

Abstract
Endocannabinoids (eCBs) have recently been identified as axon guidance cues shaping the connectivity of local GABAergic interneurons in the developing cerebrum. However, eCB functions during pyramidal cell specification and establishment of long-range axonal connections are unknown. Here, we show that eCB signaling is operational in subcortical proliferative zones from embryonic day 12 in the mouse telencephalon and controls the proliferation of pyramidal cell progenitors and radial migration of immature pyramidal cells. When layer patterning is accomplished, developing pyramidal cells rely on eCB signaling to initiate the elongation and fasciculation of their long-range axons. Accordingly, CB1 cannabinoid receptor (CB1R) null and pyramidal cell-specific conditional mutant (CB1Rf/f,NEX-Cre) mice develop deficits in neuronal progenitor proliferation and axon fasciculation. Likewise, axonal pathfinding becomes impaired after in utero pharmacological blockade of CB1Rs. Overall, eCBs are fundamental developmental cues controlling pyramidal cell development during corticogenesis.
Keywords: excitation, glutamate, layer patterning, neocortex, neurogenesis
Pyramidal cell specification follows a sequential scenario in the developing cerebrum: commitment of progenitor cells to the neuronal lineage occurs in the subcortical proliferative ventricular zone (VZ) and subventricular zone (SVZ). Immature pyramidal cells undergo radial migration to populate the cortical plate (CP) (1), where they acquire layer-specific neurochemical and morphological diversity (2). Pyramidal cell positioning and patterning of their corticofugal and intracortical axons is in part achieved via transcriptional control acting throughout cellular identification (2). However, epigenetic microenvironmental cues, provided by neural progenitors, radial glia, and immature neurons, are also fundamental in attaining cortical cell identity with particularly robust effects on pathfinding and directional growth of long-range axons (3).
Endocannabinoids [eCBs; anandamide (AEA) and 2-arachidonoylglycerol] control various forms of synaptic plasticity at cortical glutamatergic synapses in the postnatal brain (4) through functional CB1 cannabinoid receptors (CB1Rs) (5). During brain development, eCBs control neuronal fate decision (6), interneuron migration (7), and axonal specification (8). Developmental eCB actions are underpinned by a temporally defined assembly of functional eCB signaling networks with coincident expression of sn-1-diacylglycerol lipases (DAGLα/β) (9) and N-arachidonoyl-phosphatidyl ethanolamine (NAPE)-selective phospholipase D involved in eCB synthesis, fatty-acid amide hydrolase (FAAH) (an enzyme preferentially degrading AEA), and CB1Rs (8). The selective axonal targeting of CB1Rs and DAGLs in immature neurons suggests that eCBs may function in either cell-autonomous (6, 9) or target-derived (8) manner to control axonal elongation and postsynaptic target selection, respectively.
Although recent findings in both mammals (8) and nonmammalian vertebrates (10) suggest that eCB signaling is required for axonal elongation and fasciculation, eCB functions instructing distinct stages of pyramidal cell development are unknown. Here, we show that eCB signaling is operational in the subcortical VZ/SVZ and drives neural progenitor proliferation and migration, thus contributing to defining the final positions and densities of immature pyramidal cells. Subsequently, eCB signaling in immature pyramidal cells is required for axonal polarization and the formation of long-range glutamatergic axons. Accordingly, genetic and pharmacological disruption of CB1R functions reveals fasciculation deficits and axonal mistargeting. In sum, our data demonstrate that eCB signaling is indispensable for the genesis, proliferation, migration, and axonal behaviors of neocortical pyramidal cells and support the concept that eCBs act as a novel class of morphogens during corticogenesis.
Results
CB1R Expression in Developing Cerebrum.
We used in situ hybridization (11, 12) and immunofluorescence (8) histochemistry combined with high-resolution confocal microscopy [see supporting information (SI) Methods and Figs. S1 and S2] to define the identity of cells expressing CB1R and DAGLα/β expression in the developing mouse and human neocortex. CB1R mRNA expression was restricted to telencephalic differentiation zones at embryonic day (E)12.5 in mouse (Fig. 1 A and B). By E14.5, significant CB1R mRNA expression was evident in immature pyramidal cells populating the CP and hippocampal primordium (Figs. 1C and Fig. S2 A–D), reached peak expression levels by approximately E16.5 (Fig. 1D), and gradually declined during pyramidal cell morphogenesis in the late gestational embryo (Fig. S2 G–J). Considerable CB1R mRNA expression in neural progenitors exiting the cortical SVZ was sustained throughout corticogenesis, whereas the VZ was largely devoid of CB1R hybridization signal (Fig. 1 E and G). These findings are not restricted to mouse development, because a similar CB1R mRNA expression pattern was seen in the second trimester human fetal telencephalon, comparable to E18/postnatal day (P)0 rodent brain (13). In particular, robust CB1R mRNA expression was detected in the human SVZ, CP neurons, and hippocampal pyramidal cell layers (Fig. 1 H and I).
Fig. 1.

CB1R localization in the developing brain. (A–G) In situ hybridization demonstrating the spatial and temporal distribution of CB1R mRNA in the mouse brain. Arrows in C and E and G denote CB1R mRNA hybridization signal in pyramidal cells and SVZ progenitors, respectively. (H and I) Distribution of CB1R mRNA in human fetal brain. Nissl/AChE histochemistry reveals territorial boundaries. (J–M) DAGLβ and CB1R expression in mouse VZ/SVZ. Cortical Tbr2+ projection neurons migrating toward the CP express CB1Rs. (N–R) CB1Rs are selectively enriched in axons of cortical projection neurons. Arrows indicate corticothalamic axons, and arrowheads identify axons committed to the fibria. (Scale bars: E–G and K–M, 30 μm; N–R, 85 μm; A–D, H, and I, 100 μm.) See SI Text for abbreviations.
eCB Signaling During Corticogenesis.
DAGLβ-like immunoreactivity (i.r.) was present in the cortical VZ/SVZ from E12.5 until birth (Fig. 1 J and K and Fig. S3A) (10), suggesting the coincidence of local eCB synthesis with neuronal progenitor proliferation. CB1R i.r. was evident in intermediate progenitor cells (Tbr2+), known to differentiate into pyramidal cells (2), that had engaged in radial migration toward the CP (Fig. 1 L and M and Fig. S3). CB1R mRNA expression by immature pyramidal cells concurred with the targeting of CB1Rs to developing long-range axons between E13.5 and P0 (Fig. 1 N–R). Coexistence of DAGLβ and CB1Rs in developing glutamatergic axons (Fig. S2 E and F) reinforces the hypothesis (9) that eCB signaling is required for pyramidal cell development and functional specification.
eCBs Control SVZ Progenitor Proliferation.
We tested whether eCBs control neural progenitor proliferation (6) in subcortical VZ/SVZ by analyzing CB1R−/− and wild-type littermates pulsed with BrdU on E14.5. Lack of CB1Rs significantly decreased neural progenitor proliferation (Fig. 2A). Conversely, FAAH−/− (14) increased proliferation of VZ/SVZ progenitors (Fig. 2B). Here, genetic manipulation of FAAH activity was used to elevate eCB levels; however, the particular eCB mediating these phenomena was not identified.
Fig. 2.

eCBs regulate neural progenitor proliferation in cortical VZ/SVZ. (A) CB1R deletion significantly reduces the rate of neural progenitor proliferation, as defined by the density of BrdU+ cells in the VZ/SVZ. (B) Conversely, elevated eCB levels in FAAH−/− mice (14) significantly increase neural progenitor proliferation. (C) Conditional CB1R deletion in subcortical SVZ progenitors (arrows) (16) decreases the rate of Ki67+ progenitor proliferation (n = 3 per genotype). **, P < 0.01, compared with wild-type littermates. (Scale bar, 100 μm.)
eCB functions specifically underpinning pyramidal cell progenitor proliferation were elucidated in mice with conditional deletion of CB1Rs through Cre recombinase expressed under the control of regulatory sequences of NEX, a neuronal basic helix–loop–helix protein (15) (CB1Rf/f,NEX-Cre). Prominent Cre activity is observed by approximately E11.5 in cortical progenitors in NEX-Cre mice (15), ensuring the lack of CB1Rs in pyramidal cells at all developmental stages studied here. In these mutants (16), VZ/SVZ progenitor proliferation was significantly impaired, as indicated by reduced Ki67 and GOLGA5 cell density in proliferative zones (Fig. 2C and Fig. S4A). Because NEX is expressed by pyramidal progenitors in SVZ but not in VZ (16), a coincident proliferation deficit in CB1Rf/f,NEX-Cre and CB1R−/− mice suggests that eCBs exert differential control on subcortical progenitor pools. Therefore, we analyzed BrdU+ progenitor proliferation rates separately in SVZ and VZ of CB1R−/− (SVZ, 55%; VZ, 77% of control) and FAAH−/− mice (SVZ, 226%; VZ, 150% of control). Thus, progenitor proliferation in SVZ may rely more on autocrine eCB regulation, whereas non-cell-autonomous signaling predominates in VZ. Our genetic data were further validated by HU-210 (synthetic CB1R agonist) and URB597 (FAAH inhibitor) treatment of E14.5 organotypic slices revealing increased VZ/SVZ progenitor proliferation upon sustained cannabinoid receptor stimulation (Fig. S4B).
eCBs Regulate Pyramidal Precursor Migration.
eCB effects on radial migration of immature pyramidal cells was tested by injecting pregnant mice (E14.5) with BrdU and allowing the offspring to develop until P2.5, when layer-specific distribution of BrdU+ cells was determined. Profuse BrdU incorporation was found in the brains of wild-type mice with labeled cells enriched in neocortical layers (L)2 and 3 (bins 2–4 in Fig. 3A; see also Table S1). In contrast, BrdU+ cells accumulated in L4–6 (corresponding to bins 6–10; Fig. 3A) in CB1R−/− mice. Likewise, cell migration was also altered in FAAH−/− mice, with dense clusters of BrdU+ cells in L3 and L4 (bins 5–8 in Fig. 3B; see also Table S1). In vivo evidence of a role for eCBs during radial cell migration was confirmed by showing that HU-210 or URB597 application enhanced (Fig. 3C), whereas FAAH overexpression inhibited (Fig. S4C) radial cell migration from the VZ/SVZ to superficial cortical layers in organotypic cultures.
Fig. 3.

eCBs control radial migration of pyramidal cell progenitors. (A) Cortical distribution of neurons at P2.5 whose progenitors were BrdU labeled at E14.5. Note the migration arrest of a population of neural progenitors in deep cortical layers of CB1R−/− mice. (B) Cortical progenitor distribution in FAAH−/− mice was assessed as above. Cell counts were performed in grouped cortical layers defined as equal binned areas. (C) Distribution of GFP+ cells in brain slices from E14.5 mouse embryos after ex vivo electroporation of VZ progenitors with pCIG2-GFP. Slices were maintained for 48 h in the presence of HU-210 (1 μM) or URB597 (1 μM) in vitro. Cumulative cell counts were obtained in CP, IZ, and VZ/SVZs. **, P < 0.01; *, P < 0.05, compared with wild-type littermates or control treatment. (see Tables S1 and S2 for statistical analysis). (Scale bars: A and B, 75 μm; C, 35 μm.)
Pyramidal Cell Specification Relies on eCB Signaling.
Exogenous CB1R agonists have been identified (7, 8) as morphogens and chemotropic guidance cues for cortical interneurons. However, it is unknown whether eCBs affect pyramidal cell development. Therefore, we exposed pyramidal cells isolated from E14.5 neocortex to NGF (100 ng/ml), a differentiation promoting neurotrophin (17), AEA (200 nM) (8), or AM251 (1 μM), a CB1R inverse agonist. NGF increased axonal arbors of VGLUT1+ pyramidal cells (Fig. 4 A and B). In contrast, AEA induced the elongation of a leading axon while inhibiting axon branching. AM251 effects were reminiscent of those of NGF: an expansion of axonal arbors and reversal of AEA effects (Fig. 4 A and B). Disrupting CB1R signaling by coapplication of AEA and AM251 significantly inhibited neurochemical differentiation by decreasing the density of VGLUT1+ neurons (Fig. 4B).
Fig. 4.

eCB signaling controls pyramidal cell morphogenesis. (A and B) Quantitative morphometry of axon development of VGLUT1+ pyramidal cells after 3-day treatment in vitro. Bracketed numbers indicate population sizes (see also Table S3). (C–E) DAGLα and β undergo axonal targeting and exhibit differential distribution (C). Whereas DAGLα concentrates in axonal varicosities, DAGLβ is distributed uniformly along axons (arrows in D). (F) In axonal growth cones, DAGLα is targeted to filopodia (arrows), whereas DAGLβ concentrates in the axon stem with a clear demarcation from the growth cones (arrowheads). (G) DAGL inhibition by O-3841 significantly reduces VGLUT1 expression in pyramidal cells by 6 days in vitro. O-3841 effects were prevented by exogenous application of AEA. *, P < 0.05, compared with control. (Scale bars: A, 25 μm; C, 15 μm; D–F, 3 μm.)
Our data also support that pyramidal cells require an intrinsic “eCB tone” to initiate axonal polarization and neurochemical differentiation. Both DAGLs were expressed by pyramidal cells (Fig. 4C and Fig. S5A) and targeted to the axon (Fig. 4 D and E) and navigating growth cones (Fig. 4F). Notably, DAGLα was detected in elongating axon shafts and growth cones (including filopodia) and exhibited proximal localization to CB1R (Fig. S5A), whereas DAGLβ-like i.r. was limited to axonal shafts. DAGLα levels inversely correlated with axon development; initially a random DAGLα distribution was seen in quiescent axons, followed by DAGLα concentrating in axon varicosities (18) (Fig. S5C). These data reveal that axonal DAGLα levels remain high in developing axons and undergo redistribution during axon maturation and synapse specification (Fig. S5C). DAGL inhibition by O-3841 (19) significantly reduced VGLUT1 expression in pyramidal cells, thus supporting the involvement of eCB signaling during the acquisition of a glutamatergic phenotype (Fig. 4G). Moreover, O-3841-induced increased synaptogenesis in vitro (Fig. S5D) suggests that impaired temporal and spatial integrity of eCB signaling in pyramidal cells may disrupt postsynaptic targeting of glutamatergic axons.
CB1R Deletion Reveals Fasciculation Deficits.
We assessed the in vivo significance of our findings in CB1Rf/f,NEX-Cre mice (15). In new-born CB1Rf/f,NEX-Cre (Fig. 5 A and B′) but not CB1Rf/f or NEXCre/+ mice (Fig. S6), L1 neuronal cell adhesion molecule (L1-NCAM)+ pyramidal axons formed bundles with aberrant trajectories in the corpus callosum, failed to invade the dorsal striatum, and exhibited a change in their striatal paths. In CB1R−/− (null) mice, axonal fasciculation deficits were pronounced at E14–16 (Fig. 5C) with a significant degree of compensation by birth. Nevertheless, corticofugal axons invariably failed to invade the dorsal striatum in CB1R−/− neonates (Fig. 5D).
Fig. 5.
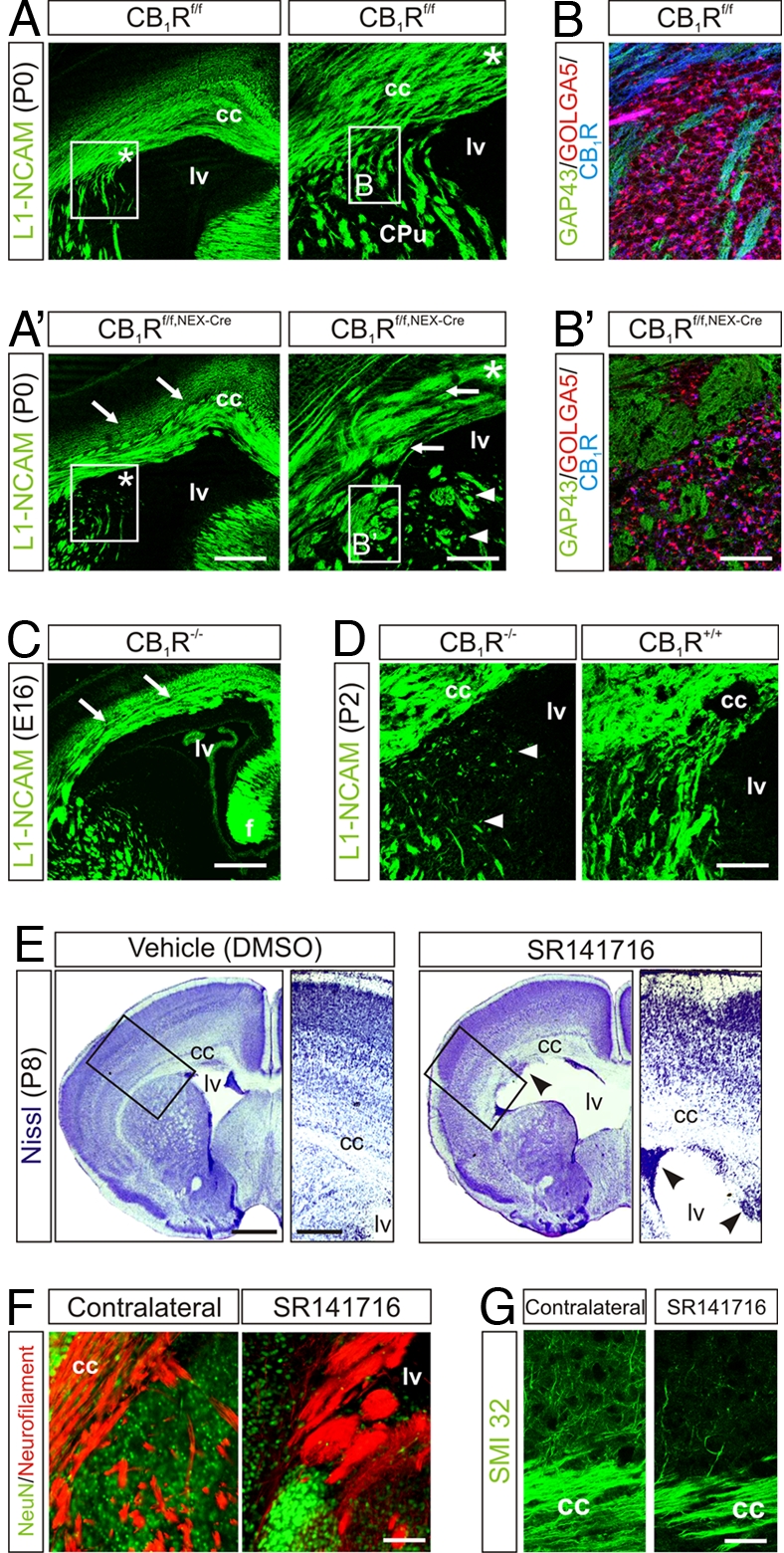
Genetic and pharmacological ablation of CB1Rs leads to axon fasciculation deficits. (A) Conditional deletion of CB1Rs in pyramidal cells evokes fasciculation deficits in L1-NCAM+ long-range axons. Arrows point to enlarged fascicles. Asterisk in open boxes denotes the general localization of insets showing the lack of fiber invasion in dorsal striatum. (B and B′) Aberrant axon bundles in dorsal striatum. (C) Axonal deficits in CB1R−/− brains at E16 are reminiscent of those seen in neonatal conditional mutants. (D) Impaired axonal targeting toward the striatum persists in CB1R−/− until P2. (E) In utero SR141716 infusion induces ventricle enlargement, migration arrest, and cortical delamination. Arrowheads indicate clusters of progenitor cells in the ventricular zone. Open boxes denote the position of insets. (F) Neurofilament M staining reveals fasciculation deficits in the subventricular corpus callosum after SR141716 infusion. (G) SR141716 decreases the commitment of pyramidal cell axons to descending projections. (Scale bars: A, C, and E, 250 μm; A Inset and D, 100 μm; B′ and G, 30 μm; F, 50 μm.)
In Utero CB1R Blockade Disrupts Neurodevelopment.
CB1R function in long-range axon fasciculation was tested also by i.c.v. injection of SR141716 (10 mM in 1 μl) in utero at E13.5, as a proof-of-concept for CB1R antagonist actions. Analysis of corticofugal connectivity at P8 revealed enlarged lateral ventricles (Fig. 5E) in conjunction with impaired progenitor proliferation in the subcortical VZ/SVZ (DMSO, 39 ± 3%; SR141716, 27 ± 3% BrdU+ cells; P < 0.05; n = 4), resulting in cortical delamination. Expansion of the corpus callosum was due to a deficit in axonal pathfinding, as indicated by a reduced commitment of pyramidal cell axons to the developing callosal trajectory (Fig. 5 F and G). Overall, data obtained in genetic models and after in utero SR141716 treatment confirm that proliferation, migration, and axonal pathfinding decisions of pyramidal cells are reliant on eCB signaling through CB1Rs.
Discussion
The molecular mechanisms of eCB actions in the adult brain are well appreciated. However, a dearth of knowledge exists on the cellular specificity of eCB actions in the developing brain (20). In this article, we show the onset and spatial restriction of eCB signaling during embryogenesis and demonstrate the cellular roles of eCB signaling on neurochemically identified cortical pyramidal cells and their progenitors. These data, together with prior results on eCB control of neuronal progenitor cell fate and GABA interneuron development, define the complexity of eCB signaling in the developing cerebrum and identify eCBs as developmental signals indispensable for cortical neuron specification and connectivity patterning (Fig. S7).
eCB Effects on VZ/SVZ Neural Progenitor Commitment.
Recent studies demonstrate that eCBs are developmental signals helping to determine neuronal identity at both the cellular and neuronal network levels (6–8, 10, 20). This article significantly expands existing knowledge on neuronal fate decisions during corticogenesis by showing that acquiring a glutamatergic neuronal phenotype, in addition to eCB actions instructing interneuron development (7, 8), is influenced by eCB signaling. First, the coincident expression of eCB synthesis enzymes and receptors in cortical progenitor zones demonstrates the existence of operational eCB signaling networks in neurogenic niches of the developing brain, where eCBs tune the rate of neural progenitor proliferation and the number of immature neurons committed to radial migration. Our findings indicate that eCB signaling is instrumental for embryonic neurogenesis, as also reported in adult brain (21), and data from knockout mice support a role for CB1Rs in committing SVZ progenitors to a pyramidal cell fate (10). Our in vitro morphometric and biochemical data also extend previous findings showing that both DAGLα/β are recruited to elongating glutamatergic axons (9) with DAGLα being the primary enzyme concentrating in axonal growth cones. Concurrent with the recent hypothesis that DAGL functions are required for axonal polarization and growth (9), we define a negative correlation between DAGLα/β levels in axons and the complexity of excitatory axon morphology (18). Also, we show that inhibition of eCB synthesis attenuates neurochemical pyramidal cell differentiation by suppressing VGLUT1 expression, a functional marker of glutamatergic synapses (22), and triggers premature synapse formation.
eCBs Exert Differential Control on Cortical Neurons.
The role of eCBs in cortical neuron specification depends on the molecular identity of neurons. Pyramidal cells express CB1Rs as postmitotic progenitors in the subcortical VZ/SVZ. However, GABAergic progenitors in ganglionic eminences lack this receptor, and immature GABAergic interneurons express CB1Rs only during and after intracortical (radial) migration (8). Postmitotic, pyramidal cell lineage-committed neurons harbor the capacity of eCB synthesis throughout their morphological and functional specification. In contrast, GABAergic interneurons seem to lack, except for sporadic NAPE-selective phospholipase D expression, eCB synthetic enzymes. Functional implications of these findings include the following. First, eCB signaling is a key determinant of the number of pyramidal cells destined to particular cortical laminae. Second, eCB control of axonal elongation in pyramidal cells is a primarily cell-autonomous mechanism, with DAGLs and CB1Rs being in close proximity predominantly along the axon stem allowing direct ligand-receptor coupling. Conversely, CB1Rs concentrate in axonal growth cones of GABAergic interneurons (8), suggesting that GABA cells use target-derived eCBs as microenvironmental directional cues for growth cone steering decisions. Third, local GABAergic and long-range glutamatergic axons are inherently different with regards to the dynamics of their development, arborization, and mechanisms of postsynaptic target selection. Because axonal arbors of cortical interneurons exhibit a high degree of complexity within volumetrically limited cellular microdomains, an autocrine eCB tone (9) would likely restrict formation of ramified, complex axonal arbors by cortical interneurons. In contrast, CB1R agonists trigger the elongation of a single pyramidal cell axon with concomitant inhibition of collateral formation in a manner reversed by CB1R inverse agonists and NGF (data not shown). Consequently, our present genetic and pharmacological findings revealing perturbed fasciculation of corticofugal glutamatergic axons endowed with CB1Rs targeted to the surface of their plasmalemma (8) and high focal DAGL activity (Fig. S5) support the hypothesis that eCB signaling has simultaneous roles in long-range axon development: Autocrine eCB signaling (9) facilitates the formation of axonal pathways (10), where an eCB gradient along individual axons propels their extension, with focal elevation of eCB concentrations among developing axons being critical to restrict premature axonal dispersion. Thus, disrupting the temporal and spatial dynamics of eCB signaling in the developing telencephalon ultimately leads to aberrant axonal behaviors.
CB1R Knock-Outs Develop Axonal Deficits.
Significant axon fasciculation deficits were observed in neonatal CB1Rf/f,NEX-Cre mice. Notably, CB1R−/− mice exhibited disrupted axon development at approximately E15 that was reminiscent of the phenotype in conditional mice; however, in CB1R−/− mice the deficit normalized by birth. These differences may demonstrate pleiotropism of the eCB system at the receptor level and suggest that the expression of non-CB1Rs on neurons or redundancy of signaling pathways (e.g., overt expression of other chemotropic/repulsive guidance cues) may compensate for the complete loss of CB1R-mediated signaling in CB1R−/− mice. The finding that pyramidal cell CB1Rs are only abundantly expressed during the restricted period of their morphological and functional specification (E14.5–E18.5) argues that targeted disruption of CB1R signaling during this period will selectively disrupt axon elongation and targeting and impose permanent deficits to a restricted set of neocortical pyramidal cells in CB1Rf/f,NEX-Cre conditional mutants.
Overall, our present and previous (6–8) data define discrete spatial and temporal niches for eCB action and identify the cellular basis of their dichotomy on glutamatergic and GABAergic cortical neurons. Although the identity of eCB(s) mediating particular cellular actions has not been elucidated, developmental effects of manipulating both AEA and 2-arachidonoylglycerol levels suggest that both ligands may control pyramidal cell specification through their promiscuity at the CB1Rs. Considering the high degree of phylogenetic conservation of eCB signaling, both at enzyme and receptor levels, across vertebrate species (23), data described herein on mammalian neurons (8) together with those on zebrafish and chick neurons (10) formulate the unifying concept that eCBs are key developmental cues establishing neuronal diversity and synaptic connectivity in the developing brain.
Methods
Animals.
The generation and genotyping of glutamate decarboxylase 67-GFP (Δneo) (GAD67gfp/+) (8), CB1R−/− (11), FAAH−/− (14), CB1Rf/f,NEX-Cre (11), and respective littermate controls has been reported elsewhere. Mouse and Sprague–Dawley rat embryos and tissues were obtained from timed matings.
Neuroanatomy.
In situ hybridization in mouse and human fetal brains was performed as described (8, 11, 12). Multiple immunofluorescence labeling, acetylcholinesterase histochemistry, Hoechst 33,258 (Sigma) and Nissl stains were performed with quality-controlled immunoreagents (6, 8) (Fig. S1) with n = 2–5 brains per group analyzed by laser-scanning microscopy.
Cell Proliferation, Migration, and ex Vivo Electroporation.
Cell proliferation and migration was determined after i.p. BrdU injection (100 μg/g) of pregnant females at E14.5. For proliferation assays, embryos were harvested 2 h after labeling. For migration experiments, pups were killed at P2 [n = 6/6 (wild type/CB1R−/−); n = 7/7 (wild type/FAAH−/−), from three litters]. Cortical layers were identified by their discrete cell densities as visualized by Hoechst 33528 (Sigma) and β-III-tubulin (TuJ1; Promega) counterstaining. Ex vivo electroporation was performed by using a pCIG2-GFP vector (24) and slice cultures were maintained in semidry conditions in wells containing neurobasal medium (1% B27/1% N2/1% glutamine/1% penicillin/streptomycin/1% Fungizone/5 μg/ml ciprofloxacine). pCIG2-GFP vector was also used to overexpress FAAH in an IRES-EGFP cassette under the control of a CMV enhancer and chicken β-actin promoter. For proliferation studies, brain slices (n ≥ 6 per condition) were cultured overnight, stimulated, and pulsed with BrdU (10 μg/ml; 2 h). Cyclohexylcarbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597) was obtained from Cayman Chemical. A minimum of six slices per treatment were analyzed (6).
Pyramidal Cell Cultures.
Rat cortices were isolated at E14.5, cells were dissociated by trypsin digestion (0.1%; 5 min) and plated at a density of 25,000 or 200,000 cells per well for morphometry or biochemical analysis, respectively. Cultured neurons were maintained in DMEM/F12, supplemented with 1% B27/1% glutamine/1% penicillin/streptomycin for 3–6 days (7). Ligands were first added 12 h after cell seeding and replenished every other day (7), except for O-3841 (1 μM) (19), which was used between days 2–6 in vitro. Density of glutamatergic neurons was defined as a ratio of VGLUT1+/TuJ1+ cells from ≥10 randomly selected view fields per coverslip. Morphometric analysis was performed as described (7, 8), with also defining the longest axon segment. Western blotting of SNAP25 and DAGLα was used to verify morphometric assessment (7).
In Utero SR141716 Treatment.
The uteruses of anesthetized pregnant mice (E13.5) were externalized, and a glass micropipette filled with 1 μl SR141716 (10 mM) or DMSO (vehicle) was advanced through the uterine wall and into the lateral ventricle of the embryo. A single hemisphere was injected, whereas the other served as an internal control. After drug infusion, the uterus was gently repositioned and the abdominal wall was sutured. Mice were transcardially perfused on P8 and processed (Fig. S1).
Statistics.
Data were expressed as means ± SEM. Statistical analysis was performed by either ANOVA with Student–Neuman–Keuls post hoc tests or two-tailed unpaired Student's t test on independent samples. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments.
We thank C. Ljungberg, E. Resel, and B. Julien for technical assistance; Sanofi–Aventis (Montpellier, France) for SR141716; R. Mechoulam (Jerusalem) for HU-210; B. Cravatt (The Scripps Research Institute, La Jolla, CA) for FAAH−/− mice; and Y. Yanagawa (Gunma University, Maebashi City, Japan) for GAD67gfp/+ mice. This work was supported by the Alzheimer's Research Trust (J.M.), Alzheimer's Association (K. Mackie and T.H.), Swedish Medical Research Council (T.H.), European Molecular Biology Organization Young Investigator Programme (T.H.), Hjärnfoden (T.H.), Scottish Universities Life Science Alliance (T.H.), and Deutsche Forschungsgemeinschaft (B.L.). This work was also supported by National Institutes of Health Grants DA11322, DA15916, and DA21696 (to K. Mackie); R01DA023214 (to Y.L.H. and T.H.); NS048884 (to H.-C.L.); and ES07332 (to C.J.B.R.). I.G.-R. was supported by Santander Complutense Grant PR27/05-13988, and M.G. was supported by Comunidad de Madrid Grants S-SAL/0261/2006 and 950344.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0803545105/DCSupplemental.
References
- 1.Rakic P. A century of progress in corticoneurogenesis: From silver impregnation to genetic engineering. Cereb Cortex. 2006;16(Suppl 1):i3–i17. doi: 10.1093/cercor/bhk036. [DOI] [PubMed] [Google Scholar]
- 2.Fishell G, Hanashima C. Pyramidal neurons grow up and change their mind. Neuron. 2008;57:333–338. doi: 10.1016/j.neuron.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Bendito G, et al. Tangential neuronal migration controls axon guidance: a role for neuregulin-1 in thalamocortical axon navigation. Cell. 2006;125:127–142. doi: 10.1016/j.cell.2006.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Domenici MR, et al. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. 2006;26:5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawamura Y, et al. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aguado T, et al. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J Neurosci. 2006;26:1551–1561. doi: 10.1523/JNEUROSCI.3101-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berghuis P, et al. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc Natl Acad Sci USA. 2005;102:19115–19120. doi: 10.1073/pnas.0509494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berghuis P, et al. Hardwiring the brain: Endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- 9.Bisogno T, et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson S, Chambers D, Hobbs C, Doherty P, Graham A. The cannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol Cell Neurosci. 2008;38:89–97. doi: 10.1016/j.mcn.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Monory K, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Dow-Edwards D, Keller E, Hurd YL. Preferential limbic expression of the cannabinoid receptor mRNA in the human fetal brain. Neuroscience. 2003;118:681–694. doi: 10.1016/s0306-4522(03)00020-4. [DOI] [PubMed] [Google Scholar]
- 13.Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- 14.Cravatt BF, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goebbels S, et al. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- 16.Wu SX, et al. Pyramidal neurons of upper cortical layers generated by NEX-positive progenitor cells in the subventricular zone. Proc Natl Acad Sci USA. 2005;102:17172–17177. doi: 10.1073/pnas.0508560102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patapoutian A, Reichardt LF. Trk receptors: Mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- 18.Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bisogno T, et al. Development of the first potent and specific inhibitors of endocannabinoid biosynthesis. Biochim Biophys Acta. 2006;1761:205–212. doi: 10.1016/j.bbalip.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 20.Harkany T, et al. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol Sci. 2007;28:83–92. doi: 10.1016/j.tips.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Jin K, et al. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol. 2004;66:204–208. doi: 10.1124/mol.66.2.204. [DOI] [PubMed] [Google Scholar]
- 22.Fremeau RT, Jr, et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 23.McPartland JM. Phylogenomic and chemotaxonomic analysis of the endocannabinoid system. Brain Res Brain Res Rev. 2004;45:18–29. doi: 10.1016/j.brainresrev.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen L, et al. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006;20:1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.