

Abstract
High-valent iron species are powerful oxidizing agents in chemical and biological catalysis. The best characterized form of an Fe(V) equivalent described in biological systems is the combination of a b-type heme with Fe(IV)=O and a porphyrin or amino acid cation radical (termed Compound I). This work describes an alternative natural mechanism to store two oxidizing equivalents above the ferric state for biological oxidation reactions. MauG is an enzyme that utilizes two covalently bound c-type hemes to catalyze the biosynthesis of the protein-derived cofactor tryptophan tryptophylquinone. Its natural substrate is a monohydroxylated tryptophan residue present in a 119-kDa precursor protein. An EPR-silent di-heme reaction intermediate of MauG was trapped. Mössbauer spectroscopy revealed the presence of two distinct Fe(IV) species. One is consistent with an Fe(IV)=O (ferryl) species (δ = 0.06 mm/s, ΔEQ = 1.70 mm/s). The other is assigned to an Fe(IV) heme species with two axial ligands from protein (δ = 0.17 mm/s, ΔEQ = 2.54 mm/s), which has never before been described in nature. This bis-Fe(IV) intermediate is remarkably stable but readily reacts with its native substrate. These findings broaden our views of how proteins can stabilize a highly reactive oxidizing species and the scope of enzyme-catalyzed posttranslational modifications.
Keywords: ferryl, posttranslational modification, spectroscopy, tryptophan tryptophylquinone (TTQ), free radical
Tryptophan tryptophylquinone (TTQ) (1, 2) is the protein-derived catalytic cofactor of methylamine dehydrogenase (MADH) from Paracoccus denitrificans, a 119-kDa heterotetrameric α2β2 protein with a TTQ present on each β subunit (3, 4). TTQ biosynthesis requires incorporation of two oxygens into βTrp-57 and cross-linking of the indole rings of βTrp-57 and βTrp-108 (Scheme 1). This event is not self-processing but requires the action of at least one processing enzyme. Deletion of mauG, a gene in the methylamine utilization (mau) gene cluster (5, 6), causes accumulation of a biosynthetic precursor of MADH in which βTrp-57 is monohydroxylated at C7 and the cross-link is absent (7, 8). MauG-dependent TTQ biosynthesis from the precursor was achieved in vitro by using either O2 plus electrons from an external donor, or H2O2 (9–11).
Scheme. 1.

MauG-dependent TTQ biosynthesis.
MauG is a 42.3-kDa enzyme containing two covalently bound c-type hemes, one low-spin and one high-spin (9). The EPR parameters of oxidized MauG are atypical of c-type cytochromes but similar to those of the noncovalently bound b-type hemes in oxygen-binding proteins and oxygenases (9). The two hemes have similar intrinsic redox potentials but exhibit cooperative redox behavior, indicating that facile equilibration of electrons between the two hemes occurs (12), even though no spin coupling is evident from the EPR spectrum (9). On the basis of the CXXCH c-type heme-binding motifs in the sequence, each heme is expected to use a histidine for the proximal axial ligand with the heme vinyl groups covalently attached to the two cysteine sulfurs (13). No additional cysteines are present in the MauG sequence, so it is not possible for cysteine to provide an axial ligand for either heme as is seen in cytochrome P450s. EPR and resonance Raman studies suggested that the high-spin heme possesses a single histidine axial ligand (9, 12). The resonance Raman spectrum exhibited marker bands associated with the low-spin heme at frequencies similar to those of c-type hemes with the axial coordination of histidines (12). However, on the basis of existing data the possibility of another amino acid residue providing the distal axial ligand cannot be excluded.
For heme and nonheme iron enzymes, it is generally believed that the O2-dependent and H2O2-dependent oxygenation mechanisms each proceed via a ferric hydroperoxy intermediate (14, 15), which may then lose water to yield a high-valent Fe(IV)=O (ferryl) species (16, 17). Ferryl heme species with a π-porphyrin or amino acid radical (known as Compound I) have been observed in several enzymes (for review, see refs. 14, 18, and 19). Here, we report EPR and Mössbauer spectroscopic analysis of MauG oxidized by H2O2 in the absence and presence of its natural substrate (i.e., the isolated biosynthetic precursor of MADH). In each case, it was possible to trap and characterize an unprecedented intermediate species. Formation of mature MADH with TTQ from the biosynthetic precursor is a six-electron oxidation process: two for insertion of the second oxygen, two for formation of the cross-link, and two for oxidation of the quinol to quinone. After stoichiometric addition of H2O2 (i.e., one-third of the total requirement) to oxidized MauG alone, a high-valent bis-Fe(IV) intermediate is trapped. When the biological substrate of MauG is mixed with this MauG-based di-heme bis-Fe(IV) intermediate, the di-ferric MauG is regenerated, and a new protein-based free radical species is concomitantly observed from the substrate. These results reveal strategies for enzyme-catalyzed and heme-dependent protein posttranslational modification reactions and for stabilization of protein-based reactive intermediates.
Results
Characterization of the Product of the Reaction of MauG with H2O2 by Visible and EPR Spectroscopy.
To investigate the mechanism of oxygen activation by MauG, the di-heme enzyme was mixed with stoichiometric H2O2. Changes in the absorption spectrum of MauG occurred within 15 ms with a decrease in intensity of the Soret peak and a shift in its maximum from 405 to 407 nm (data not shown). The spectrum slowly returns to one very similar to the ferric MauG but with a slight increase and red shift in the Soret peak relative to the original ferric MauG. Parallel changes were observed in the X-band EPR spectra (Fig. 1). The ferric MauG exhibits high-spin (g = 5.57, 1.99) and low-spin (g = 2.54, 2.19, 1.87) heme signals. A minor low-spin component with g values of 2.89, 2.32, and 1.52 is also apparent that was previously assigned to a small fraction of nonreactive low-spin heme iron (9) that remains unchanged during the course of the experiment. After addition of H2O2 to ferric MauG, the reaction was quenched at various times by rapid freezing. Both hemes become EPR-silent in the first sample (2-s reaction time). No signal characteristic of Compound I, the spin-coupled Fe(IV)=O (S = 1) and porphyrin cation radical (S = 1/2) (16, 20, 21) was observed. A weak radical-like EPR signal was observed at g = 2.003 that exhibits a peak-to-peak width of 1.3 mT. At 10 K, microwave power at half-saturation (P1/2) was estimated to be 9 μW. The combined EPR linewidth, g value, and P1/2 indicate that the g = 2.003 species is an organic free radical. This signal overlaps with the spectrum of the high-spin ferric heme at the g = 2 region, which hinders precise spin quantitation for the radical component. The best estimation derived from spin double integrations of the g = 2 region on the intermediate samples is that the radical-like signal represents ≈1% of the protein, which is far from compensation for the loss of the two ferric heme EPR signals. This strongly suggests that the majority of both the high- and low-spin heme iron was oxidized to the EPR-silent Fe(IV) state. With time the high- and low-spin Fe(III) signals returned concomitant with the disappearance of the weak radical signal. Thus, the observed g = 2 signal is not a nonspecific side product of peroxidation but an uncoupled MauG-based cation radical that is likely in equilibrium with the two hemes (Fig. 2).
Fig. 1.
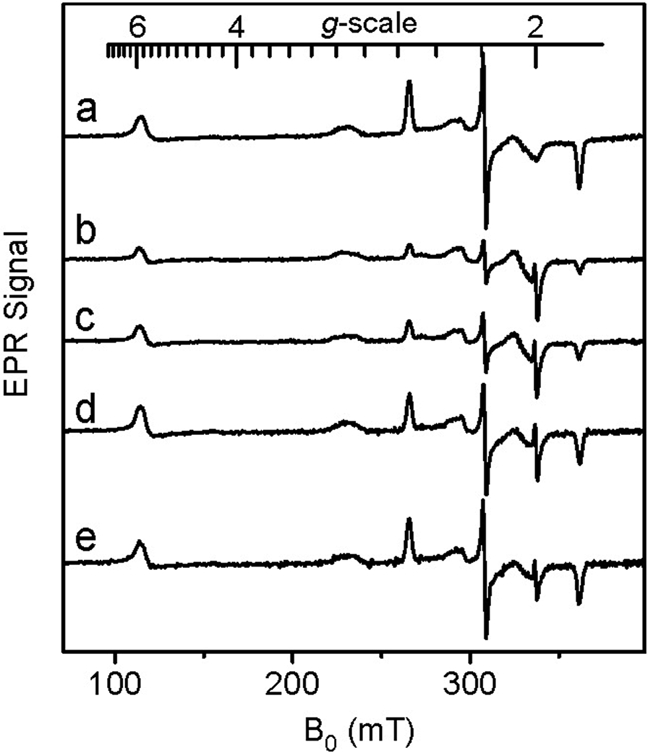
EPR analysis of the formation and decay of the intermediate formed by reaction of di-ferric MauG with a stoichiometric amount of H2O2. After mixing EPR spectra were recorded at time intervals of 0 (a), 0.03 (b), 2 (c), 8 (d), and 20 (e) minutes. Each sample contained 200 μM MauG. EPR parameters were temperature 10 K, microwave power 1 mW, modulation amplitude 5 G, time constant 40.96 ms, and sweep time 83.89 s. Each spectrum is the average of five scans.
Fig. 2.

Proposed mechanism for the formation of the bis-Fe(IV) intermediate formed by reaction of di-ferric MauG with a stoichiometric amount of H2O2.
Identification of Two Distinct Fe(IV) Heme Species in the MauG Reaction Intermediate by Mössbauer Spectroscopy.
The H2O2-generated MauG species was further characterized by Mössbauer spectroscopy. Because MauG possesses covalently bound c-type hemes, it was not possible to reconstitute purified protein with 57Fe, but necessary to isolate MauG from cells grown on 56Fe-depleted minimal medium supplemented with 57FeCl2. Fig. 3 shows 4.2 K Mössbauer spectra of ferric 57Fe-labeled MauG before (a) and 45 s after the addition of H2O2 (b). The dashed and solid lines in a are spin Hamiltonian simulations for the high- and low-spin hemes with typical parameters, respectively (22). The high- and low-spin hemes in ferric MauG are present in an ≈1:3 ratio rather than 1:1, which is likely caused by a freezing artifact often seen in which the high-spin heme converts to low-spin on freezing (9, 23). It should be noted that heme quantitation, mass spectrometry, and redox titrations of MauG indicated full presence of both hemes (9, 12). The spectrum of the H2O2-treated sample (hashed marks in Fig. 3b) contains several sharp lines that can be attributed to the intermediate, in addition to the broad, magnetically split features associated with the ferric hemes (solid line in Fig. 3b). There is also a small fraction of nonreactive ferric species present in both the resting and intermediate states of the enzyme, as was seen in the EPR experiment (Fig. 1). Removal of these ferric heme signals from Fig. 3b yields the spectral features of the EPR-silent MauG intermediate (Fig. 3c), which can be analyzed with two quadrupole doublets using the following parameters: isomer shift (δ1) of 0.06 mm/s and quadrupole splitting parameter (ΔEQ1) of 1.70 mm/s (15% of total Fe) and δ2 = 0.17 mm/s and ΔEQ2 = 2.54 mm/s (35% of total Fe). The isomer shift values are typical of Fe(IV) species (22). The quadrupole splitting parameter of species 1 is in the range typically observed for ferryl and protonated ferryl species (24). The quadrupole splitting parameter of species 2 is unusually large (ΔEQ2 = 2.54 mm/s) and likely describes the Fe(IV) state of the proposed six-coordinate heme with two axial amino acid ligands. Such a spin-uncoupled bis-Fe(IV) species has not been described. It is an Fe(V) equivalent. Furthermore, a six-coordinate Fe(IV) heme species with two axial amino acid ligands has also not been described in a protein, but analogous bis-ligated high-valent inorganic porphyrin model compounds have been generated, and in all cases a small isomer shift (δ) and large quadruple splitting (ΔEQ) parameters are observed from those model compounds (25, 26). An antiferromagnetic coupled diamagnetic Fe(III) radical has been observed in some model complexes by NMR spectroscopy (27). However, for MauG the 2-nm shift of the Soret band and the Mössbauer parameters, which are typical of Fe(IV) argue against a Fe(III)-radical resonance structure for the Fe ions of the intermediate. Our data indicate that the two Fe(IV) species in the MauG intermediate account for ≈50% of total iron contained in the sample, suggesting that some Fe(IV) heme has decayed to ferric MauG during the 45-s mixing/freezing period. Based on the fact that both the initial high- and low-spin hemes disappear upon peroxide oxidation (Fig. 1), these two distinct Fe(IV) species must arise from two distinct heme sites. The ratio between the two Fe(IV) species is 3:7, suggesting that species 1 is more reactive and thus less stable than species 2.
Fig. 3.

Mössbauer spectra of MauG recorded at 4.2 K in a 53-mT magnetic field. (a) The spectrum of ferric MauG (hashed marks) is overlaid with spin Hamiltonian simulations of a high-spin Fe(III) heme (25% of total Fe, dashed line) and of a low-spin Fe(III) heme (75% of total Fe, solid line) with the following parameters: S = 5/2, g5/2 = 2, D5/2 = 10 cm−1 (E/D)5/2 = 0, δ = 0.50 mm/s, ΔEQ = 2.0 mm/s, η = 0, A/gNβN = (−18.0, −18.0, −18.0) T and S = 1/2, g1/2 = (1.87, 2.19, 2.54), δ = 0.25 mm/s, ΔEQ = 1.97 mm/s, η = −3, A/gNβN = (−36.2, +9.3, +30.0) T. (b) Spectrum of di-ferric MauG, which was reacted with H2O2 for 45 s (hashed marks). The solid line indicates the contribution of ferric MauG (≈34% of total Fe). Removal of these features yields the reference spectrum of the intermediates (c, in which the x axis scale has been expanded), which can be simulated with two quadrupole doublets: δ1 = 0.06 mm/s and ΔEQ1 = 1.70 mm/s (15%, dashed line) and δ2 = 0.17 mm/s and ΔEQ2 = 2.54 mm/s (35%, solid line).
Reaction of the bis-Fe(IV) MauG Intermediate with Its Natural Substrate.
To determine whether the novel bis-Fe(IV) MauG intermediate was catalytically competent, it was mixed with its natural substrate, i.e., the isolated MADH biosynthetic precursor protein (Fig. 4). The MauG intermediate was generated by the addition of one equivalent of H2O2 (a) and then immediately mixed with a stoichiometric amount of the precursor protein. Whereas in the absence of substrate the EPR-silent heme species is relatively long-lived (see Fig. 1), on addition of substrate the EPR signals of the original high- and low-spin hemes returned within the rapid mixing/freezing time (b). Furthermore, an intense new radical-like EPR signal appeared with a g of 2.006 determined from the baseline crossing of a 10 K spectrum. Addition of 2 mM hydroxyurea, a known radical scavenger (28), caused an immediate decrease of the g = 2.006 EPR signal, but it had little effect on the ferric heme signals (c). This radical intermediate is not present if hydroxyurea is preincubated with MauG. This finding demonstrates that the g = 2.006 EPR component (Fig. 4 Inset) is a free radical intermediate generated during the reaction. Spin quantitation of the g = 2.006 signal by double integration and comparison with a g = 2 spin standard indicate that it corresponds to a nearly stoichiometric concentration of the added precursor protein substrate. The new EPR signal exhibits a 1.9-mT peak-to-peak splitting and partially resolved hyperfine structures typical of a protein-based free radical species with an aromatic ring (29, 30). This new radical was also relatively long-lived and remained on the MADH precursor after an anion-exchange chromatographic separation from MauG. It should be noted that this radical is observed when the reaction is initiated with stoichiometric H2O2 and MauG. Complete synthesis of TTQ from the precursor is a six-electron oxidation process. If excess oxidizing agents, either H2O2 or O2, are added, then the catalytically active mature MADH with fully synthesized TTQ is formed, which demonstrates that the bis-Fe(IV) MauG is a catalytically competent reaction intermediate in TTQ biosynthesis.
Fig. 4.

EPR analysis of the reaction of the high-valent MauG intermediate with its natural substrate. (a) High-valent MauG intermediate spectrum generated from reaction of di-ferric MauG with a stoichiometric amount of H2O2. (b) Spectrum of a further reacted with a stoichiometric amount of the biosynthetic precursor of MADH for 15 s before freezing in liquid nitrogen. (Inset) Spectrum of the g = 2 region. (c) Parallel sample of b further reacted with 2 mM hydroxyurea for 2 min. EPR parameters were temperature 10 K, microwave power 0.5 mW and modulation amplitude 5 G.
Discussion
High-valent Fe(IV)=O intermediates are frequently invoked in the catalytic cycles of Fe-dependent oxidizing enzymes (14, 31, 32). In heme-dependent enzymes, the two-electron oxidized intermediate (Compound I) consists of an Fe(IV) species (S = 1) coupled to an organic radical (S = 1/2) that is located on the porphyrin ring/axial ligand or a nearby amino acid residue (Compound ES). The unprecedented bis-Fe(IV) di-heme MauG intermediate is in essence an electronic equivalent of Compound I but with the second oxidizing equivalent stored at the second heme Fe rather than as an organic radical. A nonheme Fe(IV)2O2 species with diamond core structure and Fe–Fe distance of 2.46 Å (Intermediate Q) was trapped and characterized in methane monooxygenase (33, 34). However, previous studies of MauG yielded no evidence of spin-coupling between hemes (9, 12), and sequence homology to the structurally characterized di-heme cytochrome c peroxidases (35) suggests that the heme irons of MauG are well separated (>20 Å), with the second oxidizing equivalent transferred via an intervening amino acid residue.
MauG is an enzyme unique in several respects. It is the first Fe-dependent oxygenase to use c-type hemes to catalyze an oxygenation reaction (9–11). In contrast to cytochrome P450s, the second oxidizing equivalent is not stored as a radical but on a second Fe(IV) heme in the MauG intermediate (Fig. 2). Because the second heme is six-coordinate having two ligands provided by the protein, our results describe an example of a biological heme that is Fe(IV) without an exogenous ligand. This is also an example of a bis-Fe(IV) system in which the two irons are not in very close proximity and spin-coupled. The bis-Fe(IV) MauG intermediate is one of the most stable Fe(IV) species that has been characterized in biology. Its natural substrate is also unusual, a specific amino acid side chain within a 119-kDa tetrameric precursor protein (7). Most iron-containing oxygenases and oxidases react efficiently with oxygen only in the presence of their substrates because the generation of reactive intermediates in the absence of substrate may lead to deleterious autooxidation of the enzyme. Remarkably, the reaction of di-ferric MauG with hydrogen peroxide in the absence of its natural substrate yields a bis-Fe(IV) intermediate that is stable for minutes, yet this intermediate is chemically competent to oxidize its substrate.
Materials and Methods
The methods for homologous expression of MauG in P. denitrificans and its purification were as described in ref. 9. The concentration of MauG was determined by using its extinction coefficient of 208,000 M−1 cm−1 at 405 nm for the fully oxidized protein. The biosynthetic precursor of MADH with incompletely synthesized TTQ, which contains monohydroxylated βTrp-57 and no cross-link to βTrp-108 (7), was heterologously expressed in Rhodobacter sphaeroides (6) and purified as described in ref. 36. The concentration of the TTQ biosynthetic precursor of MADH was determined by using its extinction coefficient of 157,000 M−1 cm−1 at 280 nm.
Samples for spectroscopic analysis were prepared by rapid freezing. For EPR analysis, samples were directly injected from a rapid freeze-quench device (times <2 s) or transferred (times >2 s) to EPR tubes and frozen in liquid nitrogen to avoid packing factors. X-band EPR first-derivative spectra were recorded in perpendicular mode on an EMX spectrometer at 100-kHz modulation frequency with a 4119HS high-sensitivity resonator and Oxford ITC503S temperature controller. Spin concentration of the radical species was determined by double integration of the sample spectrum obtained under nonsaturating conditions and comparing the resulting intensity with that of a copper standard (1 mM CuSO4, 10 mM EDTA) obtained under identical conditions. Samples for Mössbauer spectroscopy were prepared in reaction vials and transferred into sample cups before immersion in liquid nitrogen.
Separation of reaction mixtures of the H2O2-oxidized MauG high-valent intermediate and the MADH biosynthetic precursor was achieved by using a Mono Q 4.6/100 PE column preequilibrated with 50 mM potassium phosphate buffer (pH 7.5) containing 25 mM NaCl and 5% glycerol (buffer A). Mixtures were applied to the column and eluted with a flow rate of 1 ml/min by using a linear NaCl gradient generated >20-column volumes from buffer A and 0–100% of buffer B [50 mM potassium phosphate buffer (pH 7.5) containing 1 M NaCl and 5% glycerol]. Chromatography was performed by using an ÅKTA-FPLC system (GE Healthcare).
Acknowledgments.
This work was supported by National Institutes of Health Grants GM41574 (to V.L.D.) and GM069618 (to A.L.), Oak Ridge Associated Universities Faculty Enhancement Award in Life Sciences (to A.L.), and a Young Investigator Award from the Arnold and Mabel Beckman Foundation and Camille Dreyfus Teacher–Scholar Award from the Camille and Henry Dreyfus Foundation (to C.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.McIntire WS, Wemmer DE, Chistoserdov A, Lidstrom ME. A new cofactor in a prokaryotic enzyme: Tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase. Science. 1991;252:817–824. doi: 10.1126/science.2028257. [DOI] [PubMed] [Google Scholar]
- 2.Davidson VL. Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry. 2007;46:5283–5292. doi: 10.1021/bi700468t. [DOI] [PubMed] [Google Scholar]
- 3.Davidson VL. Pyrroloquinoline quinone (PQQ) from methanol dehydrogenase and tryptophan tryptophylquinone (TTQ) from methylamine dehydrogenase. Adv Protein Chem. 2001;58:95–140. doi: 10.1016/s0065-3233(01)58003-1. [DOI] [PubMed] [Google Scholar]
- 4.Chen L, et al. Refined crystal structure of methylamine dehydrogenase from Paracoccus denitrificans at 1.75 Å resolution. J Mol Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 5.van der Palen CJ, et al. Mutational analysis of mau genes involved in methylamine metabolism in Paracoccus denitrificans. Eur J Biochem. 1995;230:860–871. doi: 10.1111/j.1432-1033.1995.tb20629.x. [DOI] [PubMed] [Google Scholar]
- 6.Graichen ME, et al. Heterologous expression of correctly assembled methylamine dehydrogenase in Rhodobacter sphaeroides. J Bacteriol. 1999;181:4216–4222. doi: 10.1128/jb.181.14.4216-4222.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearson AR, et al. Further insights into quinone cofactor biogenesis: Probing the role of mauG in methylamine dehydrogenase tryptophan tryptophylquinone formation. Biochemistry. 2004;43:5494–5502. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 8.Pearson AR, Marimanikkuppam S, Li X, Davidson VL, Wilmot CM. Isotope labeling studies reveal the order of oxygen incorporation into the tryptophan tryptophylquinone cofactor of methylamine dehydrogenase. J Am Chem Soc. 2006;128:12416–12417. doi: 10.1021/ja064466e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, et al. MauG, a novel diheme protein required for tryptophan tryptophylquinone biogenesis. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. Mechanistic possibilities in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:13276–13283. doi: 10.1021/bi061497d. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, et al. MauG-dependent in vitro biosynthesis of tryptophan tryptophylquinone in methylamine dehydrogenase. J Am Chem Soc. 2005;127:8258–8259. doi: 10.1021/ja051734k. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Feng M, Wang Y, Tachikawa H, Davidson VL. Evidence for redox cooperativity between c-type hemes of MauG which is likely coupled to oxygen activation during tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:821–828. doi: 10.1021/bi052000n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevens JM, Daltrop O, Allen JW, Ferguson SJ. C-type cytochrome formation: Chemical and biological enigmas. Acc Chem Res. 2004;37:999–1007. doi: 10.1021/ar030266l. [DOI] [PubMed] [Google Scholar]
- 14.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 15.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding intermediates in the O2 activation pathways of nonheme iron oxygenases. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulz CE, Rutter R, Sage JT, Debrunner PG, Hager LP. Mössbauer and electron paramagnetic resonance studies of horseradish peroxidase and its catalytic intermediates. Biochemistry. 1984;23:4743–4754. doi: 10.1021/bi00315a033. [DOI] [PubMed] [Google Scholar]
- 17.Maeda Y, Higashimura T, Morita Y. Mössbauer effect in peroxidase. Biochem Biophys Res Commun. 1967;29:362–367. doi: 10.1016/0006-291x(67)90463-9. [DOI] [PubMed] [Google Scholar]
- 18.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd Ed. New York: Plenum; 2005. [Google Scholar]
- 19.Erman JE, Hager LP, Sligar SG. Cytochrome P-450 and peroxidase chemistry. Adv Inorg Biochem. 1994;10:71–118. [PubMed] [Google Scholar]
- 20.Rutter R, Hager LP. The detection of two electron paramagnetic resonance radical signals associated with chloroperoxidase compound I. J Biol Chem. 1982;257:7958–7961. [PubMed] [Google Scholar]
- 21.Benecky MJ, Frew JE, Scowen N, Jones P, Hoffman BM. EPR and ENDOR detection of compound I from Micrococcus lysodeikticus catalase. Biochemistry. 1993;32:11929–11933. doi: 10.1021/bi00095a024. [DOI] [PubMed] [Google Scholar]
- 22.Debrunner PG. Mössbauer spectroscopy of iron porphyrins. Phys Bioinorg Chem Ser. 1989;4:137–234. [Google Scholar]
- 23.Prazeres S, et al. Mössbauer characterization of Paracoccus denitrificans cytochrome c peroxidase: Further evidence for redox and calcium binding-induced heme–heme interaction. J Biol Chem. 1995;270:24264–24269. doi: 10.1074/jbc.270.41.24264. [DOI] [PubMed] [Google Scholar]
- 24.Stone KL, Hoffart LM, Behan RK, Krebs C, Green MT. Evidence for two ferryl species in chloroperoxidase compound II. J Am Chem Soc. 2006;128:6147–6153. doi: 10.1021/ja057876w. [DOI] [PubMed] [Google Scholar]
- 25.Groves JT, et al. Preparation and characterization of a dialkoxyiron(IV) porphyrin. J Am Chem Soc. 1985;107:354–360. [Google Scholar]
- 26.Bill E, et al. Mössbauer investigations of the hexachlorantimonate salt of the phenyliron 2,3,7,8,l2,13,17,18-octaethyl-5,10,15,20-tetraphenylporphyrinate, [Fe(oetpp)Ph]SbCl6 and x-ray structure of the phenyliron(III) precursor Fe(III)(oetpp)Ph. Inorg Chim Acta. 2002;339:420–426. [Google Scholar]
- 27.Ikezaki A, Tukada H, Nakamura M. Control of electronic structure of a six-coordinate iron(III) porphyrin radical by means of axial ligands. Chem Commun. 2008;19:2257–2259. doi: 10.1039/b800674a. [DOI] [PubMed] [Google Scholar]
- 28.Karlsson M, Sahlin M, Sjöberg BM. Escherichia coli ribonucleotide reductaseL Radical susceptibility to hydroxyurea depends on the regulatory state of the enzyme. J Biol Chem. 1992;267:12622–12626. [PubMed] [Google Scholar]
- 29.Gräslund A, Sahlin M. Electron paramagnetic resonance and nuclear magnetic resonance studies of class I ribonucleotide reductase. Annu Rev Biophys Biomol Struct. 1996;25:259–286. doi: 10.1146/annurev.bb.25.060196.001355. [DOI] [PubMed] [Google Scholar]
- 30.Stubbe J, van der Donk WA. Protein radicals in enzyme catalysis. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 31.Costas M, Mehn MP, Jensen MP, Que L. Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 32.Krebs C, Galonić Fujimori G, Walsh CT, Bollinger JM., Jr Nonheme Fe(IV)-oxo intermediates. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SK, Fox BG, Froland WA, Lipscomb JD, Münck E. A transient intermediate of the methane monooxygenase catalytic cycle containing an FeIVFeIV cluster. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]
- 34.Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 35.Pettigrew GW, Echalier A, Pauleta SR. Structure and mechanism in the bacterial dihaem cytochrome c peroxidases. J Inorg Biochem. 2006;100:551–567. doi: 10.1016/j.jinorgbio.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Z, Sun D, Davidson VL. Conversion of methylamine dehydrogenase to a long-chain amine dehydrogenase by mutagenesis of a single residue. Biochemistry. 2000;39:11184–11186. doi: 10.1021/bi001568n. [DOI] [PubMed] [Google Scholar]