

Abstract
Some selective transformations of resorcinol-derived cyclohexadienone are reported. Efforts led to a structure reported to display anticancer properties. On the basis of the results, the structures for natural products reported to contain a 4,6-dihydroxy-4-alkyl-cyclohexenone nucleus are corrected.
Active ingredients from extracts of the tree, Tapirira guianensis Aublet, were assigned as structures 1 and 2 (Figure 1).1 Because of their lipophilic tails and polar head-groups, these oily compounds undoubtedly proved difficult to crystallize. Therefore, the structures were inferred from 1H NMR, 13C NMR, and MS data as well as from Cotton effects in the CD spectrum to account for the absolute stereochemistry in 1.1 In the NCI-60 panel of human cancer cell lines, these compounds were reported to display selective cytotoxicity toward prostate cancer (0.2 and 0.3 μg/mL for LCAP, respectively).
Figure 1.
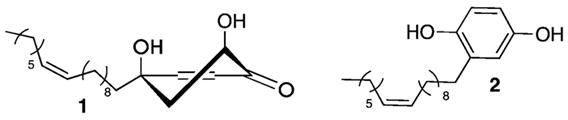
Original assignments for agents found in T. guianensis Aublet.
The geometry and location of the olefinic side chains of compounds 1 and 2 were rigorously determined using a technique developed by Francis and Veland:2 transformation into the corresponding α,β-bis(methylthio) derivative and analysis of the resulting 1H NMR and MS spectra. Boyd used the method to secure the olefin position in the related ω-9 hydroquinone called lanneaquinol.3
The architecture proposed for the cyclohexenone 1 is surprisingly unique. A literature search revealed only one other natural product displaying its 4,6-dihydroxy-4-alkyl cyclohexenone core.4 The obvious simplicity and unusual proposed biosynthesis of natural 1 among more complex anticancer agents5 provided a compelling case for total synthesis.
Our route to the reported structures 1 and 2 begins with palmitoleic acid (3), an ω-7 fatty acid found in the fruit of the sea buckhorn, in macadamia nuts, and in the 2005 Aldrich catalog. Reduction of carboxylic acid 3 (0.5 M in Et2O) with lithium aluminum hydride (1 equiv) affords the desired alcohol. Subsequent treatment of this intermediate alcohol (0.23 M in benzene) with triphenylphosphinedibromide (1.2 equiv) provides the bromide 4 in 94% overall yield (Scheme 1). After distillation, the bromide 4 is converted into a 2 M THF solution of the Grignard reagent 5 by the addition of a slight excess of magnesium powder.
Scheme 1.
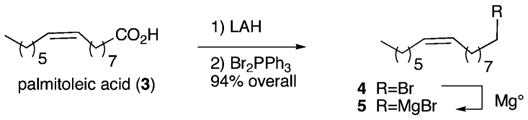
Preparation of the 16-Carbon Grignard Reagent
The aldehyde 66 (0.1 M in THF/H2O) undergoes reduction with sodium borohydride (3 equiv, 10 s) to afford the corresponding benzyl alcohol. Subsequent treatment (0.05 M in Et2O) with sodium hydride (1 equiv) and the Grignard reagent 5 provides the differentiated 1,4-hydroquinone nucleus, whereupon addition of aqueous 3 M HCl cleaves the remaining O-Boc residue and affords the hydroquinone 2 in 45% overall yield. All of the spectral data for synthetic 2 prove identical to those previously reported for natural 2. Moreover, because of a problematic demethylation in the previous synthesis of its relative lanneaquinol,3 our four-step route to this general class of hydroquinones is surprisingly efficient when compared to the alternative strategy of lithiation, alkylation, and demethylation of 1,4-dimethoxy-benzene.3
Next, we began the synthesis of the cyclohexenone 1, which is the supposed biosynthetic precursor of 2. Our strategy required that the aldehyde 77 (0.1 M in THF/H2O) undergo reduction with NaBH4 (2 equiv) to afford the corresponding benzyl alcohol, whereupon successive treatment (0.2 M in THF) with NaH (2 equiv) and a solution of 5 (2 equiv, 0.5 M in THF) affords the differentially protected resorcinol derivative 8 in 73% yield (Scheme 2).
Scheme 2.
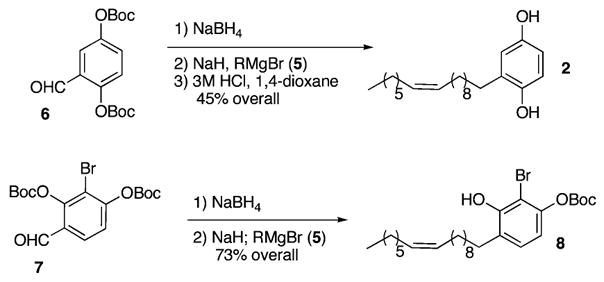
Elaboration of the o-Quinone Methide Intermediate
These two coupling processes follow the sequence shown in Figure 2.6 Reduction of the aldehyde and workup provide a benzyl alcohol. Addition of sodium hydride reinitiates the cascade by causing Boc migration. In the presence of the magnesium ion, β-elimination proceeds to an o-quinone methide, which is immediately intercepted by the Grignard reagent to form a C–C bond. Workup affords the differentiated phenol. The phenol 8 (1.7 M in DMF) is then coupled with the chiral tosylate (−)-9 (1.1 equiv) by the action of potassium carbonate (1.3 equiv). Cleavage of the remaining O-Boc residue with zinc bromide (5 equiv in CH3NO2) affords the phenol 10 in 86% overall yield displaying an S-configuration about the inverted chiral center.
Figure 2.
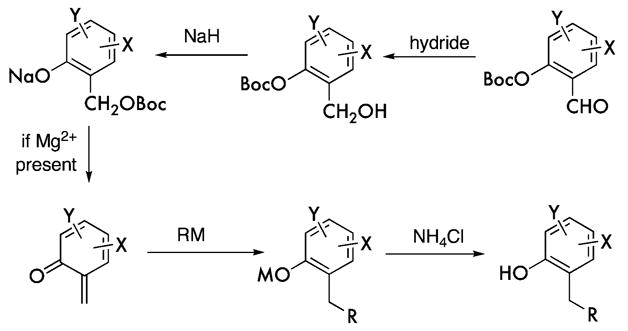
o-Quinone methide cascade leading from 6 and 7 to 2 and 8.
Our usual dearomatization conditions (0.05 M CH3NO2, 2 equiv of PhI(O2CCF3)2) afforded a 48% yield of the desired cyclohexadienone (−)-11 from 10.7 However, after considerable experimentation, we find that successive treatment of 10(0.1 M in CH2Cl2) with iodosyl benzene (2 equiv) and trimethylsilyltriflate (2 equiv) provides the cyclohexadienone 11 in 55% isolated yield (Scheme 3). We presume this transformation involves in situ formation of PhI(OTf)OTMS leading to the generation of a phenoxonium cation and cyclization with the carbonyl of the amide. The resulting iminium species then undergoes hydrolysis to afford the δ-lactone (−)-11. In this example, the other diastereomer is not evident in the 1H NMR spectrum of the crude product mixture. These modified conditions have significantly improved the versatility of this dearomatization process; other derivatives display gains in yields when compared to our previous conditions that employed PhI(O2CCF3)2 as the oxidant.7
Scheme 3.
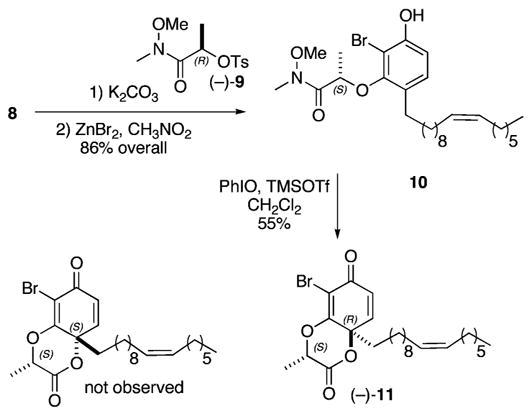
Dearomatization and Diastereoselective Synthesis of 11
Four transition states explain the two possible diastereomeric products. These include the Meax-boat, the Meax-chair shown in Figure 3, and their corresponding ring flips, the Meeq-boat and the Meeq-chair. However, the severe allylic 1,3-steric interaction between the methoxy amide residue and the methyl moiety in the Meeq-boat and the Meeq-chair precludes a substantial effect on the product distribution, and these less important transition states are not shown. The absolute stereochemistry of the major product, which has been confirmed in other examples through X-ray analysis, relay of relative stereochemistry to a known natural product, and extensive analysis of nOe’s, proves that the Meax-boat transition state is favored. Because of the number of contributing sp2 carbon atoms, the boat does not experience any destabilizing steric effects. Its dipole (μ), derived from the iminium and the phenoxonium functionality, is smaller than the corresponding (μ) for the Meax-chair transition state. Sevin has used a similar dipole stabilization argument to explain the diastereoselective addition of enamines to unsaturated aldehydes.8
Figure 3.
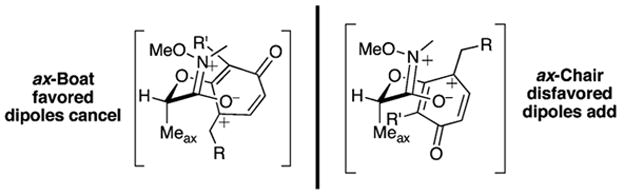
Probable transition states leading to (−)-11 and the unseen diastereomer.
Using Morrow’s protocol (Scheme 4),9 the vinylogous ester portion of compound 11 succumbs to a diastereoselective 1,4-reduction and after treatment (0.2 M in CH3CN) with iodotrimethysilane10 (2 equiv) affords the des-bromo compound 12 in an overall yield of 50%. After significant experimentation with newer procedures, we find Ojima’s 1,4-hydrosilylation method [10 equiv of triethylsilane and catalytic chloro(triphenylphosphine)rhodium in benzene] to be superior in providing the desired silyl enol ether intermediate.11 Subsequent treatment of the crude enol ether (0.4 M in CH2Cl2) with dimethyldioxirane (1.01 equiv) affords the epoxide 13 as a single diastereomer in 84% yield as established through several nOe’s. Successive treatment of the unstable epoxide (0.45 M in H2O/THF) with camphor sulfonic acid followed by protection of the resulting crude alcohol (0.1 M in DMF) affords the silyl ether 14 in 63% overall yield. β-Elimination and subsequent deprotection would seem to complete the synthesis of 1. However, this proves impossible because of the transfused ring system, which positions the leaving group in an equatorial orientation.
Scheme 4.
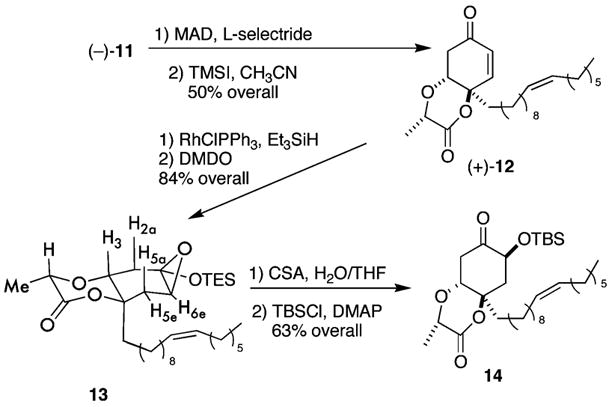
Diastereoselective Synthesis of 14
Therefore, the carbonyl moieties in 14 (0.05 M in THF) are reduced with diisobutylaluminum hydride (6 equiv) to provide a lactol intermediate that undergoes ring opening upon consecutive treatment with Me2NNH2 (2.0 equiv) to afford the hydrazone 15 in 40% overall yield (Scheme 5). An oxidation of 15 (0.1 M in CH2Cl2, 1.5 equiv of NMO, 0.2 equiv of TPAP) is followed by a noteworthy cleavage (Figure 4) of the chiral tether under mild acidic conditions (2 equiv of TMSCl, 0.1 M CH2Cl2). These conditions afford the corresponding β-hydroxy ketone. Conversion of the β-alcohol into an acetate with acetic anhydride (1.5 equiv) and base (0.4 equiv of DBU) facilitates acetylation and β-elimination thereby providing the α-siloxy enone 16 in 39% overall yield. The silyl ether of 16 succumbs to deprotection (1 equiv of TBAF, 0.02 M in THF) and affords the syn-diol (−)-1 in 90% isolated yield.
Scheme 5.
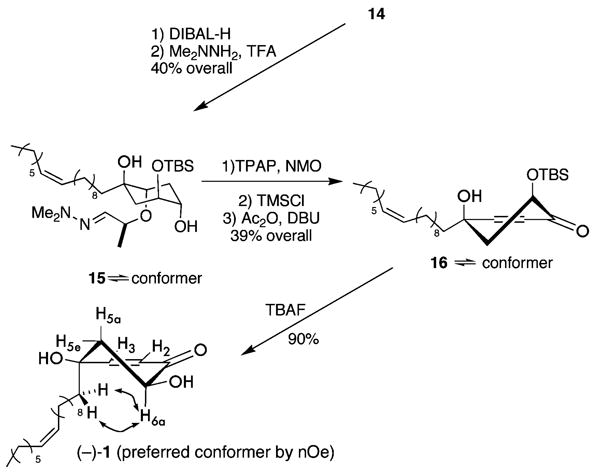
Completion of Synthetic 1
Figure 4.
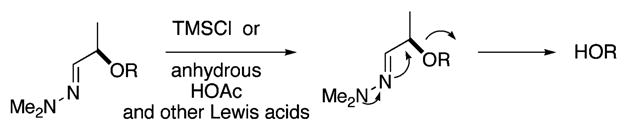
Unusual transformation that enables ether cleavage.
High-resolution mass spectroscopy [ESI+/TOF: m/z calcd for C23H40O3 ) 364.5619, found (M + Na)+ ) 387.2853] confirms the elemental composition of the synthetic 1 to be the same as that for the natural diol 1. However, the structure proposed for natural 1 had stipulated that the diol functionality adopts a 1,3-diaxial arrangement. In the case of synthetic 1, however, a sizable nOe between the methylene of the lipophilic side chain and the axial proton adjacent to the secondary alcohol proves the hydroxy residues to be disposed in 1,3-diequatorial fashion. Upon further inspection and comparison of the 1H NMR and 13C NMR data, it becomes evident that synthetic 1 and natural 1 are different compounds. After careful consideration of our intervening transformations and the integrity of our intermediates and further analysis of the data that led to the structural assignment for natural 1,1 particularly the unusually large 2.7 Hz W-coupling proposed for C3H–C5H (on a type A), it is painfully apparent that the ring architecture for natural 1 was improperly assigned.12
Three regioisomers (A–C, Table 1) accommodate the functionality that comprises the six-membered ring and includes an enone, a secondary alcohol, a tertiary alcohol, and a methylene functionality. In the case of regioisomer C, however, the C5 methylene affords an AB-quartet in the 1H NMR,3 whereas the methylene in regioisomers A and B would show some additional proton coupling. Although the natural product previously assigned as 11 does not have the C architecture, it should be noted that some related natural products possess a C architecture and are presumably related by biosynthesis to supposed natural 1.3
Table 1.
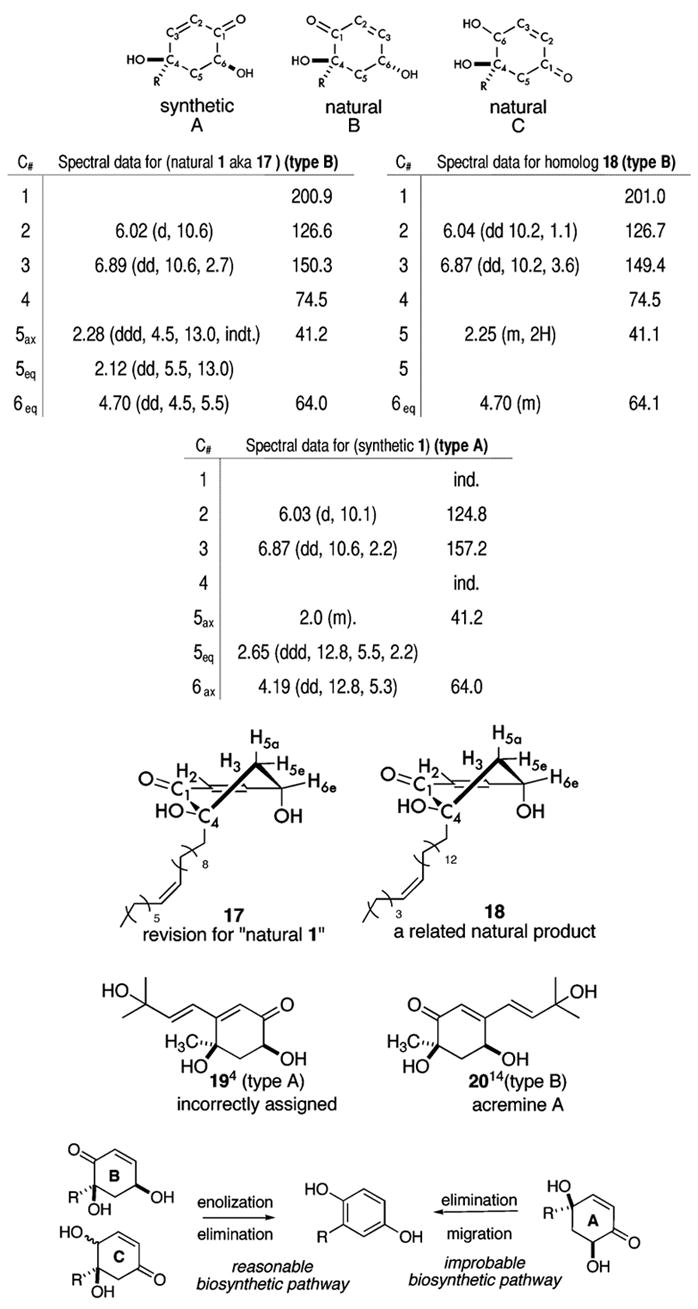
|
After careful reexamination of the reported data for natural 1, it became evident that architecture B was mistaken for architecture A. The supposed 2.7 Hz W-coupling is actually a vicinal coupling (C3–H–C6–H). In addition to our synthesis of 1, additional evidence was found in the structural assignment for compound 18, a related natural product isolated from Tapirira obtusa.13 Although this analogue displays a slightly longer side chain with an ω-5 site of unsaturation, the 1H NMR data for 18 are strikingly similar to those previously recorded for natural 1, particularly the (4.70 ppm) shift of the C6 methine. Furthermore, the 13C NMR data prove to be practically identical to those of homologue 18 in regards to the six-membered ring. We therefore propose that the natural product reported to be structure 11 is in fact its regioisomer 17. The confusion between type A and B regioisomers has led others to improper structural assignments as well. For example, the compound assigned as structure 19 was reported in extracts of Periploca aphylla.4 We propose that the natural product assigned as 194 is actually acremine A (20),14 a structure proven by X-ray.
Furthermore, from a chemist’s perspective, the B and C architectures can readily lead to a hydroquinone nucleus, such as 2, by enolization, elimination, and tautomerization. However, architecture A requires a 1,2-alkyl shift, which is unprecedented among biosynthetic transformations, to produce the corresponding 1,4-hydroquinone. Given our synthesis of 1, the considerable stability of synthetic 1 to acidic and basic conditions, as compared to natural 11 (17), and our reassignments of natural 11 as its regioisomer 17 and of natural 194 as its regioisomer 20, no evidence for a biocatalyzed dione rearrangement exists as of yet. We hope this letter helps to clarify future cyclohexenone assignments and prevents future confusion between these regiomeric cyclohexenones. At the very least, we have described several new diastereoselective transformations, introduced an innovative method for the cleavage of the chiral directing group (Figure 4), and increased the utility of chiral cyclohexadienones as building bocks15 for future enantioselective syntheses.
Acknowledgments
A Research Grant from the National Institutes of Health (GM-64831) is greatly appreciated.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for compounds 3–16 and 1 are available. This material is available free of charge via the Internet at http://pubs.acs.org.
OL061000S
References
- 1.David JM, Chávez JP, Chai HB, Cordell GA. J Nat Prod. 1998;61:287–289. doi: 10.1021/np970422v. [DOI] [PubMed] [Google Scholar]
- 2.Francis GW, Veland K. J Chromatogr. 1981;219:379–384. [Google Scholar]
- 3.Groweiss A, Cardellina JH, Pannell LK, Uyakul D, Kashman Y, Boyd MR. J Nat Prod. 1997;60:116–121. doi: 10.1021/np960435t. [DOI] [PubMed] [Google Scholar]
- 4.Aziz-ur-Rehman, Malik A, Riaz N, Nawaz HR, Ahmad H, Nawaz SA, Choudhary MI. J Nat Prod. 2004;67:1450–1454. doi: 10.1021/np030494o. [DOI] [PubMed] [Google Scholar]
- 5.Kim J. Curr Med Chem. 2002;2:485–537. doi: 10.2174/1568011023353949. [DOI] [PubMed] [Google Scholar]
- 6.Hoarau C, Pettus TRR. Synlett. 2003:127–137. doi: 10.1055/s-2003-36234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mejorado L, Hoarau C, Pettus TRR. Org Lett. 2004;6:1535–1538. doi: 10.1021/ol0498592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sevin A, Maddaluno J. J Org Chem. 1987;52:5611–5615. [Google Scholar]
- 9.Doty BJ, Morrow GW. Tetrahedron Lett. 1990;31:6125–6128. [Google Scholar]
- 10.Jung ME, Ornstein PL. Tetrahedron Lett. 1977;31:2659–2662. [Google Scholar]
- 11.Ojima I, Kogure T, Nihonyanagi M, Nagai Y. Bull Chem Soc Jpn. 1972;45:3506. [Google Scholar]
- 12.Nicolaou KC, Snyder AA. Angew Chem, Int Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864. [DOI] [PubMed] [Google Scholar]
- 13.de Jesus S, David JM, David JP, Chai HB, Pezzuto JM, Cordell GA. Phytochemistry. 2001;56:781–784. doi: 10.1016/s0031-9422(00)00476-3. [DOI] [PubMed] [Google Scholar]
- 14.Assanta ZG, Dallavalle S, Malpezzi L, Nasini G, Burrano S, Torta L. Tetrahedron. 2005;61:7686–7692. At our behest, these authors confirm that the compound previously assigned as 194 gives the same 1H NMR spectra as 20 when analyzed in the same deuterated solvent. [Google Scholar]
- 15.(a) Magdziak D, Meek S, Pettus TRR. Chem Rev. 2004;104:1383–1429. doi: 10.1021/cr0306900. [DOI] [PubMed] [Google Scholar]; (b) Van De Water RW, Hoarau C, Pettus TRR.Tetrahedron Lett 2003445109–5113.16957784 [Google Scholar]; (c) Pettus LH, Van De Water RW, Pettus TRR. Org Lett. 2001;3:905–908. doi: 10.1021/ol0155438. [DOI] [PMC free article] [PubMed] [Google Scholar]