regulate the differentiation of both α- and β-cells. To test this hypothesis we examined the effect of MafB deficiency on endocrine differentiation using krENU/krENU mice. At E15.5, MafB deficiency did not affect pancreatic appearance but insulin+ and glucagon+ cells were drastically reduced (Figure 5A, B). As a percentage of total epithelial area, insulin+ and glucagon+ area were reduced to nearly 55% and 60%, respectively (Figures 5C, D). Thus, as in SeyNeu/SeyNeu mice, MafB deficiency results in a reduction, but not complete loss, of insulin+ and glucagon+ cells.
Figure 5. MafB deficiency reduces insulin+ and glucagon+ cells.

(A, B) Pancreas from E15.5 wild type (+/+) and MafB mutant (krENU/krENU) (n = 4 wild type, 4 mutant) embryos were stained for glucagon in green and insulin in red. Immunostained sections were used to quantify the proportion of insulin (C) and glucagon (D) positive areas per pancreatic epithelial area as described in Materials and Method. Bars: 20μm.
MafB deficiency does not affect the initiation of endocrine differentiation
We next examined the mechanism underlying the reduced number of insulin+ and glucagon+ cells in krENU/krENU mice. During pancreatic development, the transcription factor Ngn3 marks the progenitors that give rise to endocrine cells, while Pax6 marks cells further along the endocrine differentiation pathway. Nkx2.2 is expressed in early epithelial progenitors and subsequently becomes restricted to endocrine cells. Pancreatic sections from E15.5 wild type and krENU/krENU mice were immunostained for Ngn3, Nkx2.2 and Pax6 (Figures 6). Although krENU/krENU mice have reduced insulin+ cells, the number of Ngn3+ cells was unaffected (Figures 6A, D), suggesting that MafB deficiency does not affect the specification of endocrine differentiation. Similarly, MafB deficiency did not affect Nkx2.2+ or Pax6+ cell number (Figures 6B, C, E, F). These results show that the krENU mutation does not affect pancreatic precursors or formation of endocrine cells and that the reduction in the number of insulin+ and glucagon+ cells (Figures 5 and 6) occurs in the absence of any change in the levels of Pax6+ cells (Figures 6C, F).
Figure 6. MafB deficiency does not inhibit the initiation of endocrine differentiation.

Sections from E15.5 pancreata from wild type and krENU/krENU embryos were stained for Ngn3, Nkx2.2 and Pax6 in green and insulin in red. Expression of insulin was reduced in krENU/krENU pancreata, but Ngn3, Nkx2.2 and Pax6 expression was similar to that in the wild type embryos. Bars: 20μm.
Effect of MafB deficiency on MafB+, PDX-1high and MafA+ cells
Whether the homozygous krENU mutation affected the maturation of insulin+ cells was addressed next. Since the krENU allele expresses a mutant protein recognized by the MafB antibody (Figure 4B), we examined the effect of MafB deficiency on MafB expression. In the krENU/krENU pancreas at E15.5 Nkx6.1+ cells were not affected by MafB deficiency (Figures 7B, F), insulin+ MafB+, insulin− MafB+ cells and occasional insulin+ MafB− cells were seen, and the total number of MafB+ cells showed a modest increase (~20%) compared to the wild type (Figure 7A, E). Unlike the effect of MafB deficiency on its own expression, the number of PDX-1high and MafA+ cells were reduced (Figure 7C, D, G, H), suggesting that deficient MafB function was sufficient to reduce, but not eliminate, cells expressing PDX-1high and MafA. At E15.5, nearly 60% of MafB+ cells (175 MafB+, 110 Ins+ and 105 MafB+Ins+ cells) expressed insulin in wild type, but this number dropped to ~25% in krENU/krENU (238 MafB+, 68 Ins+ and 65 MafB+Ins+ cells) (Figure 7I). This observation suggests that a functionally compromised MafB isoform (krENU) prevents a significant proportion of differentiating endocrine cells from expressing insulin. The deficiency of MafB function also resulted in a reduced proportion of PDX-1high cells co-expressing insulin (128 PDX-1high, 122 Ins+ and 92 PDX1highIns+ cells in wild type vs. 107 PDX-1high, 90 Ins+ and 65 PDX1highIns+ cells in krENU/krENU pancreas). This reduced ability of MafB+ and PDX-1high cells to co-express insulin in MafB deficient mice was similar to that observed in SeyNeu/SeyNeu mice (Figure 2). In krENU/krENU pancreas all MafA+ cells were insulin+, but not all insulin+ cells expressed MafA (75 MafA+, 111 Ins+ and 75 MafA+Ins+ cells in wild type vs. 60 MafA+, 92 Ins+ and 60 MafA+Ins+ cells krENU/krENU pancreas). Interestingly, the proportion of insulin+ cells co-expressing the different transcription factors was unaltered in wild type and krENU/krENU mice (Figure 7J): nearly 95%, 75% and 65% of insulin+ cells co-expressed MafB, PDX-1high and MafA, respectively.
Figure 7. MafB deficiency reduces PDX-1, MafA and insulin expression and impairs the ability of MafB+ and PDX-1+ cells to express insulin.

(A–H) Adjacent sections from E15.5 wild type and krENU/krENU (n = 4 wild type, 3 mutant) pancreata were stained for MafB, Nkx6.1, PDX-1 and MafA in green and insulin in red. In krENU/krENU sections the number of cells expressing PDX-1, MafA and insulin were reduced compared to wild type pancreata. Bars: 20μm. (
I, J) Stained E15.5 pancreatic sections from wild type and krENU/krENU embryos were quantified to determine the proportion of transcription factor+ cells expressing insulin (I), and the proportion of insulin+ cells expressing different transcription factors (J). (I) Proportions of MafB+Ins+ cells / total MafB+ cells and PDX-1+Ins+ cells / total PDX-1+ cells in krENU/krENU were significantly reduced, while all MafA+ cells expressed insulin. (J) The proportion of insulin+ cells expressing MafB, PDX-1 or MafA was not significantly different between wild type and krENU/krENU. (a) MafB and other large Maf factors can activate PDX-1 expression. A −4.5kb PDX-1 promoter:luciferase reporter construct was transfected into HeLa cells with the indicated expression plasmids and pSVβ-gal was used as an internal control. Results are presented relative to the activity of a wild-type luciferase construct transfected with the pcDNA3.1 ± S.E. (n = 3).
The ability of MafB to directly activate PDX-1 expression was examined by co-transfecting −4.5kb PDX-1:luciferase plasmid (Eto et al., 2007) and either MafA, MafB or cMaf plasmids in HeLa cells (Figure 7K). MafA, MafB and cMaf were all capable of activating PDX-1 expression, consistent with the reported ability of MafA to induce PDX-1 expression (Samaras et al., 2003).
Unlike Pax6, MafB deficiency does not affect the cell-fate decision of endocrine cells
As stated above, SeyNeu/SeyNeu and krENU/krENU mice show very similar pattern of reductions of insulin+ and glucagon+ cells. Loss of Pax6 function results in endocrine cells that do not express insulin or glucagon and that acquire an alternate cell-fate with ghrelin expression (Prado et al 2004; Heller et al., 2005). However, in E15.5 krENU/krENU the reduction in insulin+ and glucagon+ cells was not accompanied by an increase in somatostatin+ or ghrelin+ cells (Figure 8 and Artner et. al., 2007). This finding is consistent with our observation in SeyNeu/SeyNeu mice that the ghrelin+ ε cells do not express MafB (Suppl. Figure 2). Thus, MafB deficiency, unlike Pax6 deficiency, does not trigger endocrine cells to acquire alternate cell-fates.
Figure 8. MafB deficiency does not induce the formation of ε-cells.
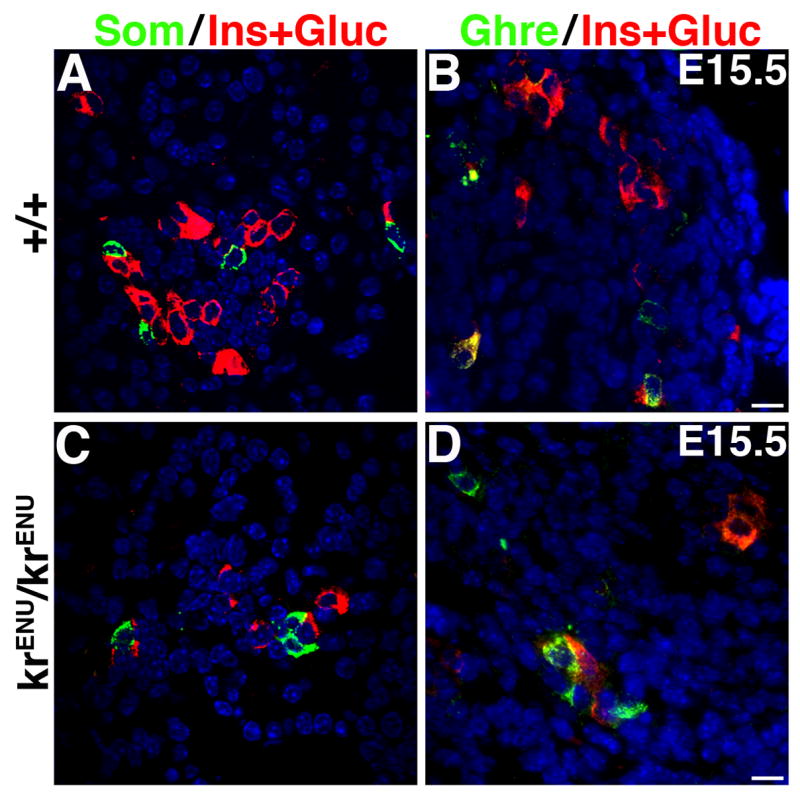
Sections from E15.5 wild type (+/+) and krENU/krENU pancreata were stained to detect somatostatin (Som) or ghrelin (Ghre) in green and a mixture of antibodies that recognize insulin and glucagon in red. In spite of reduction in the number of insulin and glucagon expressing cells, the number of somatostatin+ and ghrelin+ cells is unchanged in krENU/krENU embryos. Also, ghrelin+ glucagon− ε-cell numbers were similar in pancreata from wild type and krENU/krENU embryos.
An increased proportion of the remaining insulin+ cells in krENU/krENU pancreata express Hb9
The presence of insulin+ and glucagon+ cells in SeyNeu/SeyNeu and krENU/krENU mice suggests the induction of these hormones can occur via a pathway independent of Pax6 or MafB function. Two parallel pathways have been proposed to trigger the formation of insulin+ cells: one involving Nkx2.2 and Pax4 in regulating the Hb9 and PDX-1 dependent expression of insulin, and a second involving Nkx2.2, Pax6 and PDX-1 (Wang et al., 2004). Wang and colleagues reported that at E14.5, Pax6 deficiency had no obvious effect on Hb9 expression; we also observed cells expressing Hb9 in our Pax6 deficient mice at E15.5 (Suppl. Figure 3). Since our data suggest that MafB functions downstream of Pax6, we examined whether MafB deficiency differentially affected the formation of insulin+ cells from either the Nkx2.2-Pax6 or Pax4-Hb9 pathways by quantifying immunostained E15.5 wild type and krENU/krENU pancreata. Here, MafB deficiency increased the proportion of insulin+ cells expressing Hb9 but had no effect on the ability of Hb9+ cells to co-express insulin (220 Hb9+, 227 Ins+ and 164 Hb9+Ins+ cells in wild type vs. 139 Hb9+, 111 Ins+ and 104 Hb9+Ins+ cells krENU/krENU pancreas) (Figures 9). The increased proportion of insulin+ cells expressing Hb9 in krENU/krENU mice suggests that these remaining insulin+ cells may be specified via the Pax4-Hb9 pathway.
Figure 9. An increased proportion of insulin+ cells in the MafB deficient pancreata co-express Hb9.
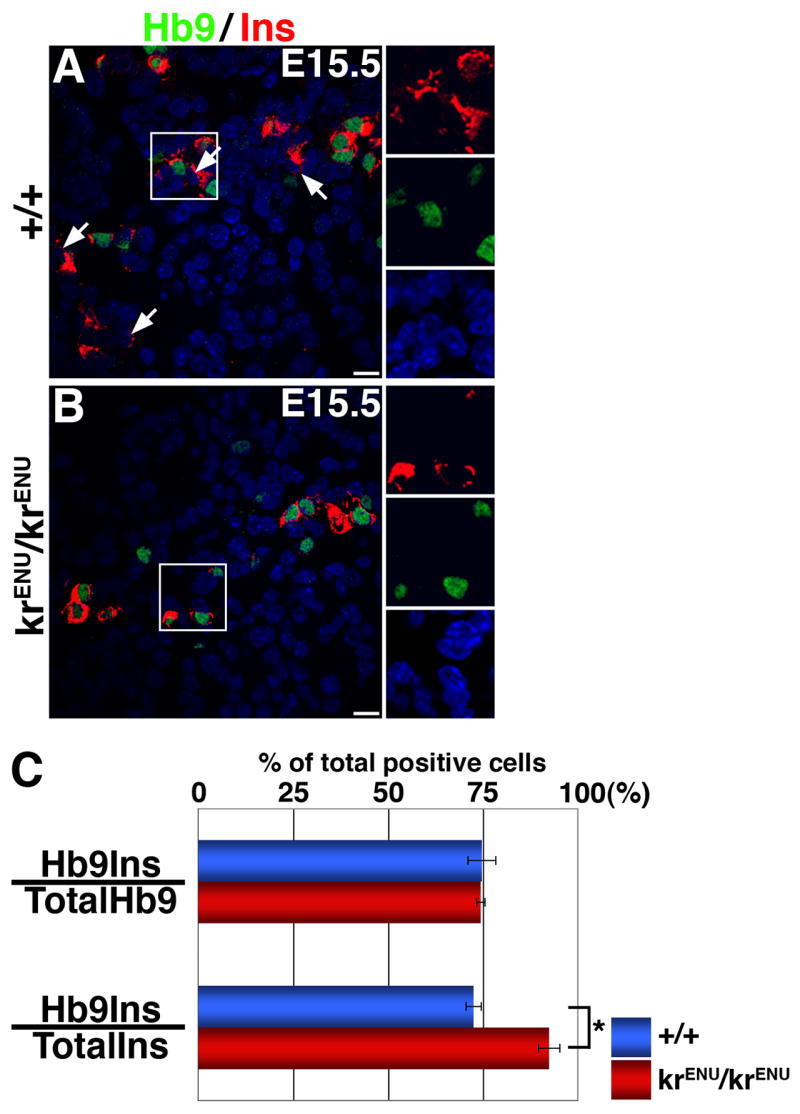
(A, B) E15.5 sections from wild type and krENU/krENU pancreata were stained for Hb9 in green and insulin in red. Hb9− insulin+ cells and Hb9+ insulin+ cells co-exist in the wild type pancreas while the number of Hb9− insulin+ cells is reduced in krENU/krENU pancreas. Bars: 20μm. (C) Quantification of Immunohistochemical data. The proportion of Hb9+ cells expressing insulin did not change between the krENU/krENU and wild type mice. However, the proportion of insulin+ cells that co-express Hb9 in these mice is significantly increased (p = 0.02).
Discussion
Previously we proposed that the maturation of insulin+ cells during embryonic development requires a switch from an insulin+ MafB+ state to an insulin+ MafA+ state via an intermediate PDX-1high stage. Here we use Pax6 and MafB deficient mice to demonstrate that the deficiency of either Pax6 or MafB function results in reduced number of cells expressing insulin, glucagon, PDX-1 and MafA (Figures 1, 3, 5, 7). Comparable proportions of Ngn3 and MafB expressing cells in krENU/krENU and wild type pancreata (Figures 6, 7) suggest that similar to Pax6 deficiency (Ashery-Padan et al., 2004), MafB deficiency does not affect the initiation of endocrine differentiation, but affects expression of markers of terminal differentiation. The reduced function of either Pax6 or MafB results in an increased proportion of MafB+ insulin− and PDX-1high insulin− cells, while the expression of MafA, although reduced, is always restricted to the cells that express insulin (Figures 2, 7), suggesting that PDX-1 and MafA function downstream of Pax6 and MafB. The loss of Pax6 function reduces the numbers of MafB expressing cells (Figures 1, 2), while the deficiency of MafB activity has no effect on Pax6 expression (Figure 6). Taken together, these results suggest that MafB can be a downstream mediator of Pax6 function or that Pax6 requires MafB for inducing the expression of insulin and glucagon. However, the conversion of endocrine precursors into ghrelin+ cells does not occur in MafB deficient mice (Figure 8, and Artner et al., 2007) as observed in the absence of Pax6 function (Heller et al., 2005; Prado et al., 2004, and Suppl. Figure 2). Thus, the suppression of ghrelin+ ε-cells occurs at the level of Pax6 expression, and MafB does not appear to have a direct role in regulating the formation of ε-cells.
Analysis of Nkx2.2, Pax6 and Pax4 knockout mice suggested that specification of insulin+ cells involves two distinct pathways; one requiring Nkx2.2, Pax4, Hb9 and PDX-1, and a second involving Nkx2.2, Pax6 and PDX-1 (Wang et al., 2004). Deficiency of MafB function in krENU/krENU mice led to reduction of insulin+ and glucagon+ cells (Figure 5) as in Pax6 deficient mice (Sander et al., 1997; Wang et al., 2004; Ashery-Padan et al., 2004; Figures 1,3). These observations suggest that insulin and glucagon expression in some endocrine precursors is independent of Pax6 and MafB function. A similar reduction in the proportion of hormone+ cells found in MafB knockout mice (Artner et al., 2007) suggests that the residual activity of the krENU allele does not regulate the formation of remaining hormone+ cells. We also observed in MafB deficient mice an increased proportion of remaining insulin+ cells expressing Hb9 (Figure 9). This increase could be due to the selective lack of insulin+ cells derived from the Pax6-MafB pathway in these krENU/krENU mice. Thus, our results provide strong support to the two parallel pathways model proposed by Sosa-Pineda and colleagues for the formation of insulin+ cells (Wang et al., 2004).
The presence of increased numbers of MafB+ insulin− cells in SeyNeu/SeyNeu mice (Figure 2) suggests that either Pax6 function is essential for MafB to activate insulin expression or Pax6 initiates insulin expression with MafB required for further maturation of insulin+ cells. However, in krENU/krENU mice the number of Pax6+ cells was unchanged while that of insulin+ cells was reduced (Figure 6). Thus, it is most likely that in the presence of Pax6 (or following its action), MafB initiates the expression of insulin in some hormone− endocrine precursors. Although Pax6 and MafB are required for the formation of only some insulin+ cells, they are expressed in most, if not all, insulin+ cells, suggesting a role for these factors even in insulin+ cells derived from a Pax6-MafB independent pathway. Since maturation of insulin+ cells accompanies their transition from MafB to MafA expression (Nishimura 2006), MafB may have two distinct roles during endocrine differentiation: one essential for regulating the differentiation of insulin+ cells via the Pax6-MafB pathway, and the other in the maturation of Pax4-Hb9 derived insulin+ cells. Additional studies are needed to confirm the consequence of a loss of MafB or Pax6 function on the maturation of these latter insulin+ cells.
The increased proportion of insulin+ Hb9+ cells in krENU/krENU mice would suggest that the induction of insulin expression in the Pax4-Hb9 pathway does not depend on MafB expression. Interestingly, a recent MafB knockout study (Artner et al., 2007) suggested that MafA was required to specify the remaining insulin+ cells. However, since MafA is expressed only in insulin+ cells in krENU/krENU mice and many insulin+ cells are MafA− (Figure 7), it is unlikely that MafA induces insulin expression in MafB knockout mice. MafA, at the most, might be responsible for a subpopulation of insulin+ cells (MafA+ cells), while another factor is required to trigger insulin expression in MafA− cells. It is important to note that MafA expression remained restricted to insulin+ cells in both SeyNeu/SeyNeu and krENU/krENU mice, and MafA knockout mice (Zhang et al., 2005) show no reduction in insulin+ cells at birth. It seems most likely that MafA expression is initiated only after the induction of insulin expression in endocrine precursors, which suggests that in krENU/krENU mice a factor other than MafA or MafB induces insulin expression in cells differentiating via the Pax4-Hb9 pathway. While our analysis of MafB deficient mice generally agrees with a recent characterization of MafB knockout mice (Artner et al., 2007), our data do not support a role for PDX-1high upstream of MafB function nor a role for MafA in inducing insulin expression. Additionally, by analyzing both MafB and Pax6 deficient mice, the current study extends our understanding of the role of MafB in pancreatic development. Our results strongly suggest that MafB functions downstream of Pax6 and is most likely responsible for the Pax6 dependent loss of insulin+ and glucagon+ cells.
Analysis of SeyNeu/SeyNeu mice demonstrates a role of Pax6 and MafB in the differentiation of glucagon+ cells. The majority of glucagon positive cells present before the secondary transition express MafB (Figure 3), consistent with the expression of Pax6 in the early glucagon+ cells reported earlier (Heller et al., 2004; Sander et al., 1997). It is likely that the MafB+ glucagon− cells present at E12.5 (Figure 3) represent the insulin+ MafB+ cells seen at this stage (Nishimura et al., 2006). At E15.5, due to the increase in insulin+ MafB+ cells, the proportion of MafB+ cells expressing glucagon decreased. Yet, the proportion of glucagon+ cells expressing MafB remained unchanged at 95% in wild type mice (Figures 2, 3), suggesting that either the induction of glucagon expression in a small proportion of endocrine precursors in wild type mice does not require MafB function or that MafB is turned on rapidly after the induction of glucagon.
Loss of Pax6 function significantly reduced the number of MafB+ cells, resulting in a significant proportion of glucagon+ cells not expressing MafB at both E12.5 and E15.5 (Figure 3). This finding suggests either the existence of a Pax6-MafB independent pathway for the induction of glucagon or a lack of sustainability of MafB in these glucagon+ cells in the absence of Pax6 function. The reduction in the proportion of glucagon+ cells expressing MafB in SeyNeu/SeyNeu mice was opposite of no effect of Pax6 deficiency on the ability of insulin+ cells to express MafB (Figures 2, 3). This observation suggests differences in the specification and maturation of glucagon+ cells and insulin+ cells. Since the specification and maturation process of glucagon+ cells does not require the function of PDX-1 and MafA, it is likely that MafB does not regulate the maturation process and only regulates initiation of glucagon expression. A recent study showing different actions of Nkx2.2 in rescuing glucagon and insulin-expressing cells in Nkx2.2 knockout mice (Doyle et al., 2007), supports the suggestion that the transcription factors that regulate differentiation of both insulin and glucagon expression, use distinct mechanisms in each endocrine cell type. Similarly, MafB may only be involved in specification of glucagon+ cells whereas it may have a dual role in the specification and maturation of insulin+ cells.
Inhibiting the proper maturation of insulin+ cells has functional consequences. It has been shown that MafA knockout mice develop diabetes due to postnatal reduction in β-cell number (Zhang et al., 2005). Inhibition of MafA function in insulin-producing cells via gene knock-down also resulted in reduced expression of genes involved in insulin synthesis and secretion (Wang et al., 2007). MafA regulates the expression of granuphilin, critical for docking the insulin vesicle to the plasma membrane (Kato et al., 2006). Furthermore, insulin+ cells derived from human embryonic stem cells expressed MafB but not PDX-1 and probably not MafA (D’Amour et al., 2006); this lack could contribute to their inability to secrete insulin in response to glucose and further highlights the importance of the maturation process.
Previously we proposed that during maturation, insulin+ cells undergo a switch from an insulin+ MafB+ state to an insulin+ MafA+ state via an intermediate PDX-1high stage. In this study, we observed that nearly 95%, 75% and 65% of insulin+ cells in wild type, SeyNeu/SeyNeu, and krENU/krENU mice expressed MafB, PDX-1 and MafA, respectively (Figures 2, 7). The observed gradation in expression of these three transcription factors in insulin+ cells as well as the ability of MafB to activate PDX-1 expression further supports our hypothesis that maturation of insulin+ cells proceeds from a MafB+ to MafA+ state after the induction of PDX-1high. Results from Pax6 and MafB deficient mice further support such sequential requirement for MafB, PDX-1 and MafA functions in the maturation process. An analysis of conditional PDX-1 knockout in developing endocrine cells will be necessary to confirm whether the MafB is upstream of PDX-1high function and MafA is downstream of this gene. Knowledge of the precise function of these transcription factors during the maturation of insulin+ cells will play a crucial role in generating glucose responsive insulin producing cells.
Supplementary Material
Suppl. Figure. 1 Pax6 deficiency dose not inhibit the initiation of endocrine differentiation. Pancreas from wild type (+/+) and Small eye (SeyNeu/SeyNeu) embryos at E15.5 stained for Pax6, Glucagon (Gluc), Neurogenin3 (Ngn3), Nkx2.2 and Nkx6.1 in green and insulin (Ins) in red. An anti-Pax6 antibody that recognizes the C-terminus of Pax6 does not detect Pax6 expression in pancreatic sections from SeyNeu/SeyNeu mice. Expression of insulin and glucagon was significantly reduced, while the expression of Ngn3, Nkx2.2 and Nkx6.1 was normal in SeyNeu/SeyNeu pancreata. Bars: 20μm.
Suppl. Figure 2. MafB is not expressed in Ghrelin+ ε cells in Pax6 deficient mice. (A, B) Pancreas from wild type (+/+) and SeyNeu/SeyNeu embryos at E15.5 stained using a mixture of insulin and glucagon antibodies in red and Ghrelin in green. Increased numbers of only ghrelin expressing ε cells can be seen in SeyNeu/SeyNeu pancreas, while in wild type mice ghrelin was co-expressed with glucagon (and insulin) expressing cells as reported earlier (Heller et al., 2005). (C–F) Pancreatic sections from wild type (+/+) and SeyNeu/SeyNeu embryos at E15.5 were stained with primary rabbit MafB antibody, using secondary biotinylated anti-rabbit antibody followed by streptavidin-conjugated Alexa flour 488. Images taken of cells expressing MafB are shown in C and E. The same imaged sections were next stained with rabbit anti ghrelin antibody and Texas red conjugated anti rabbit secondary antibody. Ghrelin and MafB stained sections are shown in D and F. Arrow depict occasional ghrelin+ cells expressing MafB in wild type pancreas (representing ghrelin+ glucagon+ cells), and arrowheads denotes cells expressing only MafB. Ghrelin expressing cells in SeyNeu/SeyNeu pancreas do not express MafB.
Suppl. Figure 3. Expression of Hb9 in Pax6 deficient mice. Pancreatic sections from wild type (+/+) and SeyNeu/SeyNeu embryos at E15.5 were stained with Hb9 in green and insulin in red. Cells expressing insulin and Hb9 were observed in both wild type and Pax6 deficient pancreas as reported earlier (Wang et al., 2004).
Acknowledgments
We thank Drs. O. Madsen, P. Serup and the NIH-funded Beta Cell Biology Consortium for Nkx6.1 and Ngn3 antibodies, Dr. Chris Wright for PDX-1 antibody, Dr. Sam Pfaff for Hb9 antibody, Drs. Dan Drucker and Melissa Thomas for glucagon and PDX-1 reporter constructs, respectively, and Dr. Greg Barsh for krENU mice. This study was supported by research grants from NIH (RO1 DK060127) and Harvard Stem Cell Institute to AS, NIH 3RO1 CA95021-04S1 to SMS, NIH ( RO1 DK065791) and Harvard Stem Cell Institute to RLM, Juvenile Diabetes Research Foundation postdoctoral fellowships (3-2005-74) to WN, Canadian Institute of Health Research Fellowship to SR, and the Media and Advanced Microscopy (Histology and Confocal facilities) Cores of the Joslin Diabetes Endocrinology Research Center (NIH DK-36836).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Artner I, Blanchi B, Raum JC, Guo M, Kaneko T, Cordes S, Sieweke M, Stein R. MafB is required for islet beta cell maturation. Proc Natl Acad Sci U S A. 2007;104:3853–8. doi: 10.1073/pnas.0700013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Le Lay J, Hang Y, Elghazi L, Schisler JC, Henderson E, Sosa-Pineda B, Stein R. MafB: an activator of the glucagon gene expressed in developing islet alpha- and beta-cells. Diabetes. 2006;55:297–304. doi: 10.2337/diabetes.55.02.06.db05-0946. [DOI] [PubMed] [Google Scholar]
- Ashery-Padan R, Zhou X, Marquardt T, Herrera P, Toube L, Berry A, Gruss P. Conditional inactivation of Pax6 in the pancreas causes early onset of diabetes. Dev Biol. 2004;269:479–488. doi: 10.1016/j.ydbio.2004.01.040. [DOI] [PubMed] [Google Scholar]
- Collombat P, Hecksher-Sorensen J, Broccoli V, Krull J, Ponte I, Mundiger T, Smith J, Gruss P, Serup P, Mansouri A. The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the alpha- and beta-cell lineages in the mouse endocrine pancreas. Development. 2005;132:2969–80. doi: 10.1242/dev.01870. [DOI] [PubMed] [Google Scholar]
- Collombat P, Hecksher-Sorensen J, Krull J, Berger J, Riedel D, Herrera PL, Serup P, Mansouri A. Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest. 2007;117:961–70. doi: 10.1172/JCI29115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Hecksher-Sorensen J, Serup P, Mansouri A. Specifying pancreatic endocrine cell fates. Mech Dev. 2006;123:501–12. doi: 10.1016/j.mod.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Cordes SP, Barsh GS. The mouse segmentation gene kr encodes a novel basic domain-leucine zipper transcription factor. Cell. 1994;79:1025–34. doi: 10.1016/0092-8674(94)90033-7. [DOI] [PubMed] [Google Scholar]
- D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24:1392–401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- Doyle MJ, Loomis ZL, Sussel L. Nkx2.2-repressor activity is sufficient to specify alpha-cells and a small number of beta-cells in the pancreatic islet. Development. 2007;134:515–23. doi: 10.1242/dev.02763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund H. Pancreatic organogenesis--developmental mechanisms and implications for therapy. Nat Rev Genet. 2002;3:524–532. doi: 10.1038/nrg841. [DOI] [PubMed] [Google Scholar]
- Eichmann A, Grapin-Botton A, Kelly L, Graf T, Le Douarin NM, Sieweke M. The expression pattern of the mafB/kr gene in birds and mice reveals that the kreisler phenotype does not represent a null mutant. Mech Develop. 1997;65:111–122. doi: 10.1016/s0925-4773(97)00063-4. [DOI] [PubMed] [Google Scholar]
- Eto K, Kaur V, Thomas MK. Regulation of pancreas duodenum homeobox-1 expression by early growth response-1. J Biol Chem. 2007;282:5973–83. doi: 10.1074/jbc.M607288200. [DOI] [PubMed] [Google Scholar]
- Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet. 1994;7:463–71. doi: 10.1038/ng0894-463. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMuer M, Guillemot F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grapin-Botton A, Melton DA. Endoderm development: from patterning to organogenesis. Trends Genet. 2000;16:124–130. doi: 10.1016/s0168-9525(99)01957-5. [DOI] [PubMed] [Google Scholar]
- Harrington RH, Sharma A. Transcription factors recognizing overlapping C1-A2 binding sites positively regulate insulin gene expression. J Biol Chem. 2001;276:104–113. doi: 10.1074/jbc.M008415200. [DOI] [PubMed] [Google Scholar]
- Heller RS, Jenny M, Collombat P, Mansouri A, Tomasetto C, Madsen OD, Mellitzer G, Gradwohl G, Serup P. Genetic determinants of pancreatic epsilon-cell development. Dev Biol. 2005 doi: 10.1016/j.ydbio.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Heller RS, Stoffers DA, Liu A, Schedl A, Crenshaw EB, 3rd, Madsen OD, Serup P. The role of Brn4/Pou3f4 and Pax6 in forming the pancreatic glucagon cell identity. Dev Biol. 2004;268:123–34. doi: 10.1016/j.ydbio.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1991;354:522–5. doi: 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn. 2004;229:176–200. doi: 10.1002/dvdy.10460. [DOI] [PubMed] [Google Scholar]
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. MafA is a glucose-regulated and pancreatic beta-cell-specific transcriptional activator for the insulin gene. J Biol Chem. 2002;277:49903–49910. doi: 10.1074/jbc.M206796200. [DOI] [PubMed] [Google Scholar]
- Kato T, Shimano H, Yamamoto T, Yokoo T, Endo Y, Ishikawa M, Matsuzaka T, Nakagawa Y, Kumadaki S, Yahagi N, Takahashi A, Sone H, Suzuki H, Toyoshima H, Hasty AH, Takahashi S, Gomi H, Izumi T, Yamada N. Granuphilin is activated by SREBP-1c and involved in impaired insulin secretion in diabetic mice. Cell Metab. 2006;4:143–54. doi: 10.1016/j.cmet.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Kim SK, MacDonald RJ. Signaling and transcriptional control of pancreatic organogenesis. Curr Opin Genet Dev. 2002;12:540–547. doi: 10.1016/s0959-437x(02)00338-6. [DOI] [PubMed] [Google Scholar]
- Leonard J, Peers B, Johnson T, Ferreri K, Lee S, Montminy MR. Characterization of somatostatin transactivating factor-1, a novel homeobox factor that stimulates somatostating expresion in pancreatic islet cells. Mol Endocrinology. 1993;7:1275–1283. doi: 10.1210/mend.7.10.7505393. [DOI] [PubMed] [Google Scholar]
- Manzanares M, Cordes S, Kwan CT, Sham MH, Barsh GS, Krumlauf R. Segmental regulation of hoxb-3 by kreisler. Nature. 1997;387:191–195. doi: 10.1038/387191a0. [DOI] [PubMed] [Google Scholar]
- Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol Cell Biol. 2003;23:6049–6062. doi: 10.1128/MCB.23.17.6049-6062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CP, McGehee RE, Habener JF. IDX-1: a new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J. 1994;13:1145–1156. doi: 10.1002/j.1460-2075.1994.tb06363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi T, Hamada M, Morito N, Terunuma T, Hasegawa K, Zhang C, Yokomizo T, Esaki R, Kuroda E, Yoh K, Kudo T, Nagata M, Greaves DR, Engel JD, Yamamoto M, Takahashi S. MafB is essential for renal development and F4/80 expression in macrophages. Mol Cell Biol. 2006;26:5715–27. doi: 10.1128/MCB.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh LC. Pancreas and beta-cell development: from the actual to the possible. Development. 2007;134:427–38. doi: 10.1242/dev.02770. [DOI] [PubMed] [Google Scholar]
- Nishimura W, Kondo T, Salameh T, El Khattabi I, Dodge R, Bonner-Weir S, Sharma A. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev Biol. 2006;293:526–539. doi: 10.1016/j.ydbio.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura W, Salameh T, Kondo T, Sharma A. Regulation of insulin gene expression by overlapping DNA-binding elements. Biochem J. 2005;392:181–189. doi: 10.1042/BJ20050970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offield MF, Jetton TL, Labosky P, Ray M, Stein R, Magnuson M, Hogan BLM, Wright CVE. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–985. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 1993;12:4251–4259. doi: 10.1002/j.1460-2075.1993.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olbrot M, Rud J, Moss LG, Sharma A. Identification of beta -cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A. 2002;99:6737–6742. doi: 10.1073/pnas.102168499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado CL, Pugh-Bernard AE, Elghazi L, Sosa-Pineda B, Sussel L. Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proc Natl Acad Sci U S A. 2004;101:2924–2929. doi: 10.1073/pnas.0308604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reza HM, Ogino H, Yasuda K. L-Maf, a downstream target of Pax6, is essential for chick lens development. Mech Dev. 2002;116:61–73. doi: 10.1016/s0925-4773(02)00137-5. [DOI] [PubMed] [Google Scholar]
- Reza HM, Yasuda K. The involvement of neural retina pax6 in lens fiber differentiation. Dev Neurosci. 2004;26:318–327. doi: 10.1159/000082273. [DOI] [PubMed] [Google Scholar]
- Sadl V, Jin F, Yu J, Cui S, Holmyard D, Quaggin S, Barsh G, Cordes S. The mouse Kreisler (Krml1/MafB) segmentation gene is required for differentiation of glomerular visceral epithelial cells. Dev Biol. 2002;249:16–29. doi: 10.1006/dbio.2002.0751. [DOI] [PubMed] [Google Scholar]
- Samaras SE, Zhao L, Means A, Henderson E, Matsuoka TA, Stein R. The islet beta cell-enriched RIPE3b1/Maf transcription factor regulates pdx-1 expression. J Biol Chem. 2003;278:12263–12270. doi: 10.1074/jbc.M210801200. [DOI] [PubMed] [Google Scholar]
- Sander M, Neubuser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997;11:1662–1673. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- Sharma A, Stein R. Glucose-induced transcription of the insulin gene is mediated by factors required for B-cell-type-specific expression. Mol Cell Biol. 1994;14:871–879. doi: 10.1128/mcb.14.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieweke MH, Tekotte H, Frampton J, Graf T. MafB is an interaction partner and repressor of Ets-1 that inhibits erythroid differentiation. Cell. 1996;85:49–60. doi: 10.1016/s0092-8674(00)81081-8. [DOI] [PubMed] [Google Scholar]
- Wang H, Brun T, Kataoka K, Sharma AJ, Wollheim CB. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia. 2007;50:348–58. doi: 10.1007/s00125-006-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Elghazi L, Parker SE, Kizilocak H, Asano M, Sussel L, Sosa-Pineda B. The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Dev Biol. 2004;266:178–189. doi: 10.1016/j.ydbio.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, Kudo T, Engel JD, Yamamoto M, Takahashi S. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005;25:4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Guo M, Matsuoka TA, Hagman DK, Parazzoli SD, Poitout V, Stein R. The islet beta cell-enriched MafA activator is a key regulator of insulin gene transcription. J Biol Chem. 2005;280:11887–11894. doi: 10.1074/jbc.M409475200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Figure. 1 Pax6 deficiency dose not inhibit the initiation of endocrine differentiation. Pancreas from wild type (+/+) and Small eye (SeyNeu/SeyNeu) embryos at E15.5 stained for Pax6, Glucagon (Gluc), Neurogenin3 (Ngn3), Nkx2.2 and Nkx6.1 in green and insulin (Ins) in red. An anti-Pax6 antibody that recognizes the C-terminus of Pax6 does not detect Pax6 expression in pancreatic sections from SeyNeu/SeyNeu mice. Expression of insulin and glucagon was significantly reduced, while the expression of Ngn3, Nkx2.2 and Nkx6.1 was normal in SeyNeu/SeyNeu pancreata. Bars: 20μm.
Suppl. Figure 2. MafB is not expressed in Ghrelin+ ε cells in Pax6 deficient mice. (A, B) Pancreas from wild type (+/+) and SeyNeu/SeyNeu embryos at E15.5 stained using a mixture of insulin and glucagon antibodies in red and Ghrelin in green. Increased numbers of only ghrelin expressing ε cells can be seen in SeyNeu/SeyNeu pancreas, while in wild type mice ghrelin was co-expressed with glucagon (and insulin) expressing cells as reported earlier (Heller et al., 2005). (C–F) Pancreatic sections from wild type (+/+) and SeyNeu/SeyNeu embryos at E15.5 were stained with primary rabbit MafB antibody, using secondary biotinylated anti-rabbit antibody followed by streptavidin-conjugated Alexa flour 488. Images taken of cells expressing MafB are shown in C and E. The same imaged sections were next stained with rabbit anti ghrelin antibody and Texas red conjugated anti rabbit secondary antibody. Ghrelin and MafB stained sections are shown in D and F. Arrow depict occasional ghrelin+ cells expressing MafB in wild type pancreas (representing ghrelin+ glucagon+ cells), and arrowheads denotes cells expressing only MafB. Ghrelin expressing cells in SeyNeu/SeyNeu pancreas do not express MafB.
Suppl. Figure 3. Expression of Hb9 in Pax6 deficient mice. Pancreatic sections from wild type (+/+) and SeyNeu/SeyNeu embryos at E15.5 were stained with Hb9 in green and insulin in red. Cells expressing insulin and Hb9 were observed in both wild type and Pax6 deficient pancreas as reported earlier (Wang et al., 2004).