

Abstract
A disparity between Caucasian and Hispanic mutation detection for cystic fibrosis continues to exist, although the carrier frequency is only moderately lower in Hispanics. We aimed to identify exonic rearrangements that remained undetected by conventional methods. In seven of 32 cystic fibrosis-affected self-identified Hispanics for whom only one or no mutations were identified by extensive molecular testing, exon deletions appeared to be present with a multiplex ligation-dependent probe amplification (MLPA) assay. Two recurrent deletions (of exons 2–3 and exons 22–23) were identified in one and three patients, respectively (12.5%, 11.1% of unidentified alleles). Two apparently novel deletions (exons 6b and 20) were identified in three additional patients. Subsequent sequencing to characterize deletion breakpoints, however, identified single nucleotide deletions at the probe binding sites close to the ligation point. All resulted in false positive MLPA deletion signals. Interestingly, these mutations were not common in Caucasians, and one (935delA) was common in U.S. Hispanics. On examination of all probe binding sites, we identified a total of 76 reported mutations and five silent variants that immediately surrounded the MLPA ligation sites, with 22 occurring in non-Caucasians. These mutations are not all rare. Thus, apparent exon deletions by MLPA may indicate the presence of both large deletions and point mutations, with important implications for pan-ethnic MLPA testing in cystic fibrosis and other genetic conditions.
Cystic fibrosis (CF; OMIM #219700) is a common and serious condition with autosomal recessive inheritance. It is caused by mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR; OMIM *602421) on chromosome 7q31 and is characterized by malfunction of chloride ion channels and of transport pathway regulation.1,2 CFTR is primarily expressed in the apical membrane of exocrine epithelial cells. Classic CF is characterized by failure to thrive, recurrent bacterial endobronchitis, progressive decline of lung function, exocrine pancreatic dysfunction, and infertility in males.3,4 The phenotype is variable, however, and ranges from mild with limited manifestations to rapid deterioration and death within the first year of life. The severity of clinical manifestations depends on the set of CFTR mutations, modifying genes, and other variables.5,6,7
To date, more than 1500 sequence variants have been reported to the Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca/cftr/). These include point mutations, deletions, insertions, frameshift mutations, and splice site variants that lead to protein truncation. The incidence of CF (approximately one in 3000 individuals) is highest in Caucasians and Ashkenazi Jews, which are the most extensively studied populations.8,9 In other ethnic backgrounds, the incidence is lower and knowledge of mutation spectra is still somewhat limited. It is clear, however, that the range and frequency of individual CFTR mutations varies considerably among different populations, ethnic backgrounds, and geographic locations.10,11 In Hispanics, the mutation spectrum remains relatively poorly defined despite a carrier frequency that is only moderately lower than that in Caucasians (∼1 in 46 compared with ∼1 in 29, respectively). “Hispanic,” although difficult to strictly define, generally refers to the self-assigned ethnicity by individuals from Latin America, Central America, South America, Spain, and Portugal.12 The population of Hispanics continues to grow both in absolute terms and relative to other subpopulations. In California, one of every two children born in 2001–2005 was Hispanic (http://ww2.cdph.ca.gov/data/statistics/Pages/default.aspx), and it is expected that approximately one-third of all new CF patients in this state will be of Hispanic origin (Martin Kharrazi, California Department of Public Health, September 14, 2007, personal communication). Identification of CFTR gene mutations in Hispanic CF patients can facilitate diagnosis, improve screening and management, and add value to genetic counseling.
After screening for 30 to 70 known mutations with commercially available DNA testing panels, temporal temperature gradient electrophoresis and direct DNA sequencing have been used to detect unknown mutations.13,14 By using a commercial panel of 87 mutations combined with temporal temperature gradient electrophoresis mutation analysis, the overall mutation detection rate for Hispanic individuals with CF in California could be raised to 94.5%.10 With other methods, a limited number of exonic rearrangements of CFTR have been identified (Table 1), suggesting that exon deletions and duplications are of clinical importance in the etiology of CF, albeit in a minority of cases. The overall frequency of large rearrangements is probably underestimated because of the testing methods commonly used, but is likely to account for several percentages of all affected alleles. Until now, large exonic rearrangements have not been studied specifically among Hispanics. To better characterize the mutation spectrum in Hispanics, we aimed to identify large deletions or duplications in the CFTR gene of Hispanic CF patients whose mutations remained undetected after conventional DNA sequence testing.
Table 1.
CFTR Rearrangements Involving Exons
| Exons | Type of rearrangment | Reported in Hispanics | References |
|---|---|---|---|
| 1–24 (all exons) | Del | 40, 47 | |
| Promoter-1 | Del | 32 | |
| Promoter-2 | Del | 32 | |
| 1 | Del | 19, 40 | |
| 2 | Del/Indel | 40, 45, 46 | |
| 2–3 | Del | X | 17, 32, 39 |
| 2–9 | Del | 45 | |
| 2–10 | Del | 41 | |
| 3–10, 14b–16 | Del | 40, 44 | |
| 4 | Del | 19 | |
| 4–6a | Del | 19, 32 | |
| 4–7, 11–18 | Del | 34 | |
| 4–8 | Dup | 40 | |
| 4–10 | Del | 41 | |
| 6b-10 | Dup | 48 | |
| 11–13 | Dup | 47 | |
| 11–16 | Del | 19 | |
| 14a | Del | 36 | |
| 14b–17b | Del | 40, 41, 45 | |
| 16–17b | Del | 41 | |
| 17a–17b | Del | 32, 45 | |
| 17a–18* | Del | X | 32, 37, 43, 47 |
| 17b | Del | 35 | |
| 20 | Del | X | 33 |
| 22–23 | Del | X | 19, 37, 47 |
| 22–24 | Del | X | 31, 32, 41, 46 |
| 24 | Del | X | 33 |
Del, genomic rearrangement involves a deletion; Dup, genomic rearrangement involves a duplication; Indel, genomic rearrangement involves an insertion and deletion.
Two different types of deletions involving exons 17a and 18 have been reported.
Materials and Methods
Subjects
The study consisted of 32 Hispanic probands with classic CF who had at least one unidentified mutation each. In a previous study,10 25 of the samples underwent comprehensive mutation analysis using a commercial screening panel of 87 mutations. If only one mutation or no mutations were identified, the DNA was subsequently used for mutation analysis by temporal temperature gradient electrophoresis. The 25 samples were enrolled in the present deletion study because molecular testing thus far had failed to identify the two expected mutations. At the time of this hemi-blinded study, it was unknown to the investigators who performed mutation analysis by multiplex ligation-dependent probe amplification (MLPA) of mutations previously identified. An additional seven samples collected from self-identified Hispanics at Stanford University were previously studied by complete DNA sequencing of the 27 CFTR exons but in none of these affected individuals were two mutations identified. Human subject protocols were approved by the California Health and Human Services Agency Committee for the Protection of Human Subjects (project 99-08-11) and by the Institutional Review Boards of the medical centers of study subjects.
MLPA
First described by Schouten et al,15 MLPA enables detection of deletions and duplications up to several kilobases in size by screening for the loss or gain of up to 45 target sequences in a single reaction. Because MLPA is a screening method, however, changes should be confirmed with another independent assay.16 MLPA is an efficient method to target all 27 exons of the CFTR gene, and we applied this method (MRC-Holland, Amsterdam, The Netherlands) to investigate deletions and duplications in the gene. Because we obtained consistent results with half the volume of polymerase chain reaction (PCR) reagents outlined in the MLPA procedure, these reagents were halved in our protocol. After completion of the MLPA reaction, amplified products were separated by capillary electrophoresis performed on an ABI-310 Genetic Analyzer (Applied Biosystems). PCR fragments were then quantified with ABI Gene Scan Analysis software. The relative peak size of the product from the probe recognition sequence was compared with the relative peak size of the product from a control; a 35 to 50% reduction indicated an exon deletion. The MLPA interpretation was facilitated by GeneMarker genotyping software (Softgenetics).
Confirmation of Suspected Deletions
The validity of deletions or duplications detected by MLPA needs to be demonstrated by confirmation with an independent method. An exception to this recommendation could perhaps be made for well characterized exon deletions identified with a validated diagnostic assay in a molecular diagnostic laboratory. The frequently contiguous exon deletions observed in Duchenne muscular dystrophy could be an example of such a scenario. The same deletions may be seen repeatedly, and the additional information from flanking exons may support the deletion findings.
The MLPA assay indicates which exons may be missing but does not specifically identify the breakpoints of a deletion. We initially applied two approaches to support and subsequently confirm the presence of deletions identified by MLPA. First, the Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca/cftr/) was searched for known single-nucleotide polymorphisms (SNPs) in the CFTR gene. Primer pairs were designed around the sites of SNPs located within the exons suspected to be deleted (Table 2). After optimization and amplification, the PCR products that encompassed these SNPs were sequenced. Homozygosity present at the SNP site(s) initially supports the notion that an exon may have been deleted but does not confirm it. The second and more direct approach involved the characterization of the actual deletion breakpoints. For each suspected deletion, forward and reverse primers were designed in the flanking introns. For the deletion of exons 2–3, a reported primer pair was used.17 For confirmation of the deletion of exons 22–23, we designed the following primer pair from GenBank reference sequence NM_000492: forward 5′-TAGCACCAAGGATGATGTCAT-3′ and reverse 5′-TCCAAGA TCAGACAATAGAGG-3′ (http://www.ncbi.nlm.nih.gov/). Genomic DNA was amplified using Amplitaq Gold Polymerase (Perkin Elmer), and the most common PCR program for CFTR exons18 was applied for the majority of PCR reactions: 1 cycle, 94°C for 10 minutes; 35 cycles, 94°C for 30 seconds, 55°C for 30 seconds, 72°C for 1 minute; 1 cycle, 72°C for 7 minutes. To capture the deletion of exons 2–3, the annealing temperature was increased to 57°C. If the size of the amplified product on the agarose gel was shorter than expected, then the product was sequenced. PCR products were either purified directly or cut out from the agarose gel and purified by using QIAquick PCR purification columns (Qiagen). The samples were sequenced with BigDye Terminator v.3.1 (Applied Biosystems) and electrophoresed on an ABI 310 or 377 sequencing instrument (Applied Biosystems).
Table 2.
SNPs Sequenced to Test the Hypothesis That Only One Copy of an Allele Is Present
| Detected exon deletion | SNPs | Primers | Subject number |
|---|---|---|---|
| 2, 3 | 296+129G/C | F: 5′-GACAGTCACATTAGTTCAGAGAT-3′* | H17 |
| R: 5′-TATCAAACTCCTGGTCTCAAGC-3′ | |||
| 2, 3 | 297-67A/C | F: 5′-CAAATATCTGGCTGAGTGTT-3′ | H17 |
| 297-50A/G | R: 5′-ATTCACCAGATTTCGTAGTC-3′* | ||
| 6b | 1001+11C/T | F: 5′-TGGAATGAGTCTGTACAGCG-3′* | 16, 31 |
| 1001+12C/T | R: 5′-CTACAGCCCATGAAAGTGAAT-3′ | ||
| 20 | 3939C/T | F: 5′-GGTCAGGATTGAAAGTGTGCA-3′* | 30 |
| 4005+28insA | R: 5′-CTATGAGAAAACTGCACTGGA-3′* | ||
| 22, 23 | 4269-139G/A | F: 5′-TTCTCAGTAAGGCGAAGAT-3′ | 1, 5, 20 |
| R: 5′-AAAATTGTTGGCATTCCAGC-3′ |
F, forward; R, reverse.
Reference 20.
Results
Identification of Large and Small Exonic Deletions
Using MLPA analysis, seven of the 32 Hispanic study subjects with CF (21.9%) demonstrated peak height reductions of 35 to 50% when compared with the associated controls, suggesting the presence of exon deletions. Four of these seven subjects were suspected to have recurrent CFTR deletions of exons 2–3 and exons 22–23, because these deletions had been reported previously (Table 1) and the same deletion pattern was detected in one (subject H17) and three (subjects 1, 5, 20) samples, respectively. Breakpoints were confirmed by direct DNA sequencing. The deletion of exons 2–3 was confirmed with a previously reported primer pair.17 For the deletion of exons 22–23, we designed a new primer pair and confirmed another previously identified breakpoint.19
Based on MLPA results, two apparently novel deletions were identified in four additional patients: a deletion of exon 6b in subjects 16 and 31 and a deletion of exon 20 in subject 30 (Figure 1). The presence of a homozygous nucleotide at a SNP site presents fast and simple, albeit indirect, support for the presence of only one copy of the exon at the site of a proposed deletion. The SNPs that were sequenced in this context were identified by a search of sequence variants in the Cystic Fibrosis Mutation Database. Relevant SNPs (Table 2) located within the potentially deleted regions included 296+129G/C (listed in the CF mutation database as 296+128), 297-67A/C, and 297-50A/G (for exon 2–3); 1001+11C/T, 1001+12C/T (for exon 6b); 3939C/T, 4005+28insA (for exon 20); and 4269-139G/A (for exon 22–23). The SNPs used to investigate the deletion of exons 2 and 3 and to assess the deletion of exons 22 and 23 were indeed homozygous (data not shown). However, the sequences for the other three study subjects were poor and appeared to harbor frameshift mutations. Single nucleotide deletions were found in all three individuals. We identified mutations 935delA and 3961delA in exons 6b and 20, respectively. On examination of the position of these small deletions relative to the MLPA probes, it was discovered that they were close to the ligation site. The CFTR sequence around the ligation site was ATTTTCAA—TCATTTCT for mutation 935delA in exon 6b, and CTTGGGAT—TCAATAAC for mutation 3961delA in exon 20, showing the nucleotides in the probe, corresponding to the position of the deleted nucleotides, in bold.
Figure 1.
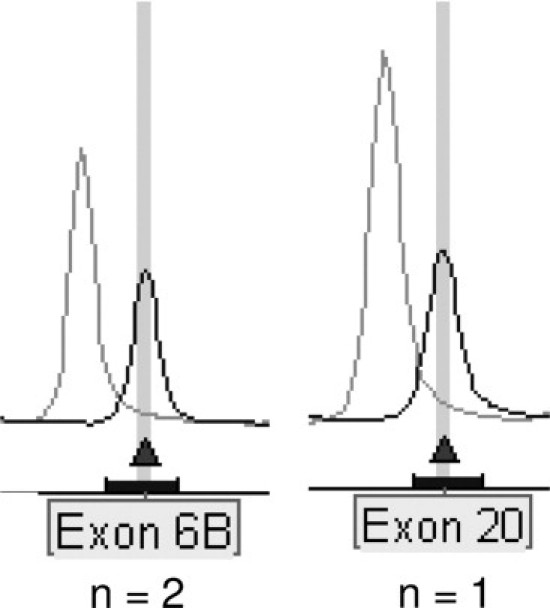
Peak pattern of three suspected exon deletions in CFTR. A 35 to 50% reduction of relative peak height of the amplified product (compared to a corresponding control) suggested two apparently novel deletions in three patients: deletion of exon 6b (two subjects) and deletion of exon 20.
Other Mutations
With the discovery of “false positive” exon deletion signals by MLPA we examined the origin of the identified single nucleotide mutations. Interestingly, neither of the two small deletions are common in Caucasians. The 935delA mutation in exon 6b was first described in two unrelated Hispanic patients who, respectively, carried the 663delT and the ΔF508 mutation on the other allele. Each had a severe course of CF.20 This frameshift mutation has been described in both U.S. Hispanics and Mexican probands13,20,21,22 and is a relatively common mutation identified in U.S. Hispanics.14 The second frameshift mutation, 3961delA, was reported in one pancreatic insufficient Hispanic patient in whom the second allele remained obscure.10
Given that none of these three mutations is common in Caucasians, we hypothesized that the probe binding and ligation sequences of the otherwise proprietary probes in this MLPA assay (MRC-Holland) may cover additional non-Caucasian mutations. If so, the assay could possibly be used to screen for small insertions and deletions in addition to exon deletions and duplications, especially in non-Caucasian populations. A total of 76 reported mutations and five silent variants listed in the Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca/cftr/) are located within the sequences that immediately surround the MLPA ligation sites (Table 3). Ethnic information is not provided for all mutations, but most of these appear to be rare or private and none are part of the ACMG/ACOG recommended CFTR carrier screening panel. A subset of 22 mutations were reported to occur in non-Caucasian individuals, including nine in Hispanics, five in individuals from the greater Middle East, four in Asians, and four in individuals of African ancestry. Interestingly, these mutations were not all rare. One such recurrent mutation was identified to occur under an MLPA probe site in this study (935delA) and seems to be prevalent in Hispanics. The 3791delC mutation in exon 19 was first reported in an African-American individual with pancreatic insufficient CF.23 The second allele, however, was not identified in this patient. In African Americans, 3791delC has been reported multiple times in both carriers and patients23,24 and has been included in some commercially available CF assays. This mutation has demonstrated a false positive MLPA result in an African-American study subject (data not shown). Deletion 936delTA, similar to 935delA, is located within the MLPA probe binding site in exon 6b. It has also been observed in multiple unrelated probands of Hispanic origin.25,26 Mutation 2766del8 in exon 14b was identified in five of 60 non-consanguineous CF chromosomes from Tunisia, suggesting that this might be a common mutation in that country.27 Mutation 444delA in exon 4 has been reported multiple times as well (23,24, http://www.genet.sickkids.on.ca/cftr/) and is considered a specific African-American mutation.24 Finally, mutation H1085R has been described repeatedly (http://www.genet.sickkids.on.ca/cftr/) and was identified in one homozygous Japanese patient who was born to consanguineous parents.28 Thus, of the 22 mutations described in non-Caucasian populations, at least six (27.3%) are recurrent and may be specific to non-Caucasian populations.
Table 3.
Mutations under MLPA Ligation Sites
| Exon | Probe length (nt) | Ligation site sequence | Mutations in area of ligation site sequence* |
|---|---|---|---|
| 1,5′ UTR | 154 | 5′-GAGCAAAT-TTGGGGCC-3′ | N/A |
| 1,5′ UTR | 238 | 5′-AAAGGGTT-GAGCGGCA-3′ | |
| 2 | 198 | 5′-TTGGTATA-TGTCTGAC-3′ | (5) |
| 3 | 136 | 5′-CTGCTAGT-GTTGCCAA-3′ | (3) |
| 3 | 220 | 5′-TTCAAAGA-AAAATCCT-3′ | |
| 4 | 247 | 5′-AGAATCAT-AGCTTCCT-3′ | 444delA, African; 451del8, Chinese; (6) |
| 5 | 346 | 5′-AAATAAGT-ATTGGACA-3′ | Q179K, Hispanic (7) |
| 6a | 274 | 5′-GAGTTGTT-ACAGGCGT-3′ | L218X, Pakistani (4) |
| 6b | 301 | 5′-ATTTTCAA-TCATTTCT-3′ | 935delA, Hispanic; 936delTA, Hispanic (3) |
| 7 | 337 | 5′-ACTTCAAT-AGCTCAGC-3′ | S307N, Turkish (9) |
| 8, IVS 8 | 364 | 5′-TTTCTAGA-TTAAGAAG-3′ | N/A |
| 9, IVS 8 | 391 | 5′-TCCATCAC-ACTGGTAG-3′ | N/A |
| 10 | 463 | 5′-TCCACTGT-GCTTAATT-3′ | H484Y, Hispanic; S485C, Chinese-Caucasian (5) |
| 11 | 418 | 5′-CAGAGAAA-GACAATAT-3′ | K536X, Iranian; 1742delAC, Japanese (5) |
| 12, IVS 12 | 292 | 5′-TGCATTTT-ACCTCTTG-3′ | N/A |
| 13 | 142 | 5′-CAGATTCT-GAGCAGGG-3′ | (1) |
| 14a | 160 | 5′-GTATGTGT-TCCATGTA-3′ | (3) |
| 14b | 178 | 5′-CTGCTTCT-TTGGTTGT-3′ | 2766del8, Tunisian (1) |
| 15 | 204 | 5′-GCTTGCTA-TGGGATTC-3′ | (1) |
| 16, IVS 16 | 229 | 5′-GATGTAAT-AGCTGTCT-3′ | N/A |
| 17a | 256 | 5′-TGCAACAA-AGATGTAG-3′ | 3171delC, Hispanic; 3173delAC, Turkish; F1016S, Hispanic (5) |
| 17b | 283 | 5′-CAGTATGT-AAATTCAG-3′ | H1085R, Japanese (4) |
| 18 | 310 | 5′-CCATGAAT-ATCATGAG-3′ | M1137R, Hispanic (6) |
| 19 | 353 | 5′-TCTGTGTA-TTTTGCTG-3′ | 3791delC, African-American (2) |
| 20 | 382 | 5′-CTTGGGAT-TCAATAAC-3′ | 3960delA, Hispanic (2) |
| 21 | 409 | 5′-TGCAACTT-TCCATATT-3′ | W1316X, African-American (2) |
| 22 | 436 | 5′-GAACAGTT-TCCTGGGA-3′ | No mutations |
| 23 | 148 | 5′-CCAGCATT-GCTTCTAT-3′ | M1407T, Turkish; E1409K, Hispanic (2) |
| 24 | 190 | 5′-ATCCAGAA-ACTGCTGA-3′ | No mutations |
| 24 | 172 | 5′-CTCCTCTT-TCAGAGCA-3′ |
UTR, untranslated region; IVS, intervening sequence; N/A, not applicable, probes not in coding region; No mutations, no reported mutations are present in the area of the ligation site sequence, regardless of ethnicity.
Without parentheses, non-Caucasian mutations. In parentheses, total number of mutations in area of ligation site sequence.
Discussion
In many Caucasians with clinical manifestations consistent with CF, the associated genotypes have been identified, but in Hispanic individuals the mutation spectrum has not yet been characterized as thoroughly. If the spectrum of mutations causing CF in the Hispanic population can be better defined, the potential to screen and diagnose CF earlier in life becomes routinely possible through the implementation of population-appropriate mutation testing methods. The discovery of novel population-specific mutations meets a clinical need for both targeted carrier testing and for diagnostic evaluation of CF.
To date, a limited number of comprehensive mutation analysis studies in Hispanics have been reported from the United States.10,13,14,20,29,30 Differences in detected mutations among these studies may be due in part to the fact that the Hispanic population is not homogeneous and is typically defined more by geographic origins in various regions of southern Europe or the Americas, rather than by one specific genetic background.12 Overall, mutation detection in the CFTR gene has largely focused on point mutations, small deletions and small insertions within or close to exons. The actual frequency of large rearrangements may still be underestimated, because the majority of methods used for routine mutation analysis are not suited for detecting gross genomic changes. In the past, deletions and duplications would only be detected through the appearance of uniparental inheritance, amplification failures in homozygously deleted samples, and changes on Southern blots. However, with the advent of techniques such as quantitative multiplex PCR of short fluorescent fragments, semiquantitative fluorescent PCR, semiquantitative fluorescent multiplex PCR, and MLPA, more genomic rearrangements in the CFTR gene have recently been identified, suggesting that exon deletions and duplications are of clinical importance in CF etiology (Table 1).17,19,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48 Our work is the first exon rearrangement study to specifically target Hispanic patients with CF.
Among CF patients who have already undergone extensive testing, up to 30% of pathogenic changes are not routinely identified. This number is influenced by the methods used and the population investigated.19 It has been estimated that the overall frequency of genomic rearrangements in the CFTR gene could approximate 3% of all affected alleles.45 Using recently developed techniques, previously unidentified alleles were elucidated in up to 24% of cases in the current literature (14%,32 16%,19,45 20%,40 24%41). These results are fairly consistent with our study of Hispanic individuals, in which four exon deletions were identified among 36 chromosomes without a characterized mutation (11.1%). Taulan et al46 studied patients with congenital bilateral absence of the vas deferens with one (n = 17) or zero (n = 15) previously identified CFTR mutations. Among these 47 alleles, two large rearrangements were identified (4.3%). These numbers may indicate that gross rearrangements are less common in patients with congenital bilateral absence of the vas deferens, although this study is potentially affected by selection bias. The infertility observed in the 15 individuals diagnosed with congenital bilateral absence of the vas deferens who had no CFTR mutations identified may have resulted from causes unrelated to this gene.
A few CFTR exon deletions have been described in Hispanic individuals with CF (Table 1). These deletions involved exons 2–3,17 exons 17a–18,32 exon 20,33 exons 22–23,32 exons 22–24, 32 and exon 24.33 The deletion of exon 20 was seen in three of 28 (10.7%) Hispanic CF chromosomes tested33 and could be relatively frequent in the Hispanic population. In our study using MLPA, we identified a deletion of exons 2–3 in one individual and confirmed that the deletion breakpoints were identical to a previous report.17 In three additional Hispanic patients we identified a deletion of exons 22–23 and determined that its breakpoints were congruent with those reported in other individuals.19 Although the combined results from this and other small studies do not yet allow an accurate estimate of CFTR exon deletion frequencies in Hispanic chromosomes, several of these deletions have been recurring (exon 20, exons 2–3, and exons 22–23) and may indicate relatively high frequencies in Hispanic CF patients. Thus, testing for genomic rearrangements appears to be a valuable addition to routine CF testing methods, both in Hispanic CF patients and in those of other ethnic groups, especially if one or both alleles remain to be identified despite comprehensive sequence analysis.
Genetic dosage determination is a valuable approach in research as well as clinical testing. MLPA15 has been successfully applied to the detection of exon deletions and duplications in a large number of conditions including CF (CFTR), Duchenne and Becker muscular dystrophy (DMD), breast cancer (BRCA1), and colon cancer (MLH1 and MSH2).49,50,51 In this study, the investigators were blinded to the results previously obtained by CFTR mutation panels and direct sequencing. The apparent deletions of exons 6b and 20 therefore were not immediately recognized as false positive deletion signals caused by sequence variations under the ligation probes. The two variants (935delA and 3961delA) were both small deletions that interfered with attachment of the probes and prevented ligation with subsequent amplification. Thus, an apparent exon deletion detected by MLPA may indicate either a large or a small deletion, potentially affecting test accuracy in any condition for which MLPA testing is applied. A review of the DNA sequence of an apparently deleted exon is prudent before extensive confirmation methods are applied or an attempt to determine the deletion breakpoints is made. The chance of small deletions under the ligation probe is always a possibility with this method, but it is even more likely in a condition such as CF, which is characterized by point mutations distributed throughout the gene. In this commercially available MLPA assay for CF, the probes were designed to avoid the most common CFTR mutations. However, we identified 76 reported mutations and five variants in the sequences under the exonic probes, and some of these changes are prevalent in populations less well studied than Caucasians (Table 3). Two of the mutations identified as false positive signals in our MLPA assay, 935delA and 3961delA, have been described in Hispanics. The 935delA mutation is a relatively common mutation in this population group. In all, at least six (444delA, 935delA, 936delTA, 2766del8, 3791delC, and H1085R) of 22 mutations identified in non-Caucasians (27.3%) are recurrent and may be specific to non-Caucasian populations. Small deletion mutations (5 of 6) are especially likely to interfere with probe hybridization. Thus, exonic deletions suggested by MLPA may indicate both point mutations and large deletions, with ramifications for pan-ethnic MLPA analysis for CF and other conditions with allelic heterogeneity. When considering potential use of MLPA as a targeted point mutation screen, it is important to realize that large sections of each exon will be missed. In addition, it is unclear how close to the ligation site an individual point deletion would have to occur before a reduction in MLPA peak height is detected. For any condition in which MLPA is considered as an early step, either because it is relatively economical compared with sequencing methods or because deletions and duplications are a common type of mutation in that disorder, sequence verification near the probe ligation site should always be performed as an initial check of data validity.
Acknowledgements
Suzanne Young was the project coordinator in charge of recruiting 25 of the subjects in this study, while Dr. Michelle Pearl provided epidemiological support and Steve Graham provided data management. We thank the physicians and staff at the Stanford Cystic Fibrosis Center and other Cystic Fibrosis Care Centers for their participation in recruiting patients.
References
- 1.Anderson MP, Rich DP, Gregory RJ, Smith AE, Welsh MJ. Generation of cAMP-activated chloride currents by expression of CFTR. Science. 1991;251:679–682. doi: 10.1126/science.1704151. [DOI] [PubMed] [Google Scholar]
- 2.Schwiebert EM, Egan ME, Hwang TH, Fulmer SB, Allen SS, Cutting GR, Guggino WB. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell. 1995;81:1063–1073. doi: 10.1016/s0092-8674(05)80011-x. [DOI] [PubMed] [Google Scholar]
- 3.Welsh MJ, Ramsey BW, Accurso F, Cutting G. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited diseases. 8th ed. McGraw-Hill; New York: 2001. pp. 5121–5188. [Google Scholar]
- 4.Robertson MB, Choe KA, Joseph PM. Review of the abdominal manifestations of cystic fibrosis in the adult patient. Radiographics. 2006;26:679–690. doi: 10.1148/rg.263055101. [DOI] [PubMed] [Google Scholar]
- 5.Mackie AD, Thornton SJ, Edenborough FP. Cystic fibrosis-related diabetes. Diabet Med. 2003;20:425–436. doi: 10.1046/j.1464-5491.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- 6.The Cystic Fibrosis Genotype-Phenotype Consortium Correlation between genotype and phenotype in patients with cystic fibrosis. N Engl J Med. 1993;329:1308–1313. doi: 10.1056/NEJM199310283291804. [DOI] [PubMed] [Google Scholar]
- 7.Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000;67:117–133. doi: 10.1159/000029497. [DOI] [PubMed] [Google Scholar]
- 8.Abeliovich D, Labon I, Cohen T, Springer C, Avital A, Cutting GR. Screening for five mutations detects 97% of cystic fibrosis (CF) chromosomes and predicts a carrier frequency of 1:29 in the Jewish Ashkenazi population. Am J Genet. 1992;51:951–956. [PMC free article] [PubMed] [Google Scholar]
- 9.Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ, Subcommittee on Cystic Fibrosis Screening Accreditation of Genetic Services Committee, ACMG American College of Medical Genetics Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med. 2001;3:149–154. doi: 10.1097/00125817-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Alper OM, Wong LJ, Young S, Pearl M, Graham S, Sherwin J, Nussbaum E, Nielson D, Platzker A, Davies Z, Lieberthal A, Chin T, Shay G, Hardy K, Kharrazi M. Identification of novel and rare mutations in California Hispanic and African American cystic fibrosis patients. Hum Mutat. 2004;24:353. doi: 10.1002/humu.9281. (Erratum: Hum Mutat 2005, 25:223) [DOI] [PubMed] [Google Scholar]
- 11.Estivill X, Bancells C, Ramos C. Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations: The Biomed CF Mutation Analysis Consortium. Hum Mutat. 1997;10:135–154. doi: 10.1002/(SICI)1098-1004(1997)10:2<135::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 12.Arzimanoglou II, Tuchman A, Li Z, Gilbert F, Denning C, Valverde K, Zar H, Quittell L, Arzimanoglou I. Cystic fibrosis carrier screening in Hispanics. Am J Hum Genet. 1995;56:544–547. [PMC free article] [PubMed] [Google Scholar]
- 13.Wong LJ, Wang J, Zhang YH, Hsu E, Heim RA, Bowman CM, Woo MS. Improved detection of CFTR mutations in Southern California Hispanic CF patients. Hum Mutat. 2001;18:296–307. doi: 10.1002/humu.1191. [DOI] [PubMed] [Google Scholar]
- 14.Schrijver I, Ramalingam S, Sankaran R, Swanson S, Dunlop CL, Keiles S, Moss RB, Oehlert J, Gardner P, Wassman ER, Kammesheidt A. Diagnostic testing by CFTR gene mutation analysis in a large group of Hispanics. J Mol Diagn. 2005;7:289–299. doi: 10.1016/S1525-1578(10)60557-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sellner L, Taylor G. MLPA and MAPH. New techniques for detection of gene deletions. Hum Mutat. 2004;23:413–419. doi: 10.1002/humu.20035. [DOI] [PubMed] [Google Scholar]
- 17.Dörk T, Macek M, Jr, Mekus F, Tümmler B, Tzountzouris J, Casals T, Krebsová A, Koudová M, Sakmaryová I, Macek M, Sr, Vávrová V, Zemková D, Ginter E, Petrova NV, Ivaschenko T, Baranov V, Witt M, Pogorzelski A, Bal J, Zékanowsky C, Wagner K, Stuhrmann M, Bauer I, Seydewitz HH, Neumann T, Jakubiczka S. Characterization of a novel 21-kb deletion, CFTRdele2,3 (21 kb), in the CFTR gene: a cystic fibrosis mutation of Slavic origin common in central and east Europe. Hum Genet. 2000;106:259–268. doi: 10.1007/s004390000246. [DOI] [PubMed] [Google Scholar]
- 18.Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics. 1991;10:214–228. doi: 10.1016/0888-7543(91)90503-7. [DOI] [PubMed] [Google Scholar]
- 19.Audrézet MP, Chen JM, Raguénès O, Chuzhanova N, Giteau K, Le Maréchal C, Quéré I, Cooper DN, Férec C. Genomic rearrangements in the CFTR gene: extensive allelic heterogeneity and diverse mutational mechanisms. Hum Mutat. 2004;23:343–357. doi: 10.1002/humu.20009. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Bowman CM, Wong LJ. A novel CFTR frame-shift mutation, 935delA, in two Hispanic cystic fibrosis patients. Mol Genet Metab. 2000;70:316–321. doi: 10.1006/mgme.2000.3021. [DOI] [PubMed] [Google Scholar]
- 21.Orozco L, Velázquez R, Zielenski J, Tsui LC, Chávez M, Lezana JL, Saldaña Y, Hernández E, Carnevale A. Spectrum of CFTR mutations in Mexican cystic fibrosis patients: identification of five novel mutations (W1098C, 846delT. P750L, 4160insGGGG and 297-1G→A) Hum Genet. 2000;106:360–365. doi: 10.1007/s004390051051. [DOI] [PubMed] [Google Scholar]
- 22.Kammesheidt A, Kharrazi M, Graham S, Young S, Pearl M, Dunlop C, Keiles S. Comprehensive genetic analysis of the cystic fibrosis transmembrane conductance regulator from dried blood specimens: implications for newborn screening. Genet Med. 2006;8:557–562. doi: 10.1097/01.gim.0000237793.19868.97. [DOI] [PubMed] [Google Scholar]
- 23.Macek M, Jr, Mackova A, Hamosh A, Hilman BC, Selden RF, Lucotte G, Friedman KJ, Knowles MR, Rosenstein BJ, Cutting GR. Identification of common cystic fibrosis mutations in African-Americans with cystic fibrosis increases the detection rate to 75% Am J Hum Genet. 1997;60:1122–1127. [PMC free article] [PubMed] [Google Scholar]
- 24.Sugarman EA, Rohlfs EM, Silverman LM, Allitto BA. CFTR mutation distribution among U.S. Hispanic and African American individuals: evaluation in cystic fibrosis patient and carrier screening populations. Genet Med. 2004;6:392–399. doi: 10.1097/01.gim.0000139503.22088.66. [DOI] [PubMed] [Google Scholar]
- 25.Chillon M, Casals T, Gimenez J, Nunes V, Estivill X. A cystic fibrosis patient homozygous for the new frameshift mutation 936delTA: description and clinical data. J Med Genet. 1994;31:369–370. doi: 10.1136/jmg.31.5.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chillón M, Casals T, Giménez J, Ramos MD, Palacio A, Morral N, Estivill X, Nunes V. Analysis of the CFTR gene confirms the high genetic heterogeneity of the Spanish population: 43 mutations account for only 78% of CF chromosomes. Hum Genet. 1994;93:447–451. doi: 10.1007/BF00201673. [DOI] [PubMed] [Google Scholar]
- 27.Messaoud T, Verlingue C, Denamur E, Pascaud O, Quere I, Fattoum S, Elion J, Ferec C. Distribution of CFTR mutations in cystic fibrosis patients of Tunisian origin: identification of two novel mutations. Eur J Hum Genet. 1996;4:20–24. doi: 10.1159/000472165. [DOI] [PubMed] [Google Scholar]
- 28.Yoshimura K, Wakazono Y, Iizuka S, Morokawa N, Tada H, Eto Y. A Japanese patient homozygous for the H1085R mutation in the CFTR gene presents with a severe form of cystic fibrosis. Clin Genet. 1999;56:173–175. doi: 10.1034/j.1399-0004.1999.560217.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Bowman MC, Hsu E, Wertz K, Wong LJ. A novel mutation in the CFTR gene correlates with severe clinical phenotype in seven Hispanic patients. J Med Genet. 2000;37:215–218. doi: 10.1136/jmg.37.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong LJ, Alper OM. Detection of CFTR mutations using temporal temperature gradient gel electrophoresis. Electrophoresis. 2004;25:2593–2601. doi: 10.1002/elps.200406015. [DOI] [PubMed] [Google Scholar]
- 31.Hantash FM, Milunsky A, Wang Z, Anderson B, Sun W, Anguiano A, Strom CM. A large deletion in the CFTR gene in CBAVD. Genet Med. 2006;8:93–95. doi: 10.1097/01.gim.0000200945.54234.d7. [DOI] [PubMed] [Google Scholar]
- 32.Hantash FM, Redman JB, Starn K, Anderson B, Buller A, McGinniss MJ, Quan F, Peng M, Sun W, Strom CM. Novel and recurrent rearrangements in the CFTR gene: clinical and laboratory implications for cystic fibrosis screening. Hum Genet. 2006;119:126–136. doi: 10.1007/s00439-005-0082-0. [DOI] [PubMed] [Google Scholar]
- 33.Ferec C, Casals T, Chuzhanova N, Macek M, Jr, Bienvenu T, Holubova A, King C, McDevitt T, Castellani C, Farrell PM, Sheridan M, Pantaleo SJ, Loumi O, Messaoud T, Cuppens H, Torricelli F, Cutting GR, Williamson R, Ramos MJ, Pignatti PF, Raguenes O, Cooper DN, Audrezet MP, Chen JM. Gross genomic rearrangements involving deletions in the CFTR gene: characterization of six new events from a large cohort of hitherto unidentified cystic fibrosis chromosomes and meta-analysis of the underlying mechanisms. Eur J Hum Genet. 2006;14:567–576. doi: 10.1038/sj.ejhg.5201590. [DOI] [PubMed] [Google Scholar]
- 34.Morral N, Nunes V, Casals T, Cobos N, Asensio O, Dapena J, Estivill X. Uniparental inheritance of microsatellite alleles of the cystic fibrosis gene (CFTR): identification of a 50 kilobase deletion. Hum Mol Genet. 1993;2:677–681. doi: 10.1093/hmg/2.6.677. [DOI] [PubMed] [Google Scholar]
- 35.Magnani C, Cremonesi L, Giunta A, Magnaghi P, Taramelli R, Ferrari M. Short direct repeats at the breakpoints of a novel large deletion in the CFTR gene suggest a likely slipped mispairing mechanism. Hum Genet. 1996;98:102–108. doi: 10.1007/s004390050167. [DOI] [PubMed] [Google Scholar]
- 36.Mickle JE, Macek M, Jr, Fulmer-Smentek SB, Egan MM, Schwiebert E, Guggino W, Moss R, Cutting GR. A mutation in the cystic fibrosis transmembrane conductance regulator gene associated with elevated sweat chloride concentrations in the absence of cystic fibrosis. Hum Mol Genet. 1998;7:729–735. doi: 10.1093/hmg/7.4.729. [DOI] [PubMed] [Google Scholar]
- 37.Lerer I, Laufer-Cahana A, Rivlin JR, Augarten A, Abeliovich D. A large deletion mutation in the CFTR gene (3120+1Kbdel8.6Kb): a founder mutation in the Palestinian Arabs (mutation in brief no. 231) Online Hum Mutat. 1999;13:337. doi: 10.1002/(SICI)1098-1004(1999)13:4<337::AID-HUMU13>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 38.Costes B, Girodon E, Vidaud D, Flori E, Ardalan A, Conteville P, Fanen P, Niel F, Vidaud M, Goossens M. Prenatal detection by real-time quantitative PCR and characterization of a new CFTR deletion, 3600+15kbdel5.3kb (or CFTRdele19) Clin Chem. 2000;46:1417–1420. [PubMed] [Google Scholar]
- 39.Kilinc MO, Ninis VN, Dagli E, Demirkol M, Ozkinay F, Arikan Z, Cogulu O, Huner G, Karakoc F, Tolun A. Highest heterogeneity for cystic fibrosis: 36 mutations account for 75% of all CF chromosomes in Turkish patients. Am J Med Genet. 2002;113:250–257. doi: 10.1002/ajmg.10721. [DOI] [PubMed] [Google Scholar]
- 40.Niel F, Martin J, Dastot-Le Moal F, Costes B, Boissier B, Delattre V, Goossens M, Girodon E. Rapid detection of CFTR gene rearrangements impacts on genetic counseling in cystic fibrosis. J Med Genet. 2004;41:e118. doi: 10.1136/jmg.2004.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chevalier-Porst F, Souche G, Bozon D. Identification and characterization of three large deletions and a deletion/polymorphism in the CFTR gene. Hum Mutat. 2005;25:504. doi: 10.1002/humu.9335. [DOI] [PubMed] [Google Scholar]
- 42.Schneider M, Hirt C, Casaulta C, Barben J, Spinas R, Buhlmann U, Spalinger J, Schwizer B, Chevalier-Porst F, Gallati S. Detection of exon deletions within an entire gene (CFTR) by relative quantification on the LightCycler. Clin Chem. 2006;52:2005–2012. doi: 10.1373/clinchem.2005.065136. [DOI] [PubMed] [Google Scholar]
- 43.Nectoux J, Audrezet MP, Viel M, Leroy C, Raguenes O, Ferec C, Lesure JF, Davy N, Renouil M, Cartault F, Bienvenu T. A frequent large rearrangement in the CFTR gene in cystic fibrosis patients from Reunion Island. Genet Test. 2006;10:208–214. doi: 10.1089/gte.2006.10.208. [DOI] [PubMed] [Google Scholar]
- 44.Niel F, Legendre M, Bienvenu T, Bieth E, Lalau G, Sermet I, Bondeux D, Boukari R, Derelle J, Levy P, Ruszniewski P, Martin J, Costa C, Goossens M, Girodon E. A new large CFTR rearrangement illustrates the importance of searching for complex alleles. Hum Mutat. 2006;27:716–717. doi: 10.1002/humu.9431. [DOI] [PubMed] [Google Scholar]
- 45.Schneider M, Hirt C, Casaulta C, Barben J, Spinas R, Buhlmann U, Spalinger J, Schwizer B, Chevalier-Porst F, Gallati S. Large deletions in the CFTR gene: clinics and genetics in Swiss patients with CF. Clin Genet. 2007;72:30–38. doi: 10.1111/j.1399-0004.2007.00820.x. [DOI] [PubMed] [Google Scholar]
- 46.Taulan M, Girardet A, Guittard C, Altieri JP, Templin C, Beroud C, des Georges M, Claustres M. Large genomic rearrangements in the CFTR gene contribute to CBAVD. BMC Med Genet. 2007;8:22. doi: 10.1186/1471-2350-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ratbi I, Legendre M, Niel F, Martin J, Soufir JC, Izard V, Costes B, Costa C, Goossens M, Girodon E. Detection of cystic fibrosis transmembrane conductance regulator (CFTR) gene rearrangements enriches the mutation spectrum in congenital bilateral absence of the vas deferens and impacts on genetic counselling. Hum Reprod. 2007;22:1285–1291. doi: 10.1093/humrep/dem024. [DOI] [PubMed] [Google Scholar]
- 48.Hantash FM, Redman JB, Goos D, Kammesheidt A, McGinniss MJ, Sun W, Strom CM. Consultations in Molecular Diagnostics. Characterization of a recurrent novel large duplication in the cystic fibrosis transmembrane conductance regulator gene. J Mol Diagn. 2007;9:556–560. doi: 10.2353/jmoldx.2007.060141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ainsworth PJ, Koscinski D, Fraser BP, Stuart JA. Family cancer histories predictive of a high risk of hereditary non-polyposis colorectal cancer associate significantly with a genomic rearrangement in hMSH2 or hMLH1. Clin Genet. 2004;66:183–188. doi: 10.1111/j.0009-9163.2004.00282.x. [DOI] [PubMed] [Google Scholar]
- 50.Pastrello C, Baglioni S, Tibiletti MG, Papi L, Fornasarig M, Morabito A, Agostini M, Genuardi M, Viel A. Stability of BAT26 in tumours of hereditary nonpolyposis colorectal cancer patients with MSH2 intragenic deletion. Eur J Hum Genet. 2006;14:63–68. doi: 10.1038/sj.ejhg.5201517. [DOI] [PubMed] [Google Scholar]
- 51.Lai KK, Lo IF, Tong TM, Cheng LY, Lam ST. Detecting exon deletions and duplications of the DMD gene using multiplex ligation-dependent probe amplification (MLPA) Clin Biochem. 2006;39:367–372. doi: 10.1016/j.clinbiochem.2005.11.019. [DOI] [PubMed] [Google Scholar]