

Abstract
Although studies have shown 17β-estradiol (E2) administration following trauma-hemorrhage (T-H) attenuates alterations in T cell cytokine production, it remains unknown whether such effects of E2 are mediated via genomic or non-genomic pathways. In this study, we determined the non-genomic effects of E2 on splenic T cell cytokine production and the role of MAPK following T-H. Male Sprague-Dawley rats underwent T-H (mean BP 40 mmHg for 90 min, then resuscitation). E2, E2 conjugated with BSA (E2-BSA, 1 mg/kg E2) with or without an estrogen receptor antagonist (ICI 182 780), or vehicle was administrated during resuscitation. Two hrs thereafter, T cell production of IL-2 and IFN-γ and activation of MAPK (p38, ERK-1/2 and JNK) were determined. The effect of selective MAPK inhibitors on cytokine production was also examined in vitro. IL-2 and IFN-γ production capacity and MAPK activation decreased in T cells following T-H. However, E2 administration normalized these parameters. Although E2-BSA administration also attenuated suppression in cytokine production, the values were lower compared to sham. In contrast, E2-BSA prevented T-H-induced suppression in MAPK activation to the same extent as E2. Co-administration of ICI 182 780 abolished E2-BSA effects. These findings suggest E2 effects on T cell cytokine production following T-H are mediated at least in part via non-genomic pathway and these non-genomic effects are likely mediated via MAPK pathways.
Keywords: shock, 17β-estradiol, E2-BSA, IL-2, IFN-γ
INTRODUCTION
Previous studies have shown that splenocyte proliferation and cytokine production are decreased in male animals following trauma-hemorrhage [1–3]. However, such suppression in splenocyte function was not observed in proestrus female animals under those conditions [1]. Additional studies have shown that male sex steroids produce deleterious effects on splenocyte function [4], but female sex steroids maintain the splenocyte functions following trauma-hemorrhage [1]. Studies have also shown that administration of a single dose of 17β-estradiol (E2) following trauma-hemorrhage normalizes splenocyte function [5].
Two major pathways, generally termed genomic and non-genomic, are known to mediate E2 effects on cells. E2 has traditionally been described to mediate its effects via intracellular receptors located in the cytoplasm or on the nuclear membrane and thus studies have investigated the effect of E2 on transcription factors in the regulation of target genes [6;7]. However, recent findings indicate that E2 also acts on the plasma membrane to initiate signaling pathways in the cytoplasm and regulate cellular functions, which is called the non-genomic pathway [8–10]. E2 conjugated to BSA (E2-BSA) has been shown to be a plasma membrane impermeable compound and thus has been used to study the role of surface E2 receptors in producing the non-genomic effects of E2 on cellular functions [11–13].
Mitogen-activated protein kinases (MAPKs) are signaling molecules that play an important role in the regulation of immune responses including T cell activation and cytokine production [14–20]. There are three major MAPK-dependent pathways: p38, extracellular-regulated protein kinase (ERK) 1/2, and c-Jun NH2-terminal kinase (JNK). All three MAPK families are activated by dual phosphorylation on adjacent threonine and tyrosine residues. However, it is not known whether these kinases are involved in mediating the salutary effects of E2 on splenic T cells following trauma-hemorrhage. The aim of our study therefore was to determine whether the salutary effects of E2 on cytokine production by splenic T cells are mediated via genomic or non-genomic pathways, and whether MAPKs play a role in mediating such effects of E2.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were used in this study. All experiments were performed in adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Trauma-hemorrhage procedure
A nonheparinized rat model of trauma-hemorrhage, as described previously, was used in this study [21]. Briefly, male Sprague-Dawley rats (275–325 g) were fasted overnight before the experiment but allowed water ad libitum. The rats were anesthetized by isoflurane (Attane, Minrad, Bethlehem, PA) inhalation before the induction of soft tissue trauma (i.e., 5-cm midline laparotomy). The abdominal incision was then closed in two layers, and polyethylene catheters (PE-50, Becton-Dickinson, Sparks, MD) were placed in both femoral arteries and the right femoral vein. The rats were then placed into a Plexiglas box (21 × 9 × 5 cm) in a prone position and allowed to awaken, following which they were bled rapidly within 10 min to a mean arterial pressure (MAP) of 35–40 mmHg. After the time at which the animals could no longer maintain a MAP of 35–40 mmHg without infusion of some fluid, MAP was maintained at 40 mmHg until 40% of the shed blood volume was returned in the form of Ringer’s lactate. Then, the animals were resuscitated with 4 × the shed blood volume with Ringer’s lactate over 60 min. Thirty minutes before the end of the resuscitation period, the rats were treated with 17β-estradiol (E2; 1 mg/kg, intravenously), E2 conjugated to BSA (E2-BSA, 1 mg/kg E2) with or without estrogen receptor (ER) antagonist ICI 182 780 (3 mg/kg, intraperitoneally at the beginning of resuscitation), or an equal volume of the vehicle (BSA) subcutaneously. E2-BSA was filtered before injection to remove free E2 [22]. Following resuscitation, the catheters were removed, the vessels ligated, and skin incisions closed with sutures. Sham-operated animals underwent laparotomy and the same groin dissection, which included the ligation of the femoral artery and vein, but neither hemorrhage nor resuscitation was carried out. At 2 hrs after trauma-hemorrhage or sham operation, the rats were anesthetized with isoflurane and exsanguinated to collect samples.
Isolation of splenic T cells
Spleens were removed aseptically and placed into 50-mL conical tubes with cold PBS [23]. The spleens were then gently ground between frosted microscope slides to produce a single-cell suspension, and centrifuged at 400 g at 4°C for 15 min. The erythrocytes were lysed with Buffer EL (Qiagen, Valencia, CA). The remaining cells were then washed and loaded into a nylon wool column. After 1 hr of incubation (37°C at 5% CO2), T cells were eluted from the column and suspended (1 × 106 cells/mL) in RPMI 1640 (Invitrogen, Grant Island, NY) containing 10% fetal bovine serum and antibiotics. T cells thus obtained were found to be >95% positive for anti-CD3 [24].
Measurement of cytokine production
The isolated T cells were cultured in 24-well plates precoated with anti-CD3 (2 µg/mL) at 37°C and 5% CO2. In additional groups of animals subjected to trauma-hemorrhage or sham operation, isolated T cells were treated with the selective inhibitors of MAPK pathway, SB203580 (5 µM) for p38, PD98059 (20µM) for ERK1/2, and SP600125 (20 µM) for JNK (Calbiochem, La Jolla, CA) 30 min before anti-CD3 stimulation. Following an incubation period of 24 hrs, the supernatants were harvested and analyzed for the concentration of IL-2 and IFN-γ using DuoSet ELISA system (R&D, Minneapolis, MN) according to the manufacturer's instructions.
Measurement of p38, ERK1/2, and JNK protein and phosphorylation levels
As described previously [23], T cells were incubated with or without 1 µg/mL anti-CD3 for 5 min and lysed in a lysis buffer. For the analysis of p38, ERK1/2, and JNK protein and phosphorylation, lysates were analyzed using SDS-PAGE and transferred to Immobilon P membranes (polyvinylidene difluoride; Millipore Bedford, MA) by using a Semi Dry Trans-Blot system (Bio-Rad, Richmond, CA). The membranes were saturated with blocking buffer (10 mM Tris, 150 mM NaCl, and 0.05% Tween 20 supplemented with 5% dry milk) for 1 hr at room temperature and incubated with the antibodies to p38 protein, phospho-p38, ERK1/2 protein, phospho-ERK1/2, JNK protein and phospho-JNK (Cell Signaling, Beverly, MA) at 4°C overnight. The membranes were then washed five times with TBST (Tris-buffered saline supplemented with 0.05% Tween 20) followed by incubation with a secondary antibody conjugated with horseradish peroxidase for 1 hr at room temperature. The membranes were washed five times with TBST and probed using enhanced chemiluminescence dye, and phosphoproteins were autoradiographed.
Statistical analysis
Data are presented as mean ± SEM. Statistical differences between groups were determined by one-way ANOVA followed by Student-Newman-Keuls Method or Dunnett's Method. The differences were considered significant if p<0.05
RESULTS
Cytokine production
IL-2 and IFN-γ production by splenic T cells decreased following trauma-hemorrhage (Fig. 1). However, E2 administration following trauma-hemorrhage normalized the production of these cytokines by T cells. Although the suppression of IL-2 and IFN-γ production by T cells was also attenuated by E2-BSA treatment following trauma-hemorrhage, the T cell capacity to produce these cytokines remained significantly lower than shams. Co-administration of ICI 182 780 abolished the effects of E2-BSA on T cell cytokine production capacity.
Figure 1.
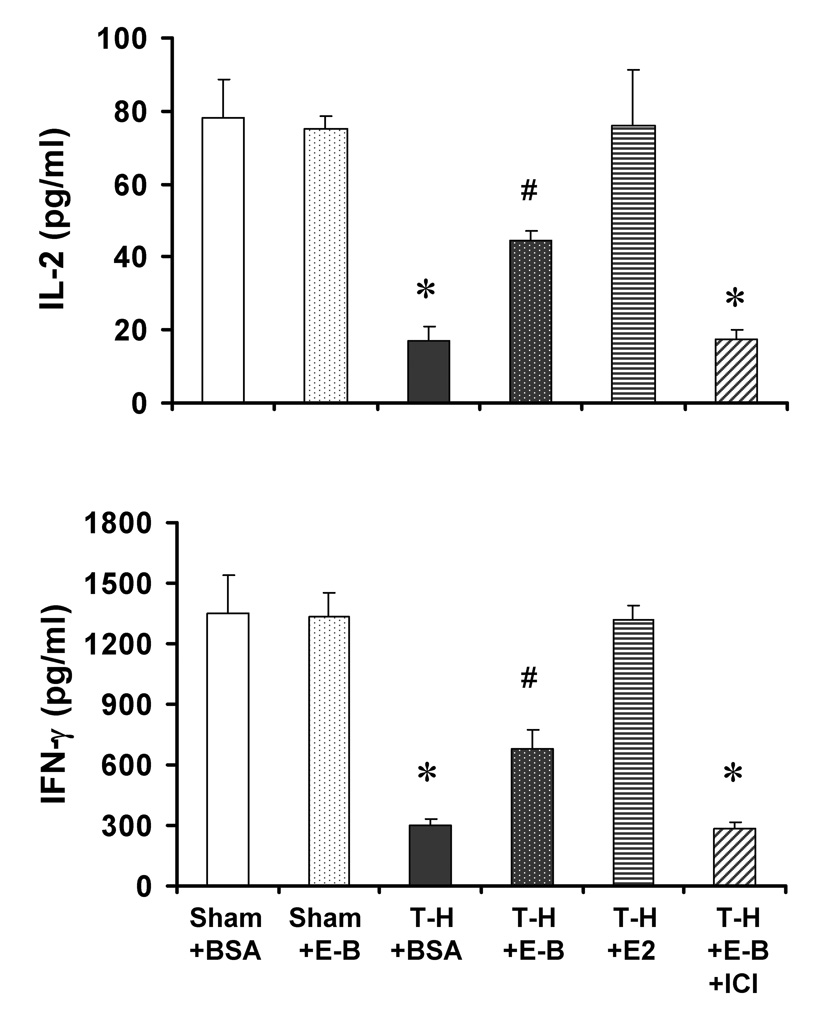
IL-2 and IFN-γ production by splenic T cells from sham animals treated with BSA (Sham+BSA), shams treated with E2-BSA (Sham+E-B), trauma-hemorrhage (T-H) treated BSA (T-H+BSA), TH treated with E2-BSA (T-H+E-B), T-H treated with E2 (T-H+E2), and T-H treated with E2-BSA and ICI 182 780 (T-H+E2+ICI). T cells were isolated 2 hrs after T-H and resuscitation and cultured in 24-well plates precoated with anti-CD3 (2 µg/mL) for 24 hrs. Cytokine levels in culture supernatants were determined by ELISA. Data are mean ± SEM of 6 to 7 animals in each group. *p<0.05 compared to respective sham; #p<0.05 compared to Sham+E-B and T-H+BSA.
p38, ERK1/2, and JNK phosphorylation
To determine MAPK activation, phosphorylation of p38, ERK1/2, and JNK in splenic T cells (Fig. 2, Fig 3, and Fig 4, respectively) with or without anti-CD3 stimulation was evaluated. The phosphorylation of p38, ERK1/2, and JNK in unstimulated T cells was found to be at basal levels which was not significantly different in cells from sham and trauma-hemorrhage animals (Fig 2–4, A). Stimulation of these cells with anti-CD3 resulted in an increase in phosphorylation of all three MAPKs in T cells from animals regardless of their injury. (Fig 2–4, A); however, anti-CD3-induced phosphorylation of p38, ERK1/2, and JNK was significantly lower in T cells from rats subjected to trauma-hemorrhage compared to those from sham-operated rats (Fig 2–4, B). Administration of E2-BSA or E2 following trauma-hemorrhage prevented the decrease in p38, ERK1/2, and JNK phosphorylation in T cells. There was no significant difference in p38, ERK1/2, and JNK protein expression in T cells from rats subjected to trauma-hemorrhage compared to rats subjected to sham operation (Fig 2–4, C). Co-administration of ICI 182 780 abolished the effects of E2-BSA on MAPK activation in T cells.
Figure 2.

p38 phosphorylation and protein expression in splenic T cells with anti-CD3 stimulation (anti-CD3 +) or without stimulation (anti-CD3 −) from sham animals treated with BSA (Sham+BSA), shams treated with E2-BSA (Sham+E-B), trauma-hemorrhage (T-H) treated BSA (T-H+BSA), TH treated with E2-BSA (T-H+E-B), T-H treated with E2 (T-H+E2), and T-H treated with E2-BSA and ICI 182 780 (T-H+E2+ICI). T cells were isolated 2 hrs after T-H and resuscitation, stimulated with 1 µg/mL anti-CD3 for 5 min, and lysed. Lysates were then analyzed for p38 phosphorylation (p-p38) and protein expression (A). Blots were reprobed for β-actin for equal protein loading in various lanes. p38 blots obtained from 5 animals were analyzed using densitometry, and densitometric values for phosphorylation and total protein were normalized to β-actin and are shown as mean ± SEM in Panels B and C respectively. *p<0.05 compared to respective sham.
Figure 3.

ERK phosphorylation and protein expression in splenic T cells with anti-CD3 stimulation (anti-CD3 +) or without stimulation (anti-CD3 −) from sham animals treated with BSA (Sham+BSA), shams treated with E2-BSA (Sham+E-B), trauma-hemorrhage (T-H) treated BSA (T-H+BSA), TH treated with E2-BSA (T-H+E-B), T-H treated with E2 (T-H+E2), and T-H treated with E2-BSA and ICI 182 780 (T-H+E2+ICI). T cells were isolated 2 hrs after T-H and resuscitation, stimulated with 1 µg/mL anti-CD3 for 5 min, and lysed. Lysates were then analyzed for ERK phosphorylation (p-ERK) and protein expression (A). Blots were reprobed for β-actin for equal protein loading in various lanes. ERK blots obtained from 5 animals were analyzed using densitometry, and densitometric values for phosphorylation and total protein were normalized to β-actin and are shown as mean ± SEM in Panels B and C respectively. *p<0.05 compared to respective sham.
Figure 4.

JNK phosphorylation and protein expression in splenic T cells with anti-CD3 stimulation (anti-CD3 +) or without stimulation (anti-CD3 −) from sham animals treated with BSA (Sham+BSA), shams treated with E2-BSA (Sham+E-B), trauma-hemorrhage (T-H) treated BSA (T-H+BSA), TH treated with E2-BSA (T-H+E-B), T-H treated with E2 (T-H+E2), and T-H treated with E2-BSA and ICI 182 780 (T-H+E2+ICI). T cells were isolated 2 hrs after T-H and resuscitation, stimulated with 1 µg/mL anti-CD3 for 5 min, and lysed. Lysates were then analyzed for JNK phosphorylation (p-JNK) and protein expression (A). Blots were reprobed for β-actin for equal protein loading in various lanes. JNK blots obtained from 5 animals were analyzed using densitometry, and densitometric values for phosphorylation and total protein were normalized to β-actin and are shown as mean ± SEM in Panels B and C respectively. *p<0.05 compared to respective sham.
DISCUSSION
Our findings demonstrate that splenic T cell IL-2 and IFN-γ productive capacity decreased following trauma-hemorrhage. However, administration of E2 following trauma-hemorrhage normalized the cytokine production and the values were similar to shams. Although E2-BSA administration also significantly attenuated the suppression in T cell cytokine production, the cytokine production capacity of these cells was lower than shams. Since E2-BSA did not mimic the total effects of E2, it appears that non-genomic effects partially contribute to the total effects of E2 on T cell cytokine production following trauma-hemorrhage.
Our results also show that MAPK activation decreased in T cells following trauma-hemorrhage. In contrast to cytokine production, administration of E2-BSA prevented the trauma-hemorrhage-induced suppression in MAPK activation in T cells to the same extent as E2 treatment. These results thus indicate that the effects of E2 on MAPK activation are mediated mainly via the non-genomic pathway. In addition, since co-administration of estrogen receptor antagonist ICI 182 780 abolished the effects of E2-BSA on T cells, such effects are likely mediated via the estrogen receptors.
T cells play a critical role in host defense against bacterial infection [25–27]. Previous studies from our laboratory have shown that the depressed T cell functions following trauma-hemorrhage were associated with an increased susceptibility to subsequent sepsis [1]. Therefore, it appears important to prevent the suppression of T cell functions following trauma-hemorrhage to maintain the immune system and avoid subsequent infection. Previous studies demonstrated that proestrus female animals, with high circulating levels of estrogen, did not show decreased cytokine production by T cells and they had normal immune functions compared to males following trauma-hemorrhage [1;28]. Furthermore, proestrus females are more resistant to subsequent sepsis than male mice and had a significantly lower mortality following trauma-hemorrhage and sepsis [1;29]. Our previous studies showed that administration of a single dose of E2 following trauma-hemorrhage improved macrophage and lymphocyte functions [29]. However, blockade of ER abolished the salutary effects of 17β-estradiol [30], suggesting that the salutary effects of 17β-estradiol are mediated via ER.
E2 has generally been considered to act through estrogen receptors located on the nuclear membrane [6]. However, recent studies revealed that some effects of E2 are mediated via plasma membrane [8;9]. These non-genomic effects modulate cell membrane-bound regulatory proteins or cytoplasmic signaling molecules. MAPK pathways are known to be involved in the non-genomic cascade of E2 in various types of cells [31–37]. For instance, ERK1/2 is activated in cardiomyocytes, colon cancer, breast cancer, and bone, and is inhibited in vascular smooth muscle cells and lung myofibroblasts by E2 in a non-genomic manner. Moreover, p38 and JNK also mediate the non-genomic pathway in cardiomyocytes, breast cancer and endothelial cells [8]. Our findings show that MAPKs are involved in the non-genomic pathway in T cells following trauma-hemorrhage. Several previous studies support a role for MAPK (i.e., p38, ERK1/2, and JNK) in T cell cytokine production [14–17]. Since the suppression in T cell cytokine production was accompanied by a decrease in MAPK activation, it is possible that E2 or E2-BSA restore T cell cytokine production by protecting this pathway. Furthermore, since the restoration of MAPK and not the T cell cytokine after E2-BSA treatment was complete as compared to cells from E2-treated animals, it is possible that MAPK are partially involved in mediating E2 actions which include both genomic and non-genomic pathways. Several pathways other than MAPK (e.g., PLC/PKC; PI3k/Akt, NO, Ca2+) are also implicated in mediating non-genomic action of E2 [38–40]. Our studies have also demonstrated that PI3K/Akt pathway plays major role in mediating the non-genomic effects of E2 on cardiac functions [41]. Similar findings were obtained by other investigators supporting the role of PI3K/Akt signaling in non-genomic effects of E2. Although we examined the role of MAPK in this study, it remains to be determined whether signaling molecules other than MAPK such as PI3K/Akt are also involved. However, it should be noted that although both genomic and non-genomic actions of estrogen are differently regulated, both genomic and non-genomic pathways work synergistically in the regulation of cell functions whether it is a cardiomyocyte or an immune cell.
There is evidence indicating the existence of cell membrane estrogen receptors [9]. Two kinds of membrane receptors have been proposed to exist: 1) membrane receptors with a molecular structure related to the classic estrogen receptors, ER-α or ER-β [12;42], and 2) non-classic estrogen receptors, such as G protein-coupled receptor 30 (GPR30) [43;44]. Our recent studies have shown that the salutary effects of E2 on T cell functions following trauma-hemorrhage are mediated predominantly via ER-α [23], and we have shown in this study that the effects of E2-BSA on T cells are abolished by co-administration of ER antagonist, ICI 182 780. It is therefore likely that E2-BSA acts via membrane ER-α on these cells.
In this study we used E2-BSA to limit estrogen’s actions to the cell membrane and to distinguish non-genomic effects from total effects of E2. Previous studies have shown that E2-BSA binds only to the plasma membrane and does not go to the inside of the cell [45;46]. Furthermore, such binding to the membrane is due specifically to E2 and not to BSA [45], and ERs on the plasma membrane contribute to such binding [46]. In functional aspect, E2-BSA does not activate estrogen receptor element-dependent transcription, indicating that this compound does not have traditional genomic effects of E2 [47]. Because of these findings, E2-BSA is generally used to investigate non-genomic effects of E2 [11–13]. Since we administer E2-BSA in vivo in the present study, there is potential risk that E2 is dissociate from E2-BSA and has some genomic effects. However, the rate of such dissociation is very low (~0.00063%/ml/hr) [48], and we collected samples just 2 hrs after trauma-hemorrhage and resuscitation. According to the previous study, the amount of free E2 in our study would be too low to show any genomic effects [11]. Therefore, we consider that the effects of E2-BSA in the present study are mediated specifically via non-genomic pathway of E2.
It can be argued that the present study utilized measurement at a single time point, i.e., at 2 hrs after treatment and thus it remains unclear whether the salutary effects of E2 or E2-BSA on macrophage signaling and cytokine production are sustained for periods of time longer than 2 hrs after treatment. Our previous studies, however, have shown that if the improvement in cell and organ functions by a pharmacological agent was observed early after treatment, those salutary effects are sustained for prolonged intervals, and it also decreased the mortality of animals from subsequent sepsis [1;4;29]. Thus, although a time point other than 2 hrs was not examined in this study, based on our previous studies it would appear that the salutary effects of E2 or E2-BSA on T cells would be evident even if one measured those effects at another time point following trauma-hemorrhage and resuscitation.
In summary, the present results showed that E2 administration following trauma-hemorrhage normalized the decreased cytokine production and MAPK activity, and that E2-BSA has partial effects of E2 on cytokine production and total effects of E2 on MAPK. In addition, cytokine production was suppressed by MAPK inhibiters. These results collectively suggest that the salutary effects of E2 on T cell cytokine production are partially mediated via non-genomic pathway and such non-genomic effects are likely mediated via normalization of MAPK activity.
ACKNOWLEDGEMENTS
This investigation was supported by NIH grant R37 GM39519. The authors would like to thank Bobbi Smith for her assistance with manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Angele MK, Schwacha MG, Ayala A, Chaudry IH. Effect of gender and sex hormones on immune responses following shock. Shock. 2000;14:81–90. doi: 10.1097/00024382-200014020-00001. [DOI] [PubMed] [Google Scholar]
- 2.Shukla A, Hashiguchi N, Chen Y, Coimbra R, Hoyt DB, Junger WG. Osmotic regulation of cell function and possible clinical applications. Shock. 2004;21:391–400. doi: 10.1097/00024382-200405000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Wichmann MW, Angele MK, Ayala A, Cioffi WG, Chaudry IH. Flutamide: a novel agent for restoring the depressed cell-mediated immunity following soft-tissue trauma and hemorrhagic shock. Shock. 1997;8:242–248. [PubMed] [Google Scholar]
- 4.Schneider CP, Nickel EA, Samy TS, Schwacha MG, Cioffi WG, Bland KI, Chaudry IH. The aromatase inhibitor, 4-hydroxyandrostenedione, restores immune responses following trauma-hemorrhage in males and decreases mortality from subsequent sepsis. Shock. 2000;14:347–353. doi: 10.1097/00024382-200014030-00019. [DOI] [PubMed] [Google Scholar]
- 5.Knoferl MW, Jarrar D, Angele MK, Ayala A, Schwacha MG, Bland KI, Chaudry IH. 17 beta-Estradiol normalizes immune responses in ovariectomized females after trauma-hemorrhage. Am J Physiol Cell Physiol. 2001;281:C1131–C1138. doi: 10.1152/ajpcell.2001.281.4.C1131. [DOI] [PubMed] [Google Scholar]
- 6.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meldrum DR. G-protein-coupled receptor 30 mediates estrogen's nongenomic effects after hemorrhagic shock and trauma. Am J Pathol. 2007;170:1148–1151. doi: 10.2353/ajpath.2007.070025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho KJ, Liao JK. Non-nuclear actions of estrogen. Arterioscler Thromb Vasc Biol. 2002;22:1952–1961. doi: 10.1161/01.atv.0000041200.85946.4a. [DOI] [PubMed] [Google Scholar]
- 9.Simoncini T, Genazzani AR. Non-genomic actions of sex steroid hormones. Eur J Endocrinol. 2003;148:281–292. doi: 10.1530/eje.0.1480281. [DOI] [PubMed] [Google Scholar]
- 10.Simoncini T, Mannella P, Fornari L, Caruso A, Varone G, Genazzani AR. Genomic and non-genomic effects of estrogens on endothelial cells. Steroids. 2004;69:537–542. doi: 10.1016/j.steroids.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Kow LM, Pfaff DW. The membrane actions of estrogens can potentiate their lordosis behavior-facilitating genomic actions. Proc Natl Acad Sci USA. 2004;101:12354–12357. doi: 10.1073/pnas.0404889101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ER-α and ER-β expressed in Chinese hamster ovary cells. Mol Endocrinol. 1999;13:307–319. doi: 10.1210/mend.13.2.0239. [DOI] [PubMed] [Google Scholar]
- 13.Russell KS, Haynes MP, Sinha D, Clerisme E, Bender JR. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc Natl Acad Sci USA. 2000;97:5930–5935. doi: 10.1073/pnas.97.11.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 15.Egerton M, Fitzpatrick DR, Kelso A. Activation of the extracellular signal-regulated kinase pathway is differentially required for TCR-stimulated production of six cytokines in primary T lymphocytes. Int Immunol. 1998;10:223–229. doi: 10.1093/intimm/10.2.223. [DOI] [PubMed] [Google Scholar]
- 16.Hardy K, Chaudhri G. Activation and signal transduction via mitogen-activated protein (MAP) kinases in T lymphocytes. Immunol Cell Biol. 1997;75:528–545. doi: 10.1038/icb.1997.84. [DOI] [PubMed] [Google Scholar]
- 17.Matthews JS, O'Neill LA. Distinct roles for p42/p44 and p38 mitogen-activated protein kinases in the induction of IL-2 by IL-1. Cytokine. 1999;11:643–655. doi: 10.1006/cyto.1998.0478. [DOI] [PubMed] [Google Scholar]
- 18.Noel JG, Osterburg A, Wang Q, Guo X, Byrum D, Schwemberger S, Goetzman H, Caldwell CC, Ogle CK. Thermal injury elevates the inflammatory monocyte subpopulation in multiple compartments. Shock. 2007 doi: 10.1097/shk.0b013e31805362ed. [Epub ahead of print; scheduled for December 2007 issue] [DOI] [PubMed] [Google Scholar]
- 19.Tsung A, McCoy SL, Klune JR, Geller DA, Billiar TR, Hefeneider SH. A novel inhibitory peptide of Toll-like receptor signaling limits lipopolysaccharide-induced production of inflammatory mediators and enhances survival in mice. Shock. 2007;27(4):364–369. doi: 10.1097/01.shk.0000239773.95280.2c. [DOI] [PubMed] [Google Scholar]
- 20.Meng X, Ao L, Song Y, Raeburn CD, Fullerton DA, Harken AH. Signaling for myocardial depression in hemorrhagic shock: roles of Toll-like receptor 4 and p55 TNF-alpha receptor. Am J Physiol Regul Integr Comp Physiol. 2005;288(3):R600–R606. doi: 10.1152/ajpregu.00182.2004. [DOI] [PubMed] [Google Scholar]
- 21.Yokoyama Y, Kuebler JF, Matsutani T, Schwacha MG, Bland KI, Chaudry IH. Mechanism of the salutary effects of 17β-estradiol following trauma-hemorrhage: direct downregulation of Kupffer cell proinflammatory cytokine production. Cytokine. 2003;21:91–97. doi: 10.1016/s1043-4666(03)00014-0. [DOI] [PubMed] [Google Scholar]
- 22.Stevis PE, Deecher DC, Suhandolnik L, Mallis LM, Frail DE. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology. 1999;140:5455–5458. doi: 10.1210/endo.140.11.7247. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki T, Shimizu T, Yu HP, Hsieh YC, Choudhry MA, Chaudry IH. Salutary effects of 17β-estradiol on T-cell signaling and cytokine production after traumahemorrhage are mediated primarily via estrogen receptor-alpha. Am J Physiol Cell Physiol. 2007;292:C2103–C2111. doi: 10.1152/ajpcell.00488.2006. [DOI] [PubMed] [Google Scholar]
- 24.Samy TS, Ayala A, Catania RA, Chaudry IH. Trauma-hemorrhage activates signal transduction pathways in mouse splenic T cells. Shock. 1998;9:443–450. doi: 10.1097/00024382-199806000-00009. [DOI] [PubMed] [Google Scholar]
- 25.Dugan AL, Thellin O, Buckley DJ, Buckley AR, Ogle CK, Horseman ND. Effects of prolactin deficiency on myelopoiesis and splenic T lymphocyte proliferation in thermally injured mice. Endocrinology. 2002;143:4147–4151. doi: 10.1210/en.2002-220515. [DOI] [PubMed] [Google Scholar]
- 26.Kerksiek KM, Pamer EG. T cell responses to bacterial infection. Curr Opin Immunol. 1999;11:400–405. doi: 10.1016/S0952-7915(99)80067-3. [DOI] [PubMed] [Google Scholar]
- 27.Lederer JA, Rodrick ML, Mannick JA. The effects of injury on the adaptive immune response. Shock. 1999;11:153–159. doi: 10.1097/00024382-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Kahlke V, Angele MK, Schwacha MG, Ayala A, Cioffi WG, Bland KI, Chaudry IH. Reversal of sexual dimorphism in splenic T-lymphocyte responses following trauma-hemorrhage with aging. Am J Physiol. 2000;278:C509–C519. doi: 10.1152/ajpcell.2000.278.3.C509. [DOI] [PubMed] [Google Scholar]
- 29.Diodato MD, Knoferl MW, Schwacha MG, Bland KI, Chaudry IH. Gender differences in the inflammatory response and survival following haemorrhage and subsequent sepsis. Cytokine. 2001;14:162–169. doi: 10.1006/cyto.2001.0861. [DOI] [PubMed] [Google Scholar]
- 30.Knoferl MW, Angele MK, Schwacha MG, Anantha Samy TS, Bland KI, Chaudry IH. Immunoprotection in proestrus females following trauma-hemorrhage: the pivotal role of estrogen receptors. Cell Immunol. 2003;222:27–34. doi: 10.1016/s0008-8749(03)00081-9. [DOI] [PubMed] [Google Scholar]
- 31.Castoria G, Barone MV, Di Domenico M, Bilancio A, Ametrano D, Migliaccio A, Auricchio F. Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. EMBO J. 1999;18:2500–2510. doi: 10.1093/emboj/18.9.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Domenico M, Castoria G, Bilancio A, Migliaccio A, Auricchio F. Estradiol activation of human colon carcinoma-derived Caco-2 cell growth. Cancer Res. 1996;56:4516–4521. [PubMed] [Google Scholar]
- 33.Endoh H, Sasaki H, Maruyama K, Takeyama K, Waga I, Shimizu T, Kato S, Kawashima H. Rapid activation of MAP kinase by estrogen in the bone cell line. Biochem Biophys Res Commun. 1997;235:99–102. doi: 10.1006/bbrc.1997.6746. [DOI] [PubMed] [Google Scholar]
- 34.Flores-Delgado G, Bringas P, Buckley S, Anderson KD, Warburton D. Nongenomic estrogen action in human lung myofibroblasts. Biochem Biophys Res Commun. 2001;283:661–667. doi: 10.1006/bbrc.2001.4827. [DOI] [PubMed] [Google Scholar]
- 35.Hwang KC, Lee KH, Jang Y. Inhibition of MEK1,2/ERK mitogenic pathway by estrogen with antiproliferative properties in rat aortic smooth muscle cells. J. Steroid Biochem Mol Biol. 2002;80:85–90. doi: 10.1016/s0960-0760(01)00169-8. [DOI] [PubMed] [Google Scholar]
- 36.Jessop HL, Sjoberg M, Cheng MZ, Zaman G, Wheeler-Jones CP, Lanyon LE. Mechanical strain and estrogen activate estrogen receptor alpha in bone cells. J Bone Miner Res. 2001;16:1045–1055. doi: 10.1359/jbmr.2001.16.6.1045. [DOI] [PubMed] [Google Scholar]
- 37.Nuedling S, Kahlert S, Loebbert K, Meyer R, Vetter H, Grohe C. Differential effects of 17β-estradiol on mitogen-activated protein kinase pathways in rat cardiomyocytes. FEBS Lett. 1999;454:271–276. doi: 10.1016/s0014-5793(99)00816-9. [DOI] [PubMed] [Google Scholar]
- 38.Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett. 2006;238:1–14. doi: 10.1016/j.canlet.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 39.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 40.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 41.Yu HP, Hsieh YC, Suzuki T, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. The PI3K/Akt pathway mediates the nongenomic cardioprotective effects of estrogen following trauma-hemorrhage. Ann Surg. 2007;245:971–977. doi: 10.1097/01.sla.0000254417.15591.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–406. doi: 10.1172/JCI5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 44.Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly ES, Jr, Nethrapalli IS, Tinnikov AA. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci. 2002;22:8391–8401. doi: 10.1523/JNEUROSCI.22-19-08391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo Z, Krucken J, Benten WP, Wunderlich F. Estradiol-induced nongenomic calcium signaling regulates genotropic signaling in macrophages. J Biol Chem. 2002;277:7044–7050. doi: 10.1074/jbc.M109808200. [DOI] [PubMed] [Google Scholar]
- 46.Razandi M, Pedram A, Levin ER. Plasma membrane estrogen receptors signal to anti-apoptosis in breast cancer. Mol Endocrinol. 2000;14:1434–1447. doi: 10.1210/mend.14.9.0526. [DOI] [PubMed] [Google Scholar]
- 47.Watters JJ, Chun TY, Kim YN, Bertics PJ, Gorski J. Estrogen modulation of prolactin gene expression requires an intact mitogen-activated protein kinase signal transduction pathway in cultured rat pituitary cells. Mol Endocrinol. 2000;14:1872–1881. doi: 10.1210/mend.14.11.0551. [DOI] [PubMed] [Google Scholar]
- 48.Binder M. Oestradiol-BSA conjugates for receptor histochemistry: problems of stability and interactions with cytosol. Histochem J. 1984;16:1003–1023. doi: 10.1007/BF01003854. [DOI] [PubMed] [Google Scholar]