

Abstract
Sensitive detection of tumor-specific point mutations is of interest in both the early detection of cancer and the monitoring of treatment at a molecular level. Recently, peptide nucleic acid (PNA) clamp real-time PCR has provided a time-sparing and sensitive method for the detection of mutations in the presence of a large excess of wild-type DNA. We present the first report that the sensitivity of PNA clamp PCR is limited by the low fidelity of Taq DNA polymerase. Replication errors introduced by Taq polymerase in the PNA-binding site were amplified during PCR due to the resulting mismatches between PNA and DNA. To reduce the frequency of polymerase-induced errors, we developed a PNA clamp PCR assay for the detection of mutations in codons 12 and 13 of the K-ras gene based on a high-fidelity DNA polymerase. The sensitivity of our assay increased approximately 10-fold, significantly detecting mutant DNA diluted 20,000-fold in wild-type DNA (P = 0.025), compared with its detection at 2000-fold dilution (P = 0.039) when Taq polymerase was used. Our data suggest that the replication errors caused by Taq polymerase must be taken into consideration for PNA clamp PCR and for other methods based on selective PCR amplification, and that these assays can be enhanced by high-fidelity DNA polymerases.
Detection of microscopic spread of tumor cells is of interest in several areas of cancer research. Common strategies to distinguish tumor cells from normal cells are based on different histological properties, specific protein and mRNA expressions, and tumor-specific mutations. Somatic mutations in the K-ras gene are present in 80 to 90% of pancreatic cancers1,2 and 35 to 50% of colorectal cancers.3,4,5 Thus, methods for sensitive detection of K-ras mutations may reveal micrometastases in both pancreatic and colorectal cancer patients.
Several strategies have been used to detect tumor-specific mutations sensitively. One of the most recent methods that has proved successful is peptide nucleic acid (PNA) clamp PCR. PNA is a synthetic DNA analogue, in which the ribose/phosphate backbone of the DNA has been replaced by N-(2-aminoethyl)-glycine units linked by peptide bonds. PNA binds strongly to complementary DNA by Watson-Crick base pairing, whereas one single mismatch will severely destabilize the complex, typically lowering the melting temperature by 13 to 20°C.6,7,8,9 In PNA clamp PCR, wild-type (wt) specific PNA oligomers are used to suppress amplification of wild-type alleles during PCR, while any mutant allele will show unhindered amplification.10 Thiede et al11 reported the first PNA clamped PCR assay to detect K-ras mutations in 1996. Subsequently, several studies using similar assays to detect microscopic cancer dissemination have been reported.12,13,14,15 Recently, the technique has been adapted to real-time PCR instrumentation, increasing the sensitivity and specificity of the assay.16,17,18
In this report we have improved an assay for detecting K-ras mutations in codons 12 and 13 by real-time PNA clamp PCR based on SYBR-Green I and a high-fidelity DNA polymerase. We show that mutations are introduced by the DNA polymerase during PNA clamp PCR, limiting the sensitivity of the assay. However, by using a high-fidelity DNA polymerase, the sensitivity of the PNA clamp PCR assay is significantly improved.
Materials and Methods
DNA Isolation from Cell Lines and Peripheral Blood
The colorectal carcinoma cell lines HT29 (wt K-ras), LS174T (heterozygous GGT>GAT codon 12 K-ras mutation[c.35G>A]), and HCT116 (heterozygous GGC→ GAC codon 13 K-ras mutation[c.38G>A]) were purchased from the European Collection of Cell Cultures (Salisbury, UK) and cultured as recommended by the provider. Cells grown to near confluence were lysed in Buffer RLT (RNeasy Mini kit; QIAGEN, Hilden, GE) and homogenized by centrifugation through QIAshredder columns (QIAGEN). DNA was isolated using a combination of the DNeasy Blood & Tissue kit (QIAGEN) and the RNeasy Mini kit (QIAGEN) that allowed isolation of both DNA and RNA from the same samples. DNA was isolated from peripheral blood by the QIAGEN DNA Blood kit by a similar combination with the RNeasy Mini kit (QIAGEN). DNA concentrations were determined by UV (260 nm) spectrophotometry.
Primers, Probes, and Peptide Nucleic Acid
The sequence of the PCR primers, probes, and PNA of the real-time PCR assay adapted from Däbritz et al17 were as described, except for the acceptor probe, which had the wild-type sequence 5′-LC Red640-TTGCCTACGCCACCAGCTCCAA-3′. The PNA sequence was 5′-CCTACGCCACCAGCTCC-3′. The sequences of the PCR primers used in our enhanced PNA clamp real-time PCR assay were 5′-GCCTGCTGAAAATGACTGAATATAA-3′ (forward) and 5′-CGTCAAGGCACTCTTGCCTAC-3′ (reverse), while the sequence of the PNA was the same.
Real-Time PCR
The LightCycler (Roche Applied Science, Penzberg, GE) PNA clamp PCR assay was performed as described by Däbritz et al,17 with the exception that we used a wild-type-specific probe where Däbritz used a mutation-specific probe. Phusion (Finnzymes, Espoo, FI) HS-based PNA clamp PCR was performed in a final volume of 25 μl containing 1× Phusion HF buffer, 0.2 mmol/L deoxynucleoside-5′-triphosphate, 0.15 μmol/L forward and reverse primer, 0.75 μl 1:200 SYBR Green I in dimethyl sulfoxide, 0.25 μmol/L PNA, 0.02 U/μl Phusion HS DNA polymerase, and 200 ng of template DNA. Thermocycling was performed in an Mx3000P (Stratagene, La Jolla, CA) real-time PCR instrument using an initial denaturation and enzyme activation step at 98°C for 30 seconds, 45 cycles of 10 seconds at 98°C (denaturation), 10 seconds at 76°C (PNA annealing), 20 seconds at 60°C (primer annealing), and 20 seconds at 72°C (elongation). Fluorescence measurements for SYBR Green I were performed at the end of the elongation step.
The Platinum Taq-based PNA clamp PCR assay was identical to the Phusion HS assay, except for the use of 1× Platinum Taq PCR buffer (Invitrogen, Carlsbad, CA), 3 mmol/L MgCl2, and 0.02 U/μl Platinum Taq DNA polymerase. The denaturation temperature was 94°C and the initial denaturation and enzyme activation step lasted 2 minutes, as recommended by the manufacturer. Platinum Taq is a recombinant Thermus aquaticus (Taq) DNA polymerase with a thermolabile inhibitor to achieve hot-start capabilities (according to the manufacturer). The HotGoldStar (Eurogentec, Seraing, Belgium)-based PNA clamp PCR was also identical to the Phusion HS assay except for the use of 10× HotGoldStar PCR buffer, 3 mmol/L MgCl2, and 0.025 U/μl HotGoldStar DNA polymerase. The denaturation temperature was 95°C, and the initial denaturation and enzyme activation step lasted 10 minutes, as recommended by the manufacturer. HotGoldStar polymerase is a modified recombinant Taq DNA polymerase with hot-start capabilities (according to the manufacturer). The purity of all PCR products was monitored by melting curve analysis. Amplification and melting curves for the Phusion HS, HotGoldStar, and Platinum Taq assays with wild-type and mutant template are shown as supplemental material (Supplemental Figure 1, see http://jmd.amjpathol.org).
Figure 1.
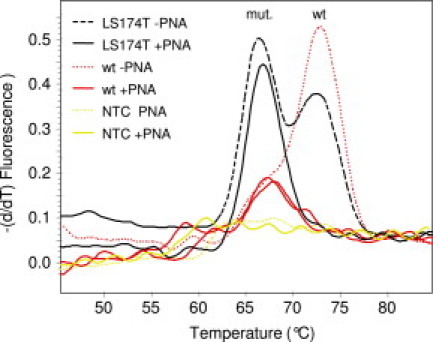
Melting curve analysis of PCR products amplified by a PNA clamp PCR assay adapted from Dabritz et al.17 Melting curves for PCR products generated from LS174T (heterozygous codon 12 mutant) DNA, wt DNA template (isolated from normal peripheral blood), and no template (NTC) in the absence (−PNA) or presence (+PNA) of PNA are shown. Mutant (mut.)- and wt-specific peaks are indicated.
The identity of the PCR products was initially also confirmed by sequencing. PCR setup and analysis were performed in separate rooms to avoid contamination.
Each sample was analyzed in triplicate both with and without PNA. For each sample, we computed ΔCt = Ct+PNA − Ct−PNA, where Ct+PNA and Ct−PNA denote the Ct value for the mean amplification curve of the triplicate with PNA and without PNA, respectively. The Ct−PNA was included to correct for varying template amount and quality.
Dilution of LS174T Cells in Peripheral Blood Lymphocytes
Peripheral blood lymphocytes were isolated from blood samples of healthy volunteers by Lymphoprep density centrifugation as recommended by the manufacturer (Axis-Shield, Dundee, Scotland, UK). The isolated lymphocytes were washed by centrifugation at 250 × g for 10 minutes and resuspended in PBS. The colon carcinoma cell line LS174T was cultured as described above, grown to near confluence, and trypsinated as suggested by the European Collection of Cell Cultures. The cell suspension was centrifuged for 5 minutes at 300 × g and the cell pellet resuspended in PBS. Cell densities were determined using a hemocytometer. The cell suspensions were repeatedly vortexed at slow speed to keep the suspensions homogeneous. Volumes corresponding to 100, 1000, 1 × 104, and 1 × 105 LS174T cells were added to 1 × 107 lymphocytes in separate tubes and mixed. DNA was isolated from the cell suspensions as described above.
Sequencing of PCR Products
DNA isolated from normal peripheral blood as described underwent PNA clamp PCR amplification by the Platinum Taq and HotGoldStar assays described above. The PCR products were purified with QIAquick PCR Purification Kit (QIAGEN) according to the protocol of the manufacturer and eluted in 40 μl of deionized water. Sequencing was performed using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) with 3 ng of purified PCR product as template. The sequencing reactions were then purified using the BigDye XTerminator Purification Kit (Applied Biosystems). The samples were analyzed on an Applied Biosystems 3130XL Genetic Analyzer using a 36-cm capillary array and POP7 polymer.
Statistics
The mean ΔCt values of two samples were compared using a two-sided, unpaired t-test with unequal variances for the two samples, assuming that the ΔCt values have normal distribution. Confidence intervals were computed using the t distribution.
Results
PNA Clamp PCR from Wild-Type Template Amplifies PCR Products with Mutation-Like Melting Temperatures
We aimed to develop a PNA-clamped real-time PCR assay for sensitive detection of all possible carcinoma-associated mutations in K-ras codons 12 and 13 in lymph nodes (wt K-ras) from colorectal cancer patients. We first adapted the assay used by Däbritz et al,17 in which they used a wild-type-specific PNA to suppress amplification from wild-type K-ras alleles and a mutation-specific probe to detect amplification of a specific K-ras mutation. To distinguish all possible codon 12 and 13 mutant PCR products from wild-type PCR products by melting curve analysis, we altered the probe sequence to be wild-type specific. With this set-up, amplifications of K-ras PCR products were observed in many samples containing wild-type DNA despite the presence of PNA. Thus, the PNA clamping seemed to be incomplete. However, melting curve analysis demonstrated that the melting temperature of the probe bound to these PCR products was similar to the melting temperature of the probe bound to PCR products with a known mutation (about 5°C lower than the melting temperature of the probe bound to wild-type PCR products) (Figure 1). This indicated that these PCR products contained mutations that were introduced during the PNA clamp PCR.
High-Fidelity DNA Polymerase Enhances the Sensitivity of a PNA Clamp PCR Assay
This led us to hypothesize that the mutations were introduced when Taq polymerase made replication errors and that any error leading to a mismatch between the PNA and DNA would be enriched during the PCR because of weaker PNA-clamping compared with wild-type template. If so, the utilization of high-fidelity DNA polymerase would reduce the frequency of new mutations and thus increase the sensitivity of the assay. Further, the use of mutation-specific hybridization probes would not enhance the assay as it would be impossible to distinguish the mutations arising during the PCR from the mutations present in the sample to be analyzed.
Thus, we developed a new SYBR Green I-based real-time PNA clamp PCR assay to test different DNA polymerases. The assay was optimized for two Taq DNA polymerases, Platinum Taq and HotGoldStar, in addition to the high-fidelity DNA polymerase Phusion HS, with new primers and emphasis on as equal conditions as possible (Figure 2). The fidelity of Phusion HS DNA polymerase has been reported (Finnzymes) to be 50 times higher than that of Taq polymerase. To compare the sensitivities of the DNA polymerases to detect minimal amounts of mutant K-ras, we analyzed dilution series of LS174T (heterozygous codon 12 GGT>GAT mutation [c.35G>A]) DNA in DNA from HT29 cells (wild-type K-ras). The sensitivity achieved was about ten times higher for Phusion HS than for the other polymerases (Figure 3), as we were able to detect a 1:20,000 dilution (P = 0.025) with Phusion HS rather than a 1:2000 dilution (P = 0.039) with Platinum Taq and only a 1:1000 dilution (P = 0.001) with HotGoldStar. We also investigated the sensitivity of the Phusion HS assay with dilution series of HCT116 (heterozygous codon 13 GGC→GAC mutation [c.38G>A]) DNA in HT29 DNA, with similar results (data not shown). The enhanced sensitivity of the PNA clamp PCR assay observed for the high-fidelity DNA polymerase Phusion HS compared with Taq DNA polymerase supported our hypothesis that replication errors in the PNA-binding site are amplified during PNA clamp PCR.
Figure 2.

Schematic pictorial of our PNA clamp assay. In our design, the primer and the PNA bind competitively to part of the same sequence. With wild-type K-ras as template, the PNA binds to DNA and blocks primer annealing and elongation (A). When a mutation is present, there is a mismatch between PNA and DNA leading to much weaker binding, allowing the primer to bind DNA, and elongation can take place (B).
Figure 3.
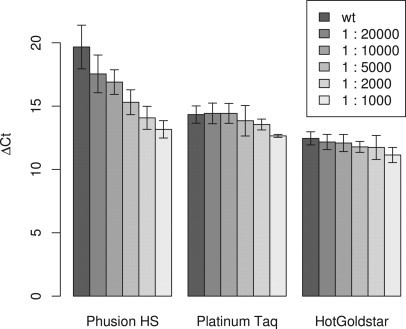
Sensitivity of the three PNA clamp PCR assays for detection of K-ras mutations in large excess of wild-type DNA. Mean ΔCt values from four independent dilution series of DNA from the colon carcinoma cell line LS174T (K-ras codon 12 mutation) in DNA from the colon carcinoma cell line HT29 (wild-type K-ras) are plotted. The error bars show 95% confidence intervals. The ΔCt values were calculated as ΔCt = Ct+PNA − Ct−PNA, where Ct+PNA and Ct−PNA denote the Ct values for the same reactions with and without PNA, respectively.
To explore the sensitivity of our assay in a cellular context, we analyzed dilution series of 102, 103, 104, and 105 LS174T cells in 107 normal lymphocytes. Our enhanced PNA clamp assay significantly distinguished the 103:107 dilution from normal lymphocytes (P = 0.0095), corresponding to a 1:104 sensitivity (Figure 4).
Figure 4.
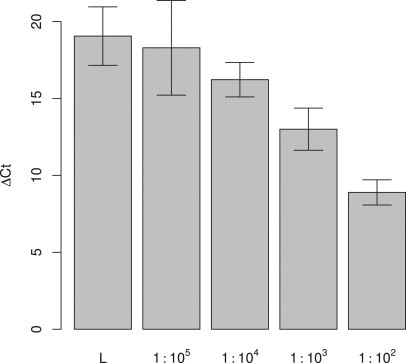
Sensitivity of the Phusion HS-based PNA clamp PCR assay for colon carcinoma cells in large excess of normal lymphocytes. Mean ΔCt values from three independent dilution series of LS174T cells (colon carcinoma cell line with codon 12 mutation) in normal lymphocytes (L) are plotted. The error bars show 95% confidence intervals.
Sequencing Confirmed New Mutations Introduced during PNA Clamp PCR
To confirm that mutations were introduced during PNA clamp PCR, we sequenced 30 of the PCR products amplified from wild-type K-ras with PNA present. We found several different mutations in the PNA-binding site (Figure 5), confirming our hypothesis.
Figure 5.
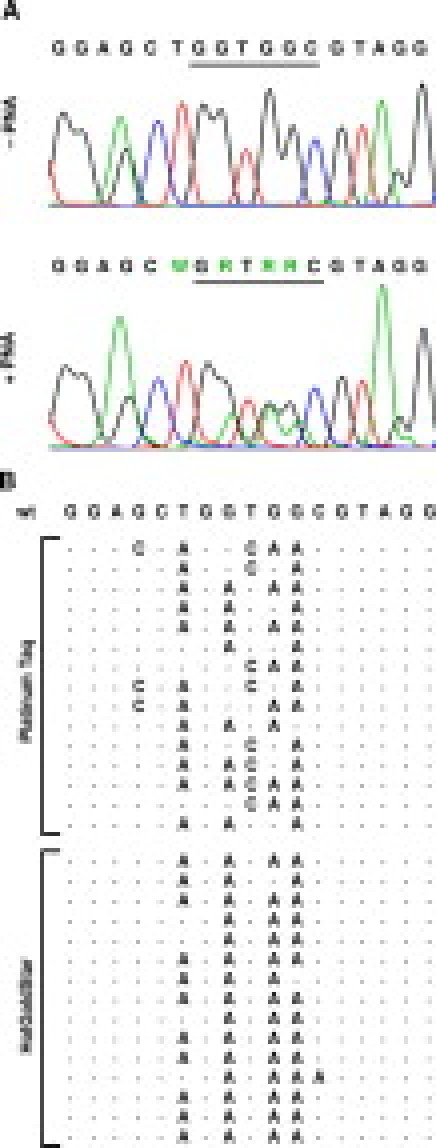
Sequencing of PCR products amplified from wild-type template by Taq-based PNA clamp PCR. A: Chromatograms showing the sequencing of a representative PCR product amplified from wild-type template with PNA present (bottom) compared with a PCR product generated from wild-type template without PNA (top). The DNA sequences are shown above the chromatograms, with codons 12 and 13 underlined. B: The DNA sequence of the PNA-binding site in 30 PCR products amplified from wild-type DNA by the Platinum Taq and HotGoldStar PNA clamp PCR assays, compared with the wild-type K-ras sequence. Only the introduced mutations are shown explicitly. Peaks higher than 10% of the mean peak height of the wild-type bases in the sequencing chromatograms were judged as introduced mutations.
Discussion
The low fidelity of Taq DNA polymerase has consequences for many applications of the PCR. PCR-induced errors may cause cloning errors and overestimation of genetic diversity.19,20 In the present study we have demonstrated that polymerization errors also influence methods for sensitive detection of genetic variation. Selective amplification of mutant templates by PNA clamping of the wild-type template also favored amplification of PCR products with polymerase-induced errors within the PNA-binding site. We were able to increase the sensitivity of our PNA clamp PCR assay for K-ras mutations 10-fold by using a high-fidelity DNA polymerase instead of Taq DNA polymerase.
The reported error rate of Taq DNA polymerase on different templates varies from 2 × 10−4 to 1 × 10−5 errors per nucleotide.21,22 The number of genomes in the template DNA added to a single PCR (here, 200 ng) can be estimated to about 6 × 104. The PNA binds to only one DNA strand, allowing linear amplification of the other strand during the PCR (Figure 2). If a fidelity of 1 × 10−5 is assumed for this template, it can be estimated that Taq polymerase would make about ten mistakes in the PNA-target region in average during the first cycle. During the next cycles, the PNA binding to the newly synthesized strands containing errors in the PNA-binding site would be reduced or completely inhibited, depending on the kind of mutation, and exponential amplification could occur. Thus, polymerization errors seem to be a plausible explanation for the mutations we observed when analyzing wild-type template with PNA present.
Studies on the importance of the position of the DNA/PNA mismatch have indicated that mismatches in the center of the PNA/DNA duplex is more destabilizing than mismatches in the ends.23 This corresponded well with our observations that the amplified errors mainly resided in the center of the PNA target (Figure 5). More interestingly, we observed an overrepresentation of G→A errors in the PCR products amplified from wild-type template with PNA present. Taq polymerase has been shown to have a preference for adenosine when adding non-templated extra nucleotides to the 3′ end of PCR products.24 A preference for adenosine insertion opposite abasic lesions in DNA has also been reported.25 We speculated whether Taq polymerase has a general preference for adenosine that could explain our observations. However, the reported base substitution specificity of Taq polymerase rather indicated a tendency toward T→C errors.22,26 Interestingly, studies of PCR mutagenesis have shown that the mutational specificity of Taq polymerase can be changed easily by using different conditions for the PCR.27 In accordance with this, we observed a somewhat wider mutational specificity for Platinum Taq than for HotGoldStar (Figure 5B).
Another explanation for the observed G→A bias could be that the C/A mismatch obtained is more destabilizing for the PNA/DNA duplex than other mismatches. Interestingly, there is evidence indicating that guanine substitutions destabilize PNA binding more than other substitutions.6,10 However, the reported studies show only minor differences among G→A, G→C, and G→T substitutions with regard to the stability of the DNA/PNA duplex.
Although the peaks corresponding to introduced mutants in the sequencing chromatograms were quite convincing, the wild-type peaks in the same position were usually higher (Figure 5A). If all mutant peaks correspond to one type of mutant PCR product, this could indicate that the main reason for exponential amplification from wild-type template is incomplete PNA-binding and not polymerase errors. However, this model would not explain why we achieved substantially higher sensitivity when applying a high-fidelity DNA polymerase. A potentially better explanation is that multiple mutants are introduced separately in the first part of the PCR and afterward compete during the amplification. Possibly, all kinds of mutations are introduced in the PNA-target, but only the most destabilizing mutants become sufficiently amplified to be observed by sequencing.
The sensitivity of our Taq-based PNA clamp PCR assays for mutant K-ras DNA in large excess of wild-type DNA were determined to be about 1:1000. This is similar to most reported studies using PNA clamp PCR for sensitive detection of point mutations.12,28,29,30 We here report the first PNA clamp PCR assay with increased sensitivity (1:20,000) due to the use of a high-fidelity DNA polymerase. Considering the fact that the cell line used to determine the sensitivity (LS174T) was heterozygous for the K-ras mutation, the sensitivity was even higher (1:40,000). However, Däbritz et al17 have reported a PNA clamp PCR assay for K-ras codon 12 mutations with sensitivity up to 1:1 × 105 despite the use of Platinum Taq DNA polymerase. The sensitivity was determined by diluting colon carcinoma cells in a large excess of peripheral blood cells (unclear whether nucleated or not). The apparent increase in sensitivity seems to be achieved by the use of a mutation-specific hybridization probe. With that approach, melting curve analysis of the PCR products can distinguish the specific mutant PCR product from wild-type and other mutants. Thus, mutation-specific probes seem to be a way to avoid the problem with polymerase-introduced errors. However, the hybridization probe cannot distinguish a real mutation in the original template from one introduced by the polymerase during PCR. In view of our results we emphasize the possibility for false-positives due to polymerase-induced errors matching the probe. Luo et al18,31 have recently reported another PNA clamp PCR assay for K-ras mutations using the wild-type PNA as a hybridization probe. In their discussion of possible limitations, they mention the possibility that errors introduced by Taq polymerase can lead to false-positive results when the amount of mutant template is less than 0.1% of the wild-type alleles present.31 They suggest using a high-fidelity DNA polymerase to reduce the problem, an idea that has now been confirmed experimentally by us.
The strategy with mutation-specific hybridization probes also suffers from the drawback that multiple hybridization probes are needed to cover all possible mutations in K-ras codon 12 and codon 13. There are 12 possible missense mutations in codons 12 and 13, of which several are common. This would require up to 12 different mutation-specific probes to cover the mutational spectrum. In contrast, our single PNA clamp PCR assay detected different mutations in codons 12 and 13 with similar efficiency (results not shown).
Using amplification curves to detect mutations introduces another challenge. The amplification curves of a sample must be compared with the amplification curve of wild-type template to judge whether it has a mutation or not. We have chosen to compute ΔCt values (see Materials and Methods) to adjust for varying DNA quality and concentration in preparations from clinical samples. Only samples with ΔCt values significantly lower than wild-type should be classified as positive for K-ras mutations (Figures 3 and 4). The apparent log-linear relationship between the ΔCt value and the dilution factor of mutant template in wild-type template suggests that the assay has a quantitative potential. In a context of minimal residual disease detection, this could be applied to obtain a relative measure for the number of tumor cells present in the clinical sample.
We have herein emphasized that DNA polymerase errors seriously affect the performance of PNA clamp PCR assays and that these assays can be enhanced by high-fidelity DNA polymerases. However, DNA polymerase errors need consideration also for other types of assays based on selective amplification of genetic variants, such as allele-specific amplification- and restriction endonuclease-mediated selective amplification-PCR. It is likely that both of these methods will benefit from high-fidelity DNA polymerases. Thus, continued effort in the development of DNA polymerase systems with higher fidelities is in the interest of several fields of molecular diagnostics.
Footnotes
Supported by the Western Norway Regional Health Authorities.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
References
- 1.Motojima K, Tsunoda T, Kanematsu T, Nagata Y, Urano T, Shiku H. Distinguishing pancreatic carcinoma from other periampullary carcinomas by analysis of mutations in the Kirsten-ras oncogene. Ann Surg. 1991;214:657–662. doi: 10.1097/00000658-199112000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghaneh P, Kawesha A, Evans JD, Neoptolemos JP. Molecular prognostic markers in pancreatic cancer. J Hepatobiliary Pancreat Surg. 2002;9:1–11. doi: 10.1007/s005340200000. [DOI] [PubMed] [Google Scholar]
- 3.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 4.Dieterle CP, Conzelmann M, Linnemann U, Berger MR. Detection of isolated tumor cells by polymerase chain reaction-restriction fragment length polymorphism for K-ras mutations in tissue samples of 199 colorectal cancer patients. Clin Cancer Res. 2004;10:641–650. doi: 10.1158/1078-0432.ccr-1355-02. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 6.Ratilainen T, Holmen A, Tuite E, Nielsen PE, Norden B. Thermodynamics of sequence-specific binding of PNA to DNA. Biochemistry. 2000;39:7781–7791. doi: 10.1021/bi000039g. [DOI] [PubMed] [Google Scholar]
- 7.Ray A, Norden B. Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for the future. FASEB J. 2000;14:1041–1060. doi: 10.1096/fasebj.14.9.1041. [DOI] [PubMed] [Google Scholar]
- 8.Thenmalarchelvi R, Yathindra N. New insights into DNA triplexes: residual twist and radial difference as measures of base triplet non-isomorphism and their implication to sequence-dependent non-uniform DNA triplex. Nucleic Acids Res. 2005;33:43–55. doi: 10.1093/nar/gki143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nielsen PE: Peptide Nucleic Acids: Protocols and Applications. Edited by Nielsen PE. Wymondham, Horizon Bioscience 2004.
- 10.Orum H, Nielsen PE, Egholm M, Berg RH, Buchardt O, Stanley C. Single base pair mutation analysis by PNA directed PCR clamping. Nucleic Acids Res. 1993;21:5332–5336. doi: 10.1093/nar/21.23.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiede C, Bayerdorffer E, Blasczyk R, Wittig B, Neubauer A. Simple and sensitive detection of mutations in the ras proto-oncogenes using PNA-mediated PCR clamping. Nucleic Acids Res. 1996;24:983–984. doi: 10.1093/nar/24.5.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taback B, Bilchik AJ, Saha S, Nakayama T, Wiese DA, Turner RR, Kuo CT, Hoon DS. Peptide nucleic acid clamp PCR: a novel K-ras mutation detection assay for colorectal cancer micrometastases in lymph nodes. Int J Cancer. 2004;111:409–414. doi: 10.1002/ijc.20268. [DOI] [PubMed] [Google Scholar]
- 13.Prix L, Uciechowski P, Bockmann B, Giesing M, Schuetz AJ. Diagnostic biochip array for fast and sensitive detection of K-ras mutations in stool. Clin Chem. 2002;48:428–435. [PubMed] [Google Scholar]
- 14.Ritter M, Kim TD, Lisske P, Thiede C, Schaich M, Neubauer A. Prognostic significance of N-RAS and K-RAS mutations in 232 patients with acute myeloid leukemia. Haematologica. 2004;89:1397–1399. [PubMed] [Google Scholar]
- 15.Su YH, Wang M, Aiamkitsumrit B, Brenner DE, Block TM. Detection of a K-ras mutation in urine of patients with colorectal cancer. Cancer Biomark. 2005;1:177–182. doi: 10.3233/cbm-2005-12-305. [DOI] [PubMed] [Google Scholar]
- 16.Chen CY, Shiesh SC, Wu SJ. Rapid detection of K-ras mutations in bile by peptide nucleic acid-mediated PCR clamping and melting curve analysis: comparison with restriction fragment length polymorphism analysis. Clin Chem. 2004;50:481–489. doi: 10.1373/clinchem.2003.024505. [DOI] [PubMed] [Google Scholar]
- 17.Dabritz J, Hanfler J, Preston R, Stieler J, Oettle H. Detection of Ki-ras mutations in tissue and plasma samples of patients with pancreatic cancer using PNA-mediated PCR clamping and hybridisation probes. Br J Cancer. 2005;92:405–412. doi: 10.1038/sj.bjc.6602319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo JD, Chan EC, Shih CL, Chen TL, Liang Y, Hwang TL, Chiou CC. Detection of rare mutant K-ras DNA in a single-tube reaction using peptide nucleic acid as both PCR clamp and sensor probe. Nucleic Acids Res. 2006;34:e12. doi: 10.1093/nar/gnj008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz MF. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl Environ Microbiol. 2005;71:8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Sahota A, Stambrook PJ, Tischfield JA. Polymerase chain reaction amplification and sequence analysis of human mutant adenine phosphoribosyltransferase genes: the nature and frequency of errors caused by Taq DNA polymerase. Mutat Res. 1991;249:169–176. doi: 10.1016/0027-5107(91)90143-c. [DOI] [PubMed] [Google Scholar]
- 21.Eckert KA, Kunkel TA. DNA polymerase fidelity and the polymerase chain reaction. PCR Methods Appl. 1991;1:17–24. doi: 10.1101/gr.1.1.17. [DOI] [PubMed] [Google Scholar]
- 22.Tindall KR, Kunkel TA. Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry. 1988;27:6008–6013. doi: 10.1021/bi00416a027. [DOI] [PubMed] [Google Scholar]
- 23.Igloi GL. Variability in the stability of DNA-peptide nucleic acid (PNA) single-base mismatched duplexes: real-time hybridization during affinity electrophoresis in PNA-containing gels. Proc Natl Acad Sci USA. 1998;95:8562–8567. doi: 10.1073/pnas.95.15.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark JM. Novel non-templated nucleotide addition reactions catalyzed by procaryotic and eucaryotic DNA polymerases. Nucleic Acids Res. 1988;16:9677–9686. doi: 10.1093/nar/16.20.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Randall SK, Eritja R, Kaplan BE, Petruska J, Goodman MF. Nucleotide insertion kinetics opposite abasic lesions in DNA. J Biol Chem. 1987;262:6864–6870. [PubMed] [Google Scholar]
- 26.Suzuki M, Yoshida S, Adman ET, Blank A, Loeb LA. Thermus aquaticus DNA polymerase I mutants with altered fidelity. Interacting mutations in the O-helix. J Biol Chem. 2000;275:32728–32735. doi: 10.1074/jbc.M000097200. [DOI] [PubMed] [Google Scholar]
- 27.Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 28.Sotlar K, Escribano L, Landt O, Mohrle S, Herrero S, Torrelo A, Lass U, Horny HP, Bultmann B. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol. 2003;162:737–746. doi: 10.1016/S0002-9440(10)63870-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hancock DK, Schwarz FP, Song F, Wong LJ, Levin BC. Design and use of a peptide nucleic acid for detection of the heteroplasmic low-frequency mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) mutation in human mitochondrial DNA. Clin Chem. 2002;48:2155–2163. [PubMed] [Google Scholar]
- 30.Tanaka T, Nagai Y, Miyazawa H, Koyama N, Matsuoka S, Sutani A, Huqun, Udagawa K, Murayama Y, Nagata M, Shimizu Y, Ikebuchi K, Kanazawa M, Kobayashi K, Hagiwara K. Reliability of the peptide nucleic acid-locked nucleic acid polymerase chain reaction clamp-based test for epidermal growth factor receptor mutations integrated into the clinical practice for non-small cell lung cancers. Cancer Sci. 2007;98:246–252. doi: 10.1111/j.1349-7006.2006.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiou CC, Luo JD, Chen TL. Single-tube reaction using peptide nucleic acid as both PCR clamp and sensor probe for the detection of rare mutations. Nat Protocols. 2006;1:2604–2612. doi: 10.1038/nprot.2006.428. [DOI] [PubMed] [Google Scholar]