

Abstract
Starting out from a brief description of the determinants of coronary blood flow (perfusion, pressure, extravascular compression, autoregulation, metabolic regulation, endothelium-mediated regulation and neurohumoral regulation) the present review highlights the overwhelming importance of metabolic regulation such that coronary blood flow is increased at increased heart rate under physiological circumstances and the overwhelming importance of extravascular compression such that coronary blood flow is decreased at increased heart rate through reduction of diastolic duration in the presence of severe coronary stenoses. The review goes on to characterize the role of heart rate in the redistribution of regional myocardial blood flow between a normal coronary vascular tree with preserved autoregulation and a poststenotic vasculature with exhausted coronary reserve. When flow is normalized by heart rate, there is a consistent close relationship of regional myocardial blood flow and contractile function for each single cardiac cycle no matter whether or not there is a coronary stenosis and what the actual blood flow is. β-Blockade improves both flow and function along this relationship. When the heart rate reduction associated with β-blockade is prevented by pacing, α-adrenergic coronary vasoconstriction is unmasked and both flow and function are deteriorated. Selective heart rate reduction, however, improves both flow and function without any residual negative effect such as unmasked α-adrenergic coronary vasoconstriction or negative inotropic action.
Keywords: coronary blood flow, regional myocardial blood flow, regional contractile function, coronary stenosis, autoregulation, β-blockade, α-adrenergic coronary vasoconstriction, myocardial ischaemia
Introduction
It is thoroughly well established that heart rate is a prognostic marker for longevity—the lower the better—and for the outcome of various cardiovascular diseases, notably ischaemic heart disease and heart failure (Guth et al., 1987a; Hjalmarson et al., 1990; Indolfi and Ross, 1993; Benetos et al., 1999).
In this article, we want to focus on (1) the mechanism(s) by which increases in heart rate are detrimental in the setting of ischaemic heart disease, (2) why and how selective heart rate reduction is beneficial and more beneficial than β-blockade, and (3) what the acute and long-term consequences of selective heart rate reduction for the ischaemic and reperfused myocardium are.
Determinants of coronary blood flow and its regulation
Coronary blood flow is very sensitive to its mechanical determinants and exquisitely well regulated (Bassenge and Heusch, 1990; Baumgart and Heusch, 1995). As any other flow of fluid through a pipe, blood flow through the coronary vasculature can be described using the law of Hagen-Poiseuille:
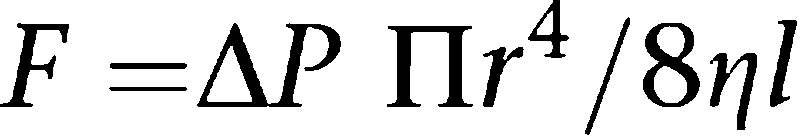 |
The length l of the coronary vasculature does not change in a given individual and changes in viscosity η can also be neglected when there are no major changes in haematocrit. Therefore, the formula can be reduced to
ΔP is the driving pressure gradient between the origin of the coronary vasculature in the aortic root and its orifice, that is, of the coronary sinus into the right atrium. Now the coronary vasculature has one particular and unique property: it is being compressed by the contracting myocardium throughout systole, such that the pipe is—at least functionally—obstructed and no flow occurs during systole; the squeezing action of the contracting myocardium can even reverse coronary blood flow in an epicardial coronary artery and even more so in the subendocardial microcirculation (Chilian and Marcus, 1982; Toyota et al., 2005). Thus, coronary blood flow occurs mostly during diastole, and the driving pressure gradient is the difference between mean diastolic pressure in the aortic root and mean right atrial pressure, which—during diastole, when the atrioventricular valves are open—is more or less equal to right and left ventricular pressure. Apart from this driving pressure gradient, the duration of diastole during which coronary blood flow occurs is of major importance. Both the driving pressure gradient and the duration of diastole are integrated into the diastolic pressure–time integral, which is the essential mechanical determinant of coronary blood flow (Figure 1; Buckberg et al., 1972). Now, anything that decreases the diastolic pressure–time integral will decrease coronary blood flow, that is, any decrease in diastolic aortic root pressure, any increase in right/left ventricular diastolic pressure, any reduction in diastolic duration and any delay in isovolumic ventricular relaxation will decrease coronary blood flow.
Figure 1.

The diastolic pressure-time integral reflects the driving force for coronary blood flow. Any decrease in diastolic aortic pressure (upper right), increase in diastolic ventricular pressure (lower left), delay in isovolumic ventricular relaxation (lower right) and decrease in diastolic duration (upper left) impedes coronary blood flow.
Having discussed the mechanical determinants to which coronary blood flow is very sensitive, its regulation will now be addressed. Regulation is achieved by changes in the diameter of the coronary microcirculation, that is, predominantly in small arteries and arterioles with a diameter of less than 100 μm (Marcus et al., 1990; DeFily and Chilian, 1995; Chilian and NHLBI Workshop Participants, 1997). From the above formula, where the radius of the pipe in its fourth power determines flow, it follows that very subtle changes in vascular diameter have a profound impact on blood flow, and therefore this is the effective site of regulation. There are several distinct regulatory mechanisms that govern coronary blood flow.
Autoregulation
Autoregulation is the response of the coronary vasculature to changes in perfusion pressure (Mosher et al., 1964). Reductions or increases, respectively, in perfusion pressure are counteracted by increases or decreases, respectively, in microvascular diameter such that coronary blood flow is maintained independent of perfusion pressure over a relatively wide range of pressures, that is, there is a plateau of coronary blood flow between 50 and 130 mm Hg. Autoregulation is a myogenic response of the vascular smooth muscle cells to transmural pressure (Bassenge and Heusch, 1990).
Metabolic vasomotion
Metabolic vasomotion is the response of the coronary vasculature to changes in myocardial metabolism, which in turn are almost entirely a consequence of changes in myocardial performance. The mechanism(s) and signal mediators of metabolic coronary vasomotion have been a matter of debate over decades and are still not clearly understood (Bassenge and Heusch, 1990). Adenosine has attracted a lot of interest and is a mediator that intuitively has a logical charme, because the formation of adenosine in cardiomyocytes is stoichiometrically coupled to ATP breakdown, and its release and vasodilator action on coronary vascular smooth muscle cells can increase coronary blood flow and the delivery of oxygen and substrates and thus restore the myocardial energetic state (Berne, 1963; Gerlach et al., 1963). Likewise, the activation of ATP-dependent potassium channels on the surface membranes of coronary vascular smooth muscle cells would imply a certain coupling to energetic state of smooth muscle cells rather than cardiomyocytes (Embrey et al., 1997; Merkus et al., 2005). Other proposed mediators of metabolic coronary vasomotion are interstitial PO2 and PCO2 per se, nitric oxide released from the endothelium, prostaglandins, potassium ions and so on. More recent studies indicate that metabolic coronary vasomotion is a complex concerted action, with indeed adenosine, adenosine-triphosphate-dependent potassium channels and nitric oxide as major players. It must be stressed that under physiological circumstances metabolic coronary vasomotion also operates in the reverse direction, that is, reduced myocardial performance and metabolism result in decreased coronary blood flow.
Endothelium
The endothelium plays a major role in coronary blood flow regulation. Tangential shear stress that goes along with increased blood flow and blood flow velocity in the longitudinal direction of the vessel as well as pulsatile stretching of the vascular wall in the transmural direction enhance the formation and release of nitric oxide from the endothelium, which acts to relax/dilate the smooth muscle cells of the vascular wall. Endothelial, nitric oxide-mediated dilation has its particular role in larger coronary conduit vessels to adapt larger coronary arterial diameter to flow and flow velocity changes, which are primarily initiated at the microvascular level. The endothelium is also the source of the vasoconstrictor endothelin (Bassenge and Heusch, 1990).
Neurohumoral regulation
Neurohumoral regulation of the coronary circulation is primarily by the release of norepinephrine from cardiac sympathetic nerves and by circulating epinephrine from the adrenal medulla. (Nor)epinephrine, apart from its β1-adrenoceptor-mediated effects on cardiomyocytes with the resulting increases in myocardial performance and metabolism and finally metabolic coronary vasodilation, exerts direct dilator actions on the coronary circulation through activation of β1- and β2-adrenoceptors (Trivella et al., 1990; Miyashiro and Feigl, 1993), and constrictor actions through activation of α-adrenoceptors. α-Adrenoceptor-mediated coronary vasoconstriction largely prevails over direct β2-adrenoceptor-mediated dilation. The constriction of large epicardial conduit coronary arteries is mediated by α1-adrenoceptors, and that of the coronary microcirculation largely by α2-adrenoceptors (Heusch, 1990; Heusch et al., 2000). Other neurotransmitters and hormones, for example, vasopressin and B-type natriuretic peptide, are also operative in the coronary circulation, but of minor functional importance.
Heart rate and the physiology of coronary blood flow
Any increase in heart rate increases the number of cardiac cycles per time frame and thus—even at unchanged work per single cardiac cycle—cardiac performance, that is, work per time, and in the consequence myocardial metabolism. In fact, an increase in heart rate does not alter myocardial oxygen consumption for any given cardiac cycle, but linearly increases myocardial oxygen consumption per minute (Figure 2) (Tanaka et al., 1990). This increase in myocardial oxygen consumption per minute results in a proportionate increase in coronary blood flow, through metabolic coronary dilation (Figure 3) (Colin et al., 2004); increased myocardial oxygen extraction contributes only to a minor extent and a very high heart rate to matching of oxygen delivery to myocardial oxygen consumption, as myocardial oxygen extraction is already near maximal under baseline resting conditions. It should be noted that the blood flow per single cardiac cycle is decreased at higher heart rate (Figure 3).
Figure 2.

Myocardial oxygen consumption increases linearly with increasing heart rate. Myocardial oxygen consumption per single cardiac cycle is independent of heart rate. From Tanaka et al. (1990).
Figure 3.

Coronary blood flow displays a typical dose–response curve to increasing heart rate. Blood flow per single cardiac cycle is reduced at increased heart rate, reflecting the decrease in diastolic duration. From Colin et al. (2004).
This is because any increase in heart rate also shortens the duration of diastole and thus creates an impediment to coronary blood flow. Increases in heart rate are associated with overproportionate decreases in diastolic duration, therefore, the relationship between heart rate and diastolic duration is hyperbolic (Colin et al., 2004).
Nevertheless, metabolic coronary vasodilation is so powerful that it largely overcompensates for decreased diastolic duration and prevails. Increased oxygen extraction also contributes at very high heart rate. Under physiological circumstances, coronary blood flow and oxygen delivery are always matched to myocardial performance and oxygen consumption.
Heart rate and the pathophysiology of coronary blood flow
Atherosclerosis
Heart rate can contribute to the development and progression of atherosclerosis. In fact, baboons when exposed to a cholesterol-rich diet develop atherosclerosis, which is blunted when their sinus node is crushed and accordingly heart rate is decreased (Beere et al., 1984). Of course, heart rate is a less dominant factor in coronary atherosclerosis development and progression than low-density lipoprotein cholesterol, blood pressure or smoking. Also, it is unclear how heart rate contributes to the progression of atherosclerosis, but aggravated shear and turbulence might be a mechanism.
Coronary stenosis: flow distribution and steal phenomena
Whereas we have discussed the mechanism(s) determining and regulating coronary blood flow under physiological circumstances above, the scenario with a significant coronary stenosis is more complex. Part of the heart is still perfused through a more or less normal coronary vasculature, whereas other parts of the heart are perfused through more or less severely stenotic coronary arteries. Distal to the stenosis, coronary perfusion pressure is reduced and the poststenotic coronary microcirculation responds to this reduction in coronary perfusion pressure with an autoregulatory (see above) vasodilation; this autoregulatory microcirculatory vasodilation can compensate for stenoses of up to about 70% diameter reduction such that coronary blood flow at rest is well maintained. Another and more long-term form of compensation for the coronary stenosis occurs through the formation of collateral vessels or increase in the diameter of pre-existing collaterals that connect the poststenotic microvascular bed with the normal surrounding coronary vasculature. Collateral blood flow then occurs along a pressure gradient between the origin of collateral vessels in the normal myocardium and the orifice of collaterals into the poststenotic myocardium. In the presence of collaterals, the poststenotic myocardium still receives its blood supply to some extent through the stenotic segment, but also to a varying extent through collaterals. Autoregulation of the poststenotic coronary microcirculation plus collateral blood flow are responsible for the fact that perfusion even of myocardium distal to severe stenoses is well maintained at rest and that myocardial ischaemia is only precipitated during stress or exercise, that is, in situations of increased heart rate.
Why is ischaemia precipitated then at increased heart rate and why does the compensation for the stenosis fail? During stress or exercise, the increase in heart rate is still met by metabolic coronary dilation, which still overcomes the effects of shortened diastole in the normal myocardium (Figure 4 left side). In the poststenotic myocardium, coronary dilator capacity is largely reduced or finally exhausted due to the autoregulatory adjustment already at rest/baseline. Further metabolic coronary vasodilation during increased heart rate is no longer possible; in contrast, the reduction in diastolic duration now even further compromises coronary blood flow. In addition, collateral blood flow is reduced because metabolic coronary vasodilation in the normal myocardium reduces pressure at the origin of collaterals (Figure 4 left side) and concomitantly the decreased coronary blood flow or increased microcirculatory resistance (secondary to reduced diastolic duration) increases pressure at the orifice of collaterals into the poststenotic myocardium (Figure 4 right side). Thus, the driving pressure gradient for collateral blood flow is largely reduced (Figure 4); it should be noted that relatively subtle change in pressure, however, in opposing directions in the donor and the recipient vascular territory, adds up to a marked reduction in the driving pressure gradient for collateral blood flow (Heusch and Yoshimoto, 1983).
Figure 4.

Schematic representation of changes in driving pressure gradient for collateral blood flow and in microvascular resistance in normal myocardium (left) and in poststenotic myocardium (right). At baseline heart rate, there is an autoregulatory decrease in microvascular resistance of the poststenotic myocardium. With increasing heart rate, there is metabolic dilation and accordingly a decrease in microvascular resistance in normal myocardium, resulting in decreased pressure at the origin of collaterals. In contrast, in poststenotic myocardium, no further dilation is possible and the reduction in diastolic duration prevails, such that microvascular resistance and pressure at the orifice of collaterals into the poststenotic coronary vasculature are increased. Note that subtle changes in perfusion pressure, yet in opposing direction in the normal and poststenotic myocardium, add up to a marked reduction in the driving pressure gradient for collateral blood flow (Heusch and Yoshimoto, 1983).
Accordingly, studies using a high spatial resolution technique for the measurement of regional myocardial blood flow demonstrated that, whereas blood flow distal to a severe coronary stenosis is well maintained and almost equal to that in remote normal myocardium at rest, stress induces metabolic coronary vasodilation and a flow increase in the normal remote myocardium, but precipitates a marked flow reduction in the poststenotic myocardium (Figure 5) (Baumgart et al., 1993). The above mechanism of regional myocardial blood flow redistribution through collaterals, where poststenotic blood flow is well maintained at rest but reduced during stress, is termed ‘collateral steal' phenomenon, somewhat inappropriately because there is no real ‘steal' of blood flow away from the poststenotic myocardium, but rather a reduced donation. In any case, a similar redistribution of myocardial blood flow occurs between the subendocardial and the subepicardial layers of the myocardial wall through transmurally penetrating coronary vessels. This happens because autoregulation is exhausted in subendocardial layers prior to that in subepicardial layers and the subendocardium is exposed to intraventricular diastolic pressure whereas the subepicardium is exposed to pericardial pressure, which is close to zero. Such ‘transmural steal' phenomenon is the basis for the greater vulnerability of the inner myocardial layers to ischaemia.
Figure 5.

At rest, the compensatory decrease in microvascular resistance in the poststenotic myocardium maintains coronary blood flow. During stress, the normal myocardium responds with metabolic dilation and increased blood flow. In contrast, blood flow in poststenotic myocardium is reduced (Baumgart et al., 1993).
Increased heart rate not only causes an unfavourable blood flow distribution between normal and poststenotic myocardium through collaterals and between subendocardial and subepicardial layers through transmural vessels, but also causes an increased turbulence and pressure loss along the stenotic segment, and this additional pressure loss along the stenotic segment also contributes to decreased poststenotic perfusion (Heusch et al., 1982).
Coronary stenosis: relationship of flow and contractile function
In detailed experimental studies that measured simultaneously regional myocardial blood flow and contractile function distal to different degrees of coronary stenosis, there was a close linear correlation of flow and function (Figure 6), that is, any reduction in regional blood flow was associated with a proportionate reduction in contractile function (Gallagher et al., 1984). Such consistent relationship between flow and function in a series of studies led to the introduction of the concept of perfusion–contraction matching (Ross, 1991). In contrast to the classical view, where ischaemia is considered as a state of imbalance between supply and demand (of either blood flow, or oxygen or energy), the concept of perfusion–contraction matching relies on the fact that contractile function is proportionate to blood flow over the entire measured range (Figure 6), that is, an imbalance between blood flow (supply) and demand (contractile function) does not exist.
Figure 6.

Systolic wall thickening and myocardial blood flow are closely and almost linearly correlated over a wide range of blood flow reduction by coronary stenoses of different severity. This is evidence for the concept of perfusion–contraction matching (Gallagher et al., 1984).
Such linear relation of blood flow and contractile function is also observed during exercise when heart rate is increased. However, the relationship observed at rest is shifted rightwards and downwards during exercise, that is, there is less contractile function at any level of blood flow (Gallagher et al., 1983). Now, when the increase in heart rate during exercise is taken into consideration by plotting blood flow per cardiac cycle rather than per minute on the x axis (in other words, by dividing blood flow per minute by heart rate), the relationships between contractile function and blood flow at rest and during exercise are superimposable (Gallagher et al., 1983). The same is true with reduction of heart rate, in this case by a pharmacological intervention: with reduced heart rate, the relationship between flow per minute and function is shifted leftwards and upwards, that is, there is a better contractile function at any level of blood flow. In this particular study, a slight curvilinear plot was better suited than a linear plot to characterize the relationship between contraction and blood flow. Nevertheless, when normalizing blood flow to a single cardiac cycle and thus considering heart rate, the relationships at two different heart rates are superimposable (Figure 7) (Indolfi et al., 1989). In a series of studies, all pharmacological agents that attenuate exercise-induced myocardial ischaemia (β-blockers, calcium antagonists, nitrates and their combinations) were demonstrated to operate along such consistent flow–function relationship, further strengthening the concept of perfusion–contraction matching (Matsuzaki et al., 1984a, 1984b, 1985; Guth et al., 1986).
Figure 7.

Left: when heart rate is reduced by a pharmacological agent, the relationship between systolic wall thickening and myocardial blood flow is displaced leftwards and upwards, indicating that contractile function for each level of blood flow is increased. Right: when myocardial blood flow is calculated for each single cardiac cycle, the relationships at two different heart rates are superimposable, again supporting the concept of perfusion–contraction matching (Indolfi et al., 1989).
Regional myocardial ischaemia also impacts on flow and function of adjacent and more remote normal myocardium. During acute coronary artery occlusion, the ischaemic region is surrounded by a narrow zone of normally perfused myocardium with depressed regional contractile function (Guth et al., 1984; Gallagher et al., 1986, 1987). This depressed contractile function in the immediate border zone surrounding the ischaemic zone is attributed to more or less well defined mechanical ‘tethering' between nonischaemic and ischaemic myocardial fibers (Bogen et al., 1980). The remote myocardium is often characterized by enhanced contractile function (Lew et al., 1985; Buda et al., 1990). Whether an increase in remote nonischaemic zone function can be considered as compensatory in that it acts to preserve global left ventricular function (Buda et al., 1990) is not entirely clear, since a major proportion of nonischaemic zone hyperfunction occurs during isovolumic systole and does not contribute to ejection (Lew et al., 1985). The increase in function in the remote nonischaemic zone is associated with a moderate, presumably metabolically mediated increase in coronary blood flow (Gascho and Beller, 1987). Both the increase in remote zone contractile function and the ensuing metabolic coronary dilation are attenuated by β-blockade with metoprolol (Vanyi et al., 2006).
Plaque rupture
Increased heart rate is associated with an increased incidence of angiographically documented plaque rupture in patients with ischaemic heart disease (Heidland and Strauer, 2001). The mechanism(s) by which heart rate contributes to plaque rupture are not clear, but increased shear and turbulence at higher heart rate in stenotic segments are plausible mechanisms. It is also possible that increased heart rate is just a marker of greater activity of the sympathetic nervous system and of a pro-inflammatory systemic condition, which then facilitates plaque rupture.
β-Blockade in myocardial ischaemia: the benefits
β-Blockade has negative chronotropic (reduction of heart rate), bathmotropic (reduction of excitation threshold of cardiomyocyte) and therefore anti-arrhythmic, inotropic (reduction of force of contraction) and lusitropic (more correctly lysitropic, reduction in the rate of isovolumic ventricular relaxation) properties.
With reference to the above schematic diagram of flow distribution through collaterals (Figures 4 and 5), β-blockade will reduce blood flow to the normal myocardium, that is, at reduced heart rate and contractile function, there is less metabolic vasodilation, such that the pressure at the origin of collaterals is better preserved. In the poststenotic myocardium, the reduced heart rate will prolong the duration of diastole, improve blood flow and reduce calculated microvascular resistance, such that the pressure at the orifice of collaterals into the poststenotic coronary vasculature is reduced. As a result of these opposing changes in pressures at the origin and at the orifice of collaterals, the driving pressure gradient for collateral blood flow is increased, and accordingly blood flow into the poststenotic myocardium is further improved. Apart from the increased diastolic duration that facilitates perfusion of the dilated poststenotic microvasculature with its exhausted autoregulatory reserve and from the favourable blood flow distribution through collaterals, there may be an additional, yet small improvement in blood flow through the stenotic segment because decreased heart rate also decreases turbulence and pressure loss along the stenotic segment. Whatever the mechanism is, clearly, as predicted by our haemodynamic considerations, blood flow to normal remote myocardium is reduced and, more importantly, blood flow to the poststenotic myocardium is increased (Figure 8), and thus the inhomogeneity of blood flow distribution is normalized (Buck et al., 1981). Again, as predicted by our considerations of the flow–function relationship and the concept of perfusion–contraction matching, β-blockade operates along such relationship, that is, improves both blood flow and contractile function proportionately (Figure 9; Matsuzaki et al., 1984b). Of note, in the poststenotic and ischaemic myocardium a negative inotropic action of β-blockade does not become apparent, because in ischaemia it is blood flow that governs contractile function, and therefore β-blockade, which increases poststenotic blood flow, increases contractile function rather than displaying its negative inotropic action.
Figure 8.

Reduced poststenotic blood flow in the ischaemic myocardium at rest. With β-blockade, blood flow in normal myocardium is reduced, whereas blood flow in ischaemic myocardium is improved (Buck et al., 1981).
Figure 9.

Relationship of systolic wall thickening and myocardial blood flow during exercise-induced ischaemia. β-Blockade moves the data point along the consistent relationship upwards and rightwards, reflecting improved blood flow and contractile function. Note that, in ischaemia, there is no negative inotropic action of β-blockade. The increase in blood flow even permits an increase in regional contractile function (Matsuzaki et al., 1984b).
Apart from the improved blood flow, which also results in improved contractile function of ischaemic myocardium, β-blockade also reduces the size of myocardial infarction, which results from sustained severe ischaemia and subsequent reperfusion. Such infarct size reduction is beyond the improvement in collateral blood flow, that is, is even seen at any given blood flow when infarct size is plotted as a function of residual blood flow (Schulz et al., 1995).
β-Blockade in myocardial ischaemia: the disadvantages
Within the framework of the present review, we focus, only on the myocardium and neglect the disadvantageous effects of β-blockade on the bronchial system, peripheral vasculature, metabolism and central nervous system, which are, of course, all of clinical relevance and depend to some extent on the β1 vs β2 selectivity, as well as on other properties of the individual β-blocker.
Although β-blockade is clearly beneficial in myocardial ischaemia, it still has disadvantages that limit its benefits. As discussed, the negative inotropic action of β-blockade is of no concern for the poststenotic ischaemic myocardium. In fact, a negative inotropic action and thus reduced need for coronary blood flow may even contribute to the more favourable blood flow distribution towards the ischaemic myocardium. Also, the potential negative inotropic effect on more remote myocardium appears not to be a problem. It is only when global left ventricular function is compromised that the negative inotropic action of β-blockade presents a problem, and again this may only be a problem with acute β-blockade as we have learned from the trials where β-blockade proved to be beneficial in the long-term treatment of heart failure. Thus, the problems of negative inotropic actions of β-blockade in the treatment of ischaemic heart disease have probably been overestimated in the past. True problems, however, exist with respect to coronary blood flow:
The heart rate reduction achieved by β-blockade does not achieve the full potential that might be expected from the increase in the duration of diastole. This is because of the negative lysitropic action of β-blockade. When we consider the diastolic pressure–time integral as the driving force for coronary blood flow, any slowing of isovolumic ventricular relaxation at any given diastolic duration will impede coronary blood flow, and this is in fact a true problem for β-blockade.
Much more importantly, the competitive displacement of norepinephrine and epinephrine from β-adrenoceptors increases their effective concentration at α-adrenoceptors. In consequence, β-blockade increases or unmasks α-vasoconstriction. Unmasking of α-adrenergic coronary vasoconstriction in situations of increased sympathetic activity such as stress and exercise then actively reduces coronary blood flow and contributes to the precipitation of acute myocardial ischaemia (Seitelberger et al., 1988). Unmasked α-adrenergic coronary vasoconstriction is particularly evident in atherosclerotic coronary vessels and seen at the level of epicardial conduit coronary arteries and mediated by α1-adrenoceptors, and it is even more prominent in the coronary microcirculation and mediated by α2-adrenoceptors (Baumgart et al., 1999; Heusch et al., 2000). Unmasked α-adrenergic coronary vasoconstriction is also manifested in the flow–function relationship, when the heart rate reduction achieved by β-blockade is artificially prevented by atrial pacing at the heart rate previously seen in the absence of β-blockade. Rather than moving upwards in the direction of improved blood flow and a proportionate improvement in contractile function as with β-blockade and heart reduction, β-blockade in the absence of heart rate reduction is characterized by decreased blood flow (consequence of α-adrenoceptor-mediated vasoconstriction and impaired isovolumic ventricular relaxation) and a proportionate decrease in contractile function (Figure 10) (Guth et al., 1987b).
Figure 10.

Relationship of systolic wall thickening and blood flow during exercise-induced ischaemia. When the reduction of heart rate by β-blockade is prevented by atrial pacing at a rate that was observed in the absence of β-blockade, the data point is moved along the consistent relationship leftwards and downwards, indicating reduced blood flow (a consequence of unmasked α-adrenergic coronary vasoconstriction and impaired isovolumic ventricular relaxation) and a proportionate reduction in regional contractile function (Guth et al., 1987b).
With respect to the distribution of regional myocardial blood flow and contractile function during acute myocardial ischaemia, no difference between β1-selective or less selective β-blockers is apparent (Buck et al., 1981; Matsuzaki et al., 1984b).
Selective heart rate reduction by ivabradine and its benefits
Mechanism of action in the sinus node
The advantage of selective bradycardic agents can only be defined against the background of the disadvantage of β-blockade.Ivabradine acts on the If-channel. The ‘f' in If stands for ‘funny' because this ion channel has ‘funny'/unusual properties. This channel is activated by hyperpolarization and is regulated not by phosphorylation through protein kinase A in response to increased intracellular cAMP, but by direct binding of cAMP. cAMP is the second messenger molecule that integrates intracellularly the balance between cardiac sympathetic nerves, which increase cAMP through the action of norepinephrine on surface β1- and β2-adrenoceptors and subsequent activation of the G-protein Gs, and cardiac vagal nerves, which decrease cAMP through the action of acetylcholine on surface muscarinic receptors and subsequent activation of the inhibitory G-protein Gi. The If-channel is genetically determined by the hyperpolarization-activated, cyclic nucleotide-gated channel gene family, which encodes four different isoforms. The hyperpolarization activated cyclic nucleotide-gated channel isoform 4 is the predominant one in the sinus node. The channel is composed of four subunits, which form a pore/channel (Zagotta et al., 2003). The If-channel is activated by the hyperpolarization in the terminal phase of action potential repolarization, and its activation increases the membrane permeability for both sodium and potassium ions. The resulting influx of sodium and potassium ions along the electrical (for sodium and potassium) and chemical (for sodium) gradient then depolarizes the cell, until the threshold for activation of the fast calcium channel is reached and an action potential is initiated. Thus, the If-channel is activated during the slow diastolic depolarization phase. The degree of activation of the If-channel determines the velocity of the diastolic depolarization and thus determines the time when the threshold for the initiation of an action potential is reached; this is the mechanism by which the If-channel controls heart rate.
Ivabradine specifically and concentration-dependently binds to the If-channel in its open/activated form, and its binding inhibits the channel, thus reducing the velocity of diastolic depolarization. Of note, while specifically prolonging the phase of diastolic depolarization, all other characteristics of the action potential remain unaltered (Figure 11). In this way, ivabradine indeed reduces heart rate selectively (Thollon et al., 1994; DiFrancesco and Camm, 2004).
Figure 11.

Typical original recording of the action potential in a sinus node cell. Ivabradine prolongs the diastolic slow depolarization phase. Note that ivabradine has no effect on action potential characteristics (DiFrancesco and Camm, 2004).
Effects of ivabradine on coronary blood flow and contractile function
The reduction of heart rate by ivabradine prolongs the duration of diastole. Of note, in contrast to β-blockade, which not only prolongs diastolic duration but also impairs isovolumic ventricular relaxation and thus offsets part of the benefit in terms of the diastolic pressure–time integral (see Figure 1 above), selective heart rate reduction by ivabradine is not at the expense of impaired isovolumic ventricular relaxation, such that the full benefit of prolonged diastole is available for coronary blood flow (Colin et al., 2002).
Ivabradine reduces myocardial oxygen consumption in normal myocardium and this effect prevails over the increase in diastolic duration in normal myocardium. In consequence, there is less metabolic vasodilation and reduced blood flow in normal myocardium. Considering our above scheme for collateral and transmural blood flow distribution (see Figure 4), a reduction in blood flow in normal myocardium together with the facilitation of blood flow through increased diastolic duration will increase blood flow to the poststenotic ischaemic myocardium and also improve its transmural distribution. In fact, ivabradine attenuates exercise-induced myocardial ischaemia in pigs (Vilaine et al., 2003).
Considering the above flow–function relationship, selective heart reduction by ivabradine during exercise-induced myocardial ischaemia improves both regional blood flow and contractile function in a proportionate fashion (Figure 12). In contrast to β-blockade, there is no residual detrimental effect when the heart rate reduction by ivabradine is artificially prevented by pacing; this lack of residual detrimental effect confirms the selectivity of ivabradine in terms of achieving a heart rate reduction and it excludes unmasking of α-adrenergic coronary vasoconstriction, as seen with β-blockade (Monnet et al., 2001).
Figure 12.

Calculated relationship between systolic wall thickening and myocardial blood flow from data (Monnet et al., 2001). Heart rate reduction by ivabradine improves both blood flow and contractile function along the consistent relationship. When a heart rate reduction is prevented by atrial pacing, ivabradine has no residual effect on either blood flow or contractile function.
Ivabradine does not only not unmask α-adrenergic coronary vasoconstriction but also preserves the endothelium-mediated vasodilation that is typically observed in large epicardial conduit vessels in response to enhanced shear stress and pulsatility when blood flow is increased following metabolic vasodilation in the coronary microcirculation (Figure 13) (Simon et al., 1995).
Figure 13.

During exercise, epicardial coronary arterial diameter is increased with increasing duration of exercise. This dilation is mediated by the endothelium in response to increased blood flow through enhanced shear stress and pulsatility. With β-blockade by propranolol, α-adrenergic coronary vasoconstriction is unmasked and the dilation is reversed to vasoconstriction. Equal reduction of heart rate by ivabradine does not interfere with the endothelium-mediated dilation. The slightly less pronounced increase in vascular diameter than with placebo is a physiological response to lower blood flow and consequently less shear stress and pulsatility at lower heart rate (Simon et al., 1995).
Thus, ivabradine offers the same advantages as β-blockade in terms of heart rate reduction, but not at the expense of impaired isovolumic ventricular relaxation and unmasking of α-adrenergic coronary vasoconstriction. The lack of a negative inotropic action of ivabradine contributes to better preservation of global left ventricular function. Also, ivabradine permits adequate increases in left ventricular function and cardiac output during exercise (Monnet et al., 2001; Colin et al., 2003; Du et al., 2004).
Benefits of ivabradine for the ischaemic/reperfused myocardium
Apart from the improvement in blood flow and contractile function in ischaemic myocardium outlined above, ivabradine improves the recovery of contractile function during reperfusion following exercise-induced myocardial ischaemia, that is, ivabradine attenuates stunning. Again, when the reduction of heart rate is prevented by atrial pacing, no residual detrimental effect on contractile function remains, that is, there is no negative inotropic action of ivabradine (Figure 14) (Monnet et al., 2004).
Figure 14.

Left: following an episode of exercise-induced ischaemia, regional contractile function only slowly recovers back to normal over several hours, that is, there is myocardial stunning. Heart rate reduction by ivabradine largely accelerates the recovery, whereas an equal reduction in heart rate by the β-blocker atenolol even impairs the recovery of contractile function, reflecting the negative inotropic action of β-blockade. Right: when changes in heart rate are eliminated by atrial pacing, ivabradine does the same as saline, whereas the negative inotropic action of atenolol is even more pronounced (Monnet et al., 2004).
Post-ejection wall thickening is a typical sign of asynchrony of ventricular contraction and relaxation, which is associated with reversible regional myocardial ischaemia and reperfusion (Heusch et al., 1987; Ehring and Heusch, 1990; Rose et al., 1993). Ivabradine, different from β-blockade (Lucats et al., 2007a), has the unique property of not only improving ischaemic and postischemic regional myocardial function but also reversing post-ejection wall thickening to wall thickening during ejection and thus making this contraction available for cardiac output (Lucats et al., 2007b). It is currently unclear how this property of ivabradine relates to its preservation of isovolumic ventricular relaxation and what the exact mechanism(s) of conversion of post-ejection to ejection wall thickening are, but in any event it is functionally beneficial.
Preliminary studies in rabbits also indicate a reduction of infarct size by ivabradine (Langenbach et al., 2006); reduced infarct size is to be expected but the preliminary studies are on their way to be confirmed in a clinically more relevant large mammal model in pigs where ivabradine's effects on coronary blood flow are also taken into consideration.
Benefits of ivabradine for the long-term outcome after myocardial ischaemia
A study in a rat model of permanent coronary ligation with subsequent myocardial infarction, ventricular remodelling and finally heart failure revealed not only a dose-dependent improvement in left ventricular function by ivabradine, which was associated with dose-dependent reductions in heart rate (Figure 15), but also structural benefits, that is, reduced fibrosis and collagen deposition and increased vascularity in the surviving myocardium (Mulder et al., 2004). Again, these promising findings must be confirmed in larger mammals.
Figure 15.

Heart failure develops following permanent coronary ligation in rats. Dose-dependent decreases in heart rate by ivabradine (right) are associated with proportionate improvements in fractional left ventricular shortening (left). At the highest dose of ivabradine, fractional shortening remains entirely normal (Mulder et al., 2004).
Conclusions and perspectives
There is solid pathophysiological evidence that selective heart rate reduction by ivabradine improves blood flow to, and its distribution within, ischaemic myocardium. This improvement in ischaemic myocardial blood flow is associated with proportionate improvements in ischaemic myocardial contractile function. Different from β-blockade, ivabradine does not impair isovolumic ventricular relaxation, does not unmask α-adrenergic coronary vasoconstriction and does not exert a negative inotropic action. In consequence, endothelium-mediated coronary vasodilation and left ventricular function, in particular during exercise, are better preserved than with β-blockade.
More mechanistic analyses on the benefits from ivabradine in settings of myocardial ischaemia/reperfusion must be carried out and are underway. Nevertheless, a proof of concept study has unequivocally confirmed the anti-ischaemic action of ivabradine in patients with chronic stable angina and also demonstrated its safety (Borer et al., 2003). Other studies have also documented its non-inferiority to atenolol or amlodipine in the treatment of chronic stable angina (Ruzyllo et al., 2004; Tardif et al., 2005). We are looking forward to the results of the Beautiful-Study (morBidity-mortality EvAlUaTion of the If inhibitor ivabradine in patients with coronary disease and left ventricULar dysfunction), which assesses the morbidity and mortality benefits of ivabradine in patients with coronary artery disease.
Conflict of interest
Professor Heusch has received unrestricted educational grant support from, held lectures and served as consultant for Servier. No honorarium was paid for the present article.
References
- Bassenge E, Heusch G. Endothelial and neuro-humoral control of coronary blood flow in health and disease. Rev Physiol Biochem Pharmacol. 1990;116:77–165. doi: 10.1007/3540528806_4. [DOI] [PubMed] [Google Scholar]
- Baumgart D, Ehring T, Krajcar M, Heusch G. A proischemic action of nisoldipine: relationship to a decrease in perfusion pressure and comparison to dipyridamole. Cardiovasc Res. 1993;27:1254–1259. doi: 10.1093/cvr/27.7.1254. [DOI] [PubMed] [Google Scholar]
- Baumgart D, Haude M, Goerge G, Liu F, Ge J, Große-Eggebrecht C, et al. Augmented alpha-adrenergic constriction of atherosclerotic human coronary arteries. Circulation. 1999;99:2090–2097. doi: 10.1161/01.cir.99.16.2090. [DOI] [PubMed] [Google Scholar]
- Baumgart D, Heusch G. Neuronal control of coronary blood flow. Basic Res Cardiol. 1995;90:142–159. doi: 10.1007/BF00789444. [DOI] [PubMed] [Google Scholar]
- Beere PA, Glagov S, Zarins CK. Retarding effect of lowered heart rate on coronary atherosclerosis. Science. 1984;226:180–182. doi: 10.1126/science.6484569. [DOI] [PubMed] [Google Scholar]
- Benetos A, Rudnichi A, Thomas F, Safar M, Guize L. Influence of heart rate on mortality in a French population: role of age, gender, and blood pressure. Hypertension. 1999;33:44–52. doi: 10.1161/01.hyp.33.1.44. [DOI] [PubMed] [Google Scholar]
- Berne RM. Cardiac nucleotides in hypoxia: possible role in regulation of coronary blood flow. Am J Physiol. 1963;204:317–322. doi: 10.1152/ajplegacy.1963.204.2.317. [DOI] [PubMed] [Google Scholar]
- Bogen DK, Rabinowitz SA, Needleman A, McMahon TA, Abelmann WH. An analysis of the mechanical disadvantage of myocardial infarction in the canine left ventricle. Circ Res. 1980;47:728–741. doi: 10.1161/01.res.47.5.728. [DOI] [PubMed] [Google Scholar]
- Borer JS, Fox K, Jaillon P, Lerebours G. Antianginal and antiischemic effects of ivabradine, an I(f) inhibitor, in stable angina. Circulation. 2003;107:817–823. doi: 10.1161/01.cir.0000048143.25023.87. [DOI] [PubMed] [Google Scholar]
- Buck JD, Hardman HF, Warltier DC, Gross GJ. Changes in ischemic blood flow distribution and dynamic severity of a coronary stenosis induced by beta blockade in the canine heart. Circulation. 1981;64:708–715. doi: 10.1161/01.cir.64.4.708. [DOI] [PubMed] [Google Scholar]
- Buckberg GD, Fixler DE, Archie JP, Jr, Hoffman JIE. Experimental subendocardial ischemia in dogs with normal coronary arteries. Circ Res. 1972;30:67–81. doi: 10.1161/01.res.30.1.67. [DOI] [PubMed] [Google Scholar]
- Buda AJ, Lefkowitz CA, Gallagher KP. Augmentation of regional function in nonischemic myocardium during coronary occlusion measured with two-dimensional echocardiography. J Am Coll Cardiol. 1990;16:175–180. doi: 10.1016/0735-1097(90)90476-6. [DOI] [PubMed] [Google Scholar]
- Chilian WM, Marcus ML. Phasic coronary blood flow velocity in intramural and epicardial coronary arteries. Circ Res. 1982;50:775–781. doi: 10.1161/01.res.50.6.775. [DOI] [PubMed] [Google Scholar]
- Chilian WM, NHLBI Workshop Participants Coronary microcirculation in health and disease. Summary of an NHLBI workshop. Circulation. 1997;95:522–528. doi: 10.1161/01.cir.95.2.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin P, Ghaleh B, Hittinger L, Monnet X, Slama M, Giudicelli J-F, et al. Differential effects of heart rate reduction and beta-blockade on left ventricular relaxation during exercise. Am J Physiol Heart Circ Physiol. 2002;282:H672–H679. doi: 10.1152/ajpheart.00547.2001. [DOI] [PubMed] [Google Scholar]
- Colin P, Ghaleh B, Monnet X, Hittinger L, Berdeaux A. Effect of graded heart rate reduction with ivabradine on myocardial oxygen consumption and diastolic time in exercising dogs. J Pharmacol Exp Ther. 2004;308:236–240. doi: 10.1124/jpet.103.059717. [DOI] [PubMed] [Google Scholar]
- Colin P, Ghaleh B, Monnet X, Su J, Hittinger L, Giudicelli J-F, et al. Contributions of heart rate and contractility to myocardial oxygen balance during exercise. Am J Physiol Heart Circ Physiol. 2003;284:H676–H682. doi: 10.1152/ajpheart.00564.2002. [DOI] [PubMed] [Google Scholar]
- DeFily DV, Chilian WM. Coronary microcirculation: autoregulation and metabolic control. Basic Res Cardiol. 1995;90:112–118. doi: 10.1007/BF00789441. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Camm JA. Heart rate lowering by specific and selective I(f) current inhibition with ivabradine: a new therapeutic perspective in cardiovascular disease. Drugs. 2004;64:1757–1765. doi: 10.2165/00003495-200464160-00003. [DOI] [PubMed] [Google Scholar]
- Du X-J, Feng X, Gao X-M, Tan TP, Kiriazis H, Dart AM. I(f) channel inhibitor ivabradine lowers heart rate in mice with enhanced sympathoadrenergic activities. Br J Pharmacol. 2004;142:107–112. doi: 10.1038/sj.bjp.0705696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehring T, Heusch G. Left ventricular asynchrony: an indicator of regional myocardial dysfunction. Am Heart J. 1990;120:1047–1057. doi: 10.1016/0002-8703(90)90116-f. [DOI] [PubMed] [Google Scholar]
- Embrey RP, Brooks LA, Dellsperger KC. Mechanism of coronary microvascular responses to metabolic stimulation. Cardiovasc Res. 1997;35:148–157. doi: 10.1016/s0008-6363(97)00096-5. [DOI] [PubMed] [Google Scholar]
- Gallagher KP, Gerren RA, Ning X-H, McManimon SP, Stirling MC, Shlafer M, et al. The functional border zone in conscious dogs. Circulation. 1987;76:929–942. doi: 10.1161/01.cir.76.4.929. [DOI] [PubMed] [Google Scholar]
- Gallagher KP, Gerren RA, Stirling MC, Choy M, Dysko RC, McManimon SP, et al. The distribution of functional impairment across the lateral border of acutely ischemic myocardium. Circ Res. 1986;58:570–583. doi: 10.1161/01.res.58.4.570. [DOI] [PubMed] [Google Scholar]
- Gallagher KP, Matsuzaki M, Koziol JA, Kemper WS, Ross J., Jr Regional myocardial perfusion and wall thickening during ischemia in conscious dogs. Am J Physiol Heart Circ Physiol. 1984;247:H727–H738. doi: 10.1152/ajpheart.1984.247.5.H727. [DOI] [PubMed] [Google Scholar]
- Gallagher KP, Matsuzaki M, Osakada G, Kemper WS, Ross J., Jr Effect of exercise on the relationship between myocardial blood flow and systolic wall thickening in dogs with acute coronary stenosis. Circ Res. 1983;52:716–729. doi: 10.1161/01.res.52.6.716. [DOI] [PubMed] [Google Scholar]
- Gascho JA, Beller GA. Adverse effects of circumflex coronary artery occlusion on blood flow to remote myocardium supplied by stenosed left anterior descending coronary artery in anesthetized open-chest dogs. Am Heart J. 1987;113:679–683. doi: 10.1016/0002-8703(87)90706-x. [DOI] [PubMed] [Google Scholar]
- Gerlach E, Deuticke B, Dreisbach RH. Der Nucleotid-Abbau im Herzmuskel bei Sauerstoffmangel und seine mögliche Bedeutung für die Coronardurchblutung. Naturwissenschaften. 1963;6:228–229. [Google Scholar]
- Guth BD, Heusch G, Seitelberger R, Matsuzaki M, Ross J., Jr Role of heart rate reduction in the treatment of exercise-induced myocardial ischemia. Eur Heart J. 1987a;8 Suppl L:61–68. doi: 10.1093/eurheartj/8.suppl_l.61. [DOI] [PubMed] [Google Scholar]
- Guth BD, Heusch G, Seitelberger R, Ross J., Jr Mechanism of beneficial effect of beta-adrenergic blockade on exercise-induced myocardial ischemia in conscious dogs. Circ Res. 1987b;60:738–746. doi: 10.1161/01.res.60.5.738. [DOI] [PubMed] [Google Scholar]
- Guth BD, Tajimi T, Seitelberger R, Lee JD, Matsuzaki M, Ross J., Jr Experimental exercise-induced ischemia: drug therapy can eliminate regional dysfunction and oxygen supply–demand imbalance. J Am Coll Cardiol. 1986;7:1036–1046. doi: 10.1016/s0735-1097(86)80221-2. [DOI] [PubMed] [Google Scholar]
- Guth BD, White FC, Gallagher KP, Bloor CM. Decreased systolic wall thickening in myocardium adjacent to ischemic zones in conscious swine during brief coronary artery occlusion. Am Heart J. 1984;107:458–464. doi: 10.1016/0002-8703(84)90086-3. [DOI] [PubMed] [Google Scholar]
- Heidland UE, Strauer BE. Left ventricular muscle mass and elevated heart rate are associated with coronary plaque disruption. Circulation. 2001;104:1477–1482. doi: 10.1161/hc3801.096325. [DOI] [PubMed] [Google Scholar]
- Heusch G. Alpha-adrenergic mechanisms in myocardial ischemia. Circulation. 1990;81:1–13. doi: 10.1161/01.cir.81.1.1. [DOI] [PubMed] [Google Scholar]
- Heusch G, Baumgart D, Camici P, Chilian W, Gregorini L, Hess O, et al. Alpha-adrenergic coronary vasoconstriction and myocardial ischemia in humans. Circulation. 2000;101:689–694. doi: 10.1161/01.cir.101.6.689. [DOI] [PubMed] [Google Scholar]
- Heusch G, Guth BD, Widmann T, Peterson KL, Ross J., Jr Ischemic myocardial dysfunction assessed by temporal Fourier transform of regional myocardial wall thickening. Am Heart J. 1987;113:116–124. doi: 10.1016/0002-8703(87)90018-4. [DOI] [PubMed] [Google Scholar]
- Heusch G, Yoshimoto N. Effects of heart rate and perfusion pressure on segmental coronary resistances and collateral perfusion. Pflügers Arch. 1983;397:284–289. doi: 10.1007/BF00580262. [DOI] [PubMed] [Google Scholar]
- Heusch G, Yoshimoto N, Müller-Ruchholtz ER. Effects of heart rate on hemodynamic severity of coronary artery stenosis in the dog. Basic Res Cardiol. 1982;77:562–573. doi: 10.1007/BF01907947. [DOI] [PubMed] [Google Scholar]
- Hjalmarson A, Gilpin EA, Kjekshus J, Schieman G, Nicod P, Henning H, et al. Influence of heart rate on mortality after acute myocardial infarction. Am J Cardiol. 1990;65:547–553. doi: 10.1016/0002-9149(90)91029-6. [DOI] [PubMed] [Google Scholar]
- Indolfi C, Guth BD, Miura T, Miyazaki S, Schulz R, Ross J., Jr Mechanisms of improved ischemic regional dysfunction by bradycardia. Studies on UL-FS 49 in swine. Circulation. 1989;80:983–993. doi: 10.1161/01.cir.80.4.983. [DOI] [PubMed] [Google Scholar]
- Indolfi C, Ross J., Jr The role of heart rate in myocardial ischemia and infarction: implications of myocardial perfusion–contraction matching. Prog Cardiovasc Dis. 1993;36:61–74. doi: 10.1016/0033-0620(93)90022-6. [DOI] [PubMed] [Google Scholar]
- Langenbach MR, Schmitz-Spanke S, Brockert M, Schepan M, Pomblum VJ, Gams E, et al. Comparison of a beta-blocker and an if current inhibitor in rabbits with myocardial infarction. J Cardiovasc Surg (Torino) 2006;47:719–725. [PubMed] [Google Scholar]
- Lew WYW, Chen Z, Guth BD, Covell JW. Mechanisms of augmented segment shortening in nonischemic areas during acute ischemia of the canine left ventricle. Circ Res. 1985;56:351–358. doi: 10.1161/01.res.56.3.351. [DOI] [PubMed] [Google Scholar]
- Lucats L, Ghaleh B, Colin P, Monnet X, Bize A, Berdeaux A. Heart rate reduction by inhibition of I(f) or by beta-blockade has different effects on postsystolic wall thickening. Br J Pharmacol. 2007a;150:335–341. doi: 10.1038/sj.bjp.0706996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucats L, Ghaleh B, Monnet X, Colin P, Bize A, Berdeaux A. Conversion of post-systolic wall thickening into ejectional thickening by selective heart rate reduction during myocardial stunning. Eur Heart J. 2007b;28:872–879. doi: 10.1093/eurheartj/ehm030. [DOI] [PubMed] [Google Scholar]
- Marcus ML, Chilian WM, Kanatsuka H, Dellsperger KC, Eastham CL, Lamping KG. Understanding the coronary circulation through studies at the microvascular level. Circulation. 1990;82:1–7. doi: 10.1161/01.cir.82.1.1. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Gallagher KP, Patritti J, Tajimi T, Kemper WS, White FC, et al. Effects of a calcium-entry blocker (diltiazem) on regional myocardial flow and function during exercise in conscious dogs. Circulation. 1984a;69:801–814. doi: 10.1161/01.cir.69.4.801. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Guth BD, Tajimi T, Kemper WS, Ross J., Jr Effects of the combination of diltiazem and atenolol on exercise-induced regional myocardial ischemia in conscious dogs. Circulation. 1985;72:233–243. doi: 10.1161/01.cir.72.1.233. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Patritti J, Tajimi T, Miller M, Kemper WS, Ross J., Jr Effects of beta-blockade on regional myocardial flow and function during exercise. Am J Physiol. 1984b;247:H52–H60. doi: 10.1152/ajpheart.1984.247.1.H52. [DOI] [PubMed] [Google Scholar]
- Merkus D, Houweling B, van Vliet M, Duncker DJ. Contribution of K+ATP channels to coronary vasomotor tone regulation is enhanced in exercising swine with a recent myocardial infarction. Am J Physiol Heart Circ Physiol. 2005;288:H1306–H1313. doi: 10.1152/ajpheart.00631.2004. [DOI] [PubMed] [Google Scholar]
- Miyashiro JK, Feigl EO. Feedforward control of coronary blood flow via coronary beta-receptor stimulation. Circ Res. 1993;73:252–263. doi: 10.1161/01.res.73.2.252. [DOI] [PubMed] [Google Scholar]
- Monnet X, Colin P, Ghaleh B, Hittinger L, Giudicelli J-F, Berdeaux A. Heart rate reduction during exercise-induced myocardial ischaemia and stunning. Eur Heart J. 2004;25:579–586. doi: 10.1016/j.ehj.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Monnet X, Ghaleh B, Colin P, Parent De Curzon O, Giudicelli J-F, Berdeaux A. Effects of heart rate reduction with ivabradine on exercise induced myocardial ischemia and stunning. J Pharmacol Exp Ther. 2001;299:1133–1139. [PubMed] [Google Scholar]
- Mosher P, Ross J, Jr, McFate PA, Shaw RF. Control of coronary blood flow by an autoregulatory mechanism. Circ Res. 1964;14:250–259. doi: 10.1161/01.res.14.3.250. [DOI] [PubMed] [Google Scholar]
- Mulder P, Barbier S, Chagraoui A, Richard V, Henry JP, Lallemand F, et al. Long-term heart rate reduction induced by the selective I(f) current inhibitor ivabradine improves left ventricular function and intrinsic myocardial structure in congestive heart failure. Circulation. 2004;109:1674–1679. doi: 10.1161/01.CIR.0000118464.48959.1C. [DOI] [PubMed] [Google Scholar]
- Rose J, Schulz R, Martin C, Heusch G. Post-ejection wall thickening as a marker of successful short term hibernation. Cardiovasc Res. 1993;27:1306–1311. doi: 10.1093/cvr/27.7.1306. [DOI] [PubMed] [Google Scholar]
- Ross J., Jr Myocardial perfusion–contraction matching. Implications for coronary heart disease and hibernation. Circulation. 1991;83:1076–1083. doi: 10.1161/01.cir.83.3.1076. [DOI] [PubMed] [Google Scholar]
- Ruzyllo W, Ford IF, Tendera MT, Fox KF. Antianginal and antiischaemic effects of the I(f) current inhibitor ivabradine compared to amlodipine as monotherapies in patients with chronic stable angina. Randomised, controlled, double-blind trial. Eur Heart J. 2004;25:A878. [Google Scholar]
- Schulz R, Rose J, Skyschally A, Heusch G. Bradycardic agent UL-FS 49 attenuates ischemic regional dysfunction and reduces infarct size in swine: comparison with the β-blocker atenolol. J Cardiovasc Pharmacol. 1995;25:216–228. doi: 10.1097/00005344-199502000-00006. [DOI] [PubMed] [Google Scholar]
- Seitelberger R, Guth BD, Heusch G, Lee JD, Katayama K, Ross J., Jr Intracoronary alpha 2-adrenergic receptor blockade attenuates ischemia in conscious dogs during exercise. Circ Res. 1988;62:436–442. doi: 10.1161/01.res.62.3.436. [DOI] [PubMed] [Google Scholar]
- Simon L, Ghaleh B, Puybasset L, Giudicelli J-F, Berdeaux A. Coronary and hemodynamic effects of S 16257, a new bradycardic agent, in resting and exercising conscious dogs. J Pharmacol Exp Ther. 1995;275:659–666. [PubMed] [Google Scholar]
- Tanaka N, Nozawa T, Yasumura Y, Futaki S, Hiramori K, Suga H. Heart-rate-proportional oxygen consumption for constant cardiac work in dog heart. Jpn J Physiol. 1990;40:503–521. doi: 10.2170/jjphysiol.40.503. [DOI] [PubMed] [Google Scholar]
- Tardif J-C, Ford I, Tendera M, Bourassa MG, Fox K. Efficacy of ivabradine, a new selective If inhibitor, compared with atenolol in patients with chronic stable angina. Eur Heart J. 2005;26:2529–2536. doi: 10.1093/eurheartj/ehi586. [DOI] [PubMed] [Google Scholar]
- Thollon C, Cambarrat C, Vian J, Prost JF, Peglion JL, Vilaine JP. Electrophysiological effects of S 16257, a novel sino-atrial node modulator, on rabbit and guinea-pig cardiac preparations: comparisons with UL-FS 49. Brit J Pharmacol. 1994;112:37–42. doi: 10.1111/j.1476-5381.1994.tb13025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota E, Ogasawara Y, Hiramatsu O, Tachibana H, Kajiya F, Yamamori S, et al. Dynamics of flow velocities in endocardial and epicardial coronary arterioles. Am J Physiol Heart Circ Physiol. 2005;288:H1598–H1603. doi: 10.1152/ajpheart.01103.2003. [DOI] [PubMed] [Google Scholar]
- Trivella MG, Broten TP, Feigl EO. Beta-receptor subtypes in the canine coronary circulation. Am J Physiol. 1990;259:H1575–H1585. doi: 10.1152/ajpheart.1990.259.5.H1575. [DOI] [PubMed] [Google Scholar]
- Vanyi J, Parratt RJ, Vegh A. Metoprolol reduces ‘compensatory' coronary blood flow following occlusion of an adjacent branch without altering post-occlusion hyperaemia. Life Sci. 2006;78:2384–2390. doi: 10.1016/j.lfs.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Vilaine J-P, Bidouard J-P, Lesage L, Reure H, Péglion J-L. Anti-ischemic effects of ivabradine, a selective heart rate-reducing agent, in exercise-induced myocardial ischemia in pigs. J Cardiovasc Pharmacol. 2003;42:688–696. doi: 10.1097/00005344-200311000-00016. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]