

Summary
We examined the role of the microtubule cytoskeleton in the localization and store-operated Ca2+ entry (SOCE) function of the endoplasmic reticulum Ca2+ sensor, Stim1, in HEK 293 cells. Stim1 tagged with an enhanced yellow fluorescent protein (EYFP-Stim1) exhibited a fibrillar localization that co-localized with endogenous α-tubulin. Depolymerization of microtubules with nocodazole caused a change from a fibrillar EYFP-Stim1 localization to one that was similar to the endoplasmic reticulum. Treatment of HEK 293 cells with nocodazole had a detrimental impact on SOCE and the associated Ca2+ release-activated Ca2+ current (Icrac). This inhibition was significantly reversed in cells overexpressing EYFP-Stim1, implying that the primary inhibitory effect of nocodazole is related to Stim1 function. Surprisingly, nocodazole treatment alone induced significant SOCE and Icrac in EYFP-Stim1-expressing cells, and this was accompanied by an increase in near-plasma membrane EYFP-Stim1 fluorescence. We conclude that microtubules play a facillatory role in the SOCE signaling pathway by optimizing Stim1 localization.
Introduction
Stromal interaction molecule 1 (Stim1) is a pivotal molecular component of the store-operated Ca2+ entry (SOCE) pathway (Roos et al. 2005; Liou et al. 2005). A major fraction of cellular Stim1 resides within the membrane of the endoplasmic reticulum (ER) (Dziadek and Johnstone 2007), the primary Ca2+ storage compartment of mammalian cells; upon depletion of Ca2+ from the ER stores, Stim1 relocates to distinct punctae located in close proximity to the plasma membrane (Liou et al. 2005; Zhang et al. 2005). Once at the plasma membrane, Stim1 directly or indirectly activates members of the Orai class of SOCE channels, resulting in Ca2+ influx into the cell (Zhang et al. 2006; Mercer et al. 2006; Peinelt et al. 2006; Xu et al. 2006).
Molecular and functional data indicate that Stim1 acts as the ER Ca2+ sensor that responds to Ca2+ store depletion. First and foremost, Stim1 contains an N-terminal EF hand domain that is situated within the lumen of the ER (Dziadek and Johnstone 2007). Mutation of Ca2+ binding residues within the EF hand domain render Stim1 constitutively active; i.e., EF-hand mutated Stim1 localizes in plasma membrane punctae even in the presence of replete Ca2+ stores, and SOCE is always active (Liou et al. 2005). Furthermore, Orai1 forms clusters within the plasma membrane in response to Ca2+ store depletion that colocalize with Stim1 punctae (Luik et al. 2006; Xu et al. 2006), and it has been shown that Stim1 and Orai1 co-immunoprecipitate ((Yeromin et al. 2006; Vig et al. 2006), but see (Gwack et al. 2007)). However, direct interaction between Stim1 and Orai channels has not been demonstrated. Thus, although it is clear that Stim1 responds to Ca2+ store depletion and subsequently activates SOCE via Orai channels, the mechanism by which this activation occurs, including the mechanism of Stim1 rearrangement, remains unclear.
The precise localization and movements of intracellular signaling proteins is often governed by cytoskeletal elements, the major components being the actin cytoskeleton and the microtubule cytoskeleton. Most studies report that depolymerization of the actin cytoskeleton with cytochalasins does not negatively impact SOCE (Ribeiro et al. 1997; Patterson et al. 1999). Thus, the actin cytoskeleton does not appear to play an obligate role per se in the SOCE pathway. The role of the microtubule cytoskeleton in SOCE has also been investigated. However, as was also shown for the actin cytoskeleton (Ribeiro et al. 1997; Patterson et al. 1999), several studies have reported that nocodazole, a drug which causes depolymerization of the microtubular cytoskeleton, fails to inhibit SOCE or the Ca2+ release-activated Ca2+ current (Icrac), the current most often associated with SOCE, in RBL, NIH 3T3, and DT40 cells (Ribeiro et al. 1997; Bakowski et al. 2001; Baba et al. 2006).
Despite these negative reports, we (Mercer et al. 2006) and others (Baba et al. 2006) have shown that when human Stim1 tagged with an N-terminal enhanced yellow fluorescent protein (EYFP-Stim1) is overexpressed, it adopts an organization that is strikingly similar to that of the microtubule cytoskeleton. The microtubular cytoskeleton is known to be a major regulator of endoplasmic reticulum structure and function (Terasaki et al. 1986). Thus, microtubules may direct or influence the organization or movements of Stim1 in some way. In this study we investigated the role of microtubules in the SOCE signaling pathway, with particular emphasis on the dependence of Stim1 function on microtubules. Our results indicate that microtubules may play a facillatory role in organizing Stim1 for optimal Ca2+ sensing and/or communication with Orai channels.
Materials and Methods
Cell culture and transfections
HEK 293 cells were obtained from ATCC and were cultured and transfected as previously described (DeHaven et al. 2007). The EYFP-Stim1 plasmid was obtained from Dr. Tobias Meyer (Stanford University), mCherry-α-tubulin was obtained from Dr. Roger Tsien (University of California, San Diego), and ER-targeted GFP and CFP constructs were purchased from Clontech. The amounts of cDNA used in transfections were as follows: EYFP-Stim1, 0.5 μg; mCherry-α-tubulin, 0.05 μg; ER-CFP, 0.5 μg. The Orai1 siRNA used for knockdown of Orai1 expression was purchased from Invitrogen and had the sequence cccuucggccugaucuuuaucgucu; 100 nM was used per transfection.
Live cell confocal and TIRFM imaging
Cells were grown on glass coverslips for 24-48 hours, and coverslips were mounted in Teflon chambers for imaging. Cells were maintained in Hepes-buffered saline solution (HBSS; in mm: 120 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgCl2, 11 glucose, and 20 Hepes, pH 7.4) at room temperature. Confocal imaging was performed using a Zeiss LSM 510 laser scanning system, and either a 40x water-immersion (N.A. 1.2) or a 63x oil-immersion (N.A. 1.4) objective was used. All confocal images were collected with the pinhole set at 1 Airy Unit. For EYFP-Stim1, 488 nm or 514 nm illumination was provided by an Argon laser and emission was selected with a 530-600 nm bandpass filter. The excitation for mCherry-tubulin was 543 nm from a HeNe laser and emission was selected with a 560 nm low-pass filter. The excitation for ER-CFP was 458 nm from an Argon laser and emission was selected with a 470-510 bandpass filter. When cells expressing multiple probes were imaged, lack of bleed-through between channels was verified by imaging cells that expressed each of the probes individually.
TIRFM was performed essentially as previously described (Smyth et al. 2005).
Fixation, immunostaining, and colocalization analysis
Cells for fixation were grown in LabTekII chamber slides (Nalge Nunc) for 24 to 48 hours. At the time of fixation, slides were rinsed in phosphate-buffered saline (PBS; in mM: 137 NaCl, 2.68 KCl, 1.47 KH2PO4, 14.9 Na2HPO4) and were extracted in Karesnti’s buffer (in mM: 80 Pipes, 1.0 MgSO4, 5.0 EGTA, 0.5% Triton X-100) for 8 seconds at room temperature. Cells were then fixed in 100% methanol on ice for 10 min and rehydrated in PBS containing 0.1% Tween-20 and 3.0 mM NaN3 (PBS-Tw-Az). Cells were incubated in primary antibodies diluted in PBS-Tw-Az containing 1% bovine serum albumin (BSA) overnight at room temperature and with secondary antibodies in PBS-Tw-Az plus BSA for 45 min at room temperature. Cells were washed 3 times in PBS-Tw-Az after each antibody incubation, and after the final wash coverslips were mounted onto the slides using Vectashield Hard Set Mounting Medium (Vector Laboratories). For detection of α-tubulin the primary antibody used was DM1A (AbCam) at a dilution of 1:100, and the secondary antibody was Alexa Fluor 633 goat anti-mouse IgG1 (Invitrogen) at a dilution of 1:1000. For detection of EYFP-Stim1, the primary antibody was a polyclonal against GFP (AbCam) used at a dilution of 1:2000, and the secondary was Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen) at a dilution of 1:1000. Slides were imaged by confocal microscopy as described above.
Colocalization analysis was performed using the Zeiss LSM 510 software. For each image analyzed, the percentage of pixels that exhibited a signal above a predetermined threshold for each channel was recorded.
Intracellular Ca2+ measurements
Intracellular Ca2+ measurements were performed as previously described (Mercer et al. 2006). Briefly, cells were loaded with 1 μM Fura-5F/AM (Invitrogen) for 25 min at 37°C. Fura-5F fluorescence was measured when cells were excited consecutively at 340 nm and 380 nm, and relative Ca2+ concentrations are reported as the ratio of fluorescence emission at the two excitation wavelengths. Cells transfected with EYFP or EYFP-Stim1 were chosen based on their fluorescence when excited at 477 nm. Typically, 20-30 cells were measured on a single coverslip per experiment.
Electrophysiology
Whole-cell currents were investigated at room temperature (20-25°C) in HEK293 cells using the patch-clamp technique in the whole-cell configuration. The standard HEPES buffered saline solution contained (mM): 145 NaCl, 3 KCl, 10 CsCl, 1.2 MgCl2, 10.0 CaCl2, 10 glucose, and 10 HEPES (pH to 7.4 with NaOH). The standard divalent free (DVF) solution contained (in mM): 155 Na-methanesulfonate, 10 HEDTA, 1 EDTA and 10 HEPES (pH = 7.4). Fire-polished pipettes fabricated from borosilicate glass capillaries (WPI, Sarasota, FL) with 3-5 MΩ resistance were filled with (in mM): 145 Cs-methanesulfonate, 20 BAPTA, 10 HEPES, and 8 MgCl2 (pH to 7.2 with CsOH). In indicated experiments, the pipette also contained 25 μM inositol 1,4,5-trisphosphate (IP3, hexasodium salt, Sigma) to actively deplete intracellular Ca2+ pools. Voltage ramps (-100 mV to +100 mV) of 250 ms were recorded every two sec immediately after gaining access to the cell from a holding potential of 0 mV, and the currents were normalized based on cell capacitance. Leak currents were subtracted by taking an initial ramp current before ICRAC developed and subtracting this from all subsequent ramp currents. Access resistance was typically between 5-10 MΩ. The currents were acquired with pCLAMP-10 (Axon Instruments) and analyzed with Clampfit (Axon Instruments) and Origin 6 (Microcal) software. All solutions were applied by means of a gravity based multi-barrel local perfusion system with an extremely low dead volume common delivery port (Perfusion Pencil, Automate Scientific, Inc.).
Drug treatments
Cells were treated with nocodazole or colchicine (both from Calbiochem) in HBSS at room temperature. Because nocodazole treatment caused activation of Ca2+ entry (see Results), all nocodazole incubations were performed in nominally Ca2+-free HBSS unless otherwise noted. Ionomycin and cyclopiazonic acid were obtained from Calbiochem and thapsigargin was obtained from Alexis Biochemicals.
Results
As previously reported (Mercer et al. 2006; Baba et al. 2006), when EYFP-Stim1 is overexpressed in HEK293 cells, the protein adopts a configuration that is strikingly similar in appearance to a microtubule array. EYFP-Stim1 is organized in long filament-like structures, many of which appear to emanate from a central organizing center near the nucleus (Figure 1A); this EYFP-Stim1 “organizing center” is highly reminiscent of a centrosome, which functions as the microtubule organizing center in interphase cells. For comparison, Figure 1B shows an HEK293 cell that was fixed and immunostained with an anti-α-tubulin antibody. By comparison, as shown in Figure 1C, the ER in HEK293 cells has a lattice-like organization throughout the cell, which is quite distinct in appearance from the microtubule or EYFP-Stim1 arrays seen in these cells. We therefore hypothesized that Stim1 may occupy only a subset of the total cellular ER at any given time, and that this specialized localization of Stim1 within the ER may be directed by microtubules in some way.
Figure 1. EYFP-Stim1 is organized in a microtubule-like array in HEK293 cells.

A) Confocal image of HEK293 cells overexpressing EYFP-Stim1. The arrowhead indicates what appears to be an “organizing center” of EYFP-Stim1 localization. B) Confocal image of an HEK293 cell that was fixed and immunostained for α-tubulin. The arrowhead indicates the microtubule organizing center near the nucleus, presumably the centrosome. C) Confocal image of an HEK293 cell overexpressing an ER-targeted CFP construct (ER-CFP). Scalebars = 10 μm.
EYFP-Stim1 Colocalizes with α-tubulin
It has previously been reported that Stim1 colocalizes with microtubules in HeLa cells that were fixed and immunostained for α-tubulin (Baba et al. 2006). We initially performed our analysis on live HEK293 cells co-overexpressing EYFP-Stim1 and an mCherry-α-tubulin construct (mCherry-tub). As shown in Figure 2A, expression of the mCherry-tub construct resulted in prominent fluorescence in the cytoplasm, and there was only a limited number of well-defined microtubules that were clearly visible above this background. It is clear, however, that for many of the filaments of mCherry-tub that are clearly discernible, there is an EYFP-Stim1 filament that exactly matches its localization (Figure 2A, upper panel, arrowheads), indicating an association between the overexpressed mCherry-tub and EYFP-Stim1. We also performed colocalization analysis on EYFP-Stim1 overexpressing cells that were fixed and immunostained with an antibody against α-tubulin and an antibody against GFP (which recognizes EYFP-Stim1). These cells exhibited a well-defined α-tubulin network, as well as EYFP-Stim1 localization that was similar to that seen in live cells (Figure 2B). In the fixed and immunostained samples, there was a high degree of colocalization between EYFP-Stim1 and α-tubulin (Figure 2B, upper panel, merged image. Note, interestingly, EYFP-Stim1 appears to be excluded from the centrosome region.). Statistical analysis of this colocalization revealed that 85.4 ± 0.02 % (n = 13 cells) of pixels that were positive for EYFP-Stim1 immunofluorescence were also positive for α-tubulin immunofluorescence. Conversely, 74.1 ± 0.02 % of α-tubulin positive pixels were also positive for EYFP-Stim1. Thus, the majority of EYFP-Stim1 colocalizes with α-tubulin, which supports the idea that Stim1 is organized into a fibrillar distribution through direct or indirect association with microtubules.
Figure 2. EYFP-Stim1 colocalizes with α-tubulin.
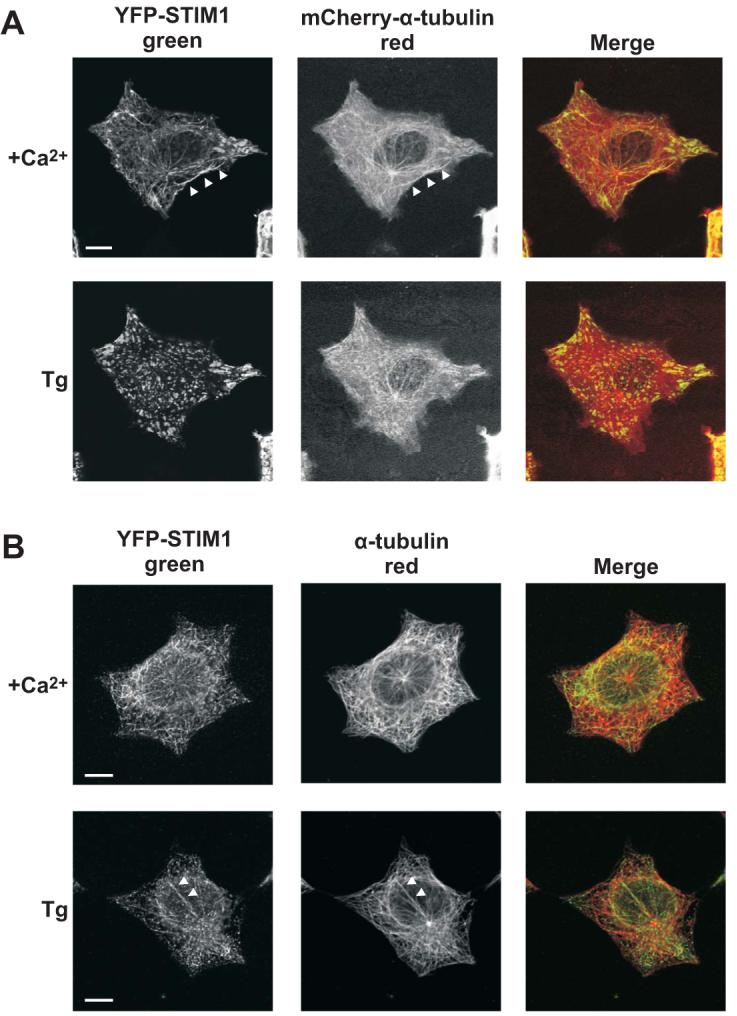
A) Upper panel: confocal images of an HEK293 cell co-overexpressing EYFP-Stim1 (left panel; green) and mCherry-α-tubulin (middle panel; red) in the presence of 1.8 mM extracellular Ca2+. The arrowheads indicate a track of EYFP-Stim1 fluorescence that exactly matches a corresponding track of mCherry-α-tubulin fluorescence. Lower panel: EYFP-Stim1 and mCherry-α-tubulin localization 15 min following store depletion with thapsigargin (Tg; 2 μM) in nominally Ca2+-free extracellular solution. B) Upper panel: Confocal images of a EYFP-Stim1 overexpressing HEK293 cell that was fixed and immunostained with antibodies against EYFP (to enhance detection of EYFP-Stim1) (left panel; green) and α-tubulin (middle panel; red). Lower panel: An HEK293 cell that was store depleted with thapsigargin for 15 min prior to fixation and immunostaining. Arrowheads indicate a track of α-tubulin along which several EYFP-Stim1 punctae appear to associate. Scalebars = 10 μm.
We also analyzed the localization of EYFP-Stim1 and α-tubulin in cells in which intracellular Ca2+ stores were depleted with thapsigargin (Figures 2A and B, lower panels). As has previously been shown (Liou et al. 2005; Zhang et al. 2005; Mercer et al. 2006; Baba et al. 2006; Wu et al. 2006), store depletion induces a rearrangement of EYFP-Stim1 from fibrillar structures to punctae that are located in close proximity to the plasma membrane. In the fixed and immunostained cells, it was possible to detect several tracks of α-tubulin along which EYFP-Stim1 punctae appear to associate (Figure 2B, bottom panel, arrowheads). Thus, microtubules may direct the relocalization of Stim1 into punctae near the plasma membrane.
Nocodazole Disrupts EYFP-Stim1 Organization and Interferes with SOCE and Icrac
To more directly test whether Stim1 associates with microtubules, we monitored EYFP-Stim1 localization following microtubule depolymerization with nocodazole. As shown in Figure 3, treatment of cells for 20 min with nocodazole (10 μM) caused EYFP-Stim1 to significantly rearrange from a filamentous configuration to one that looks strikingly similar to the configuration of the ER. In the same cell, nocodazole treatment induced only a marginal change in the ER conformation as measured by the localization of an ER-targeted cyan fluorescent protein (CFP-ER). Accordingly, it is apparent in the merged image that EYFP-Stim1 was fairly evenly distributed throughout the ER following nocodazole treatment. EYFP-Stim1 overexpressing cells that were fixed and immunostained for α-tubulin demonstrated that the 20 min treatment with nocodazole was sufficient to induce a significant disruption of the microtubule cytoskeleton (Figure 3B, compare to Figure 2B), although some long filaments were still evident in these cells. Interestingly, EYFP-Stim1 did not colocalize to any significant degree with these α-tubulin filaments that remained following nocodazole treatment.
Figure 3. Microtubule depolymerization causes dispersion of EYFP-Stim1 throughout the ER.
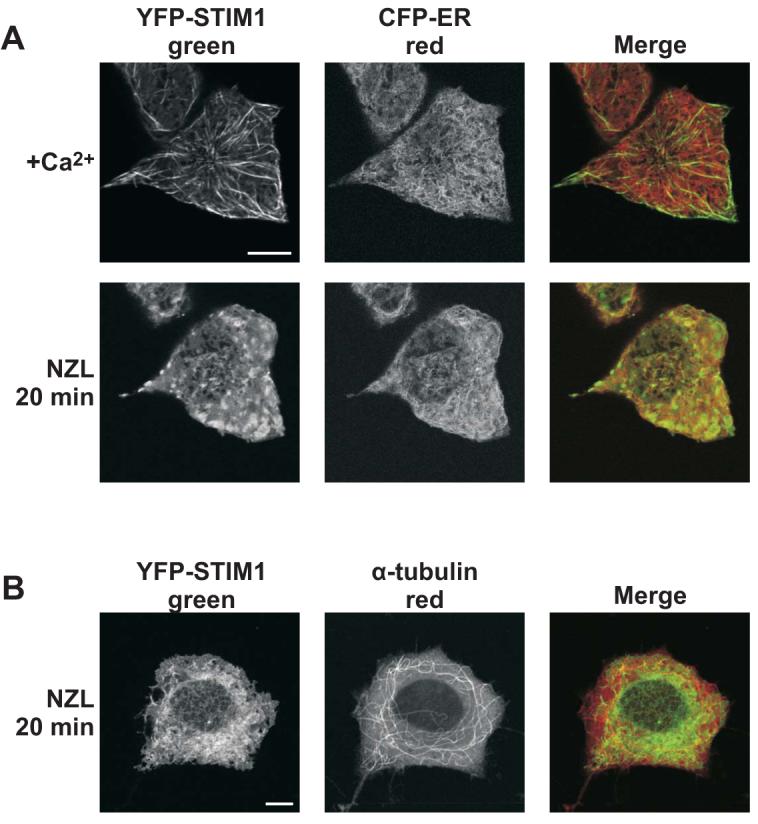
A) Confocal images of HEK293 cells co-overexpressing EYFP-Stim1 (left panel; green) and an ER-targeted CFP (CFP-ER) (middle panel; red) in the presence of 1.8 mM extracellular Ca2+ (upper panel) and following a 20 min treatment with nocodazole (NZL; 10 μM; lower panel). B) Images of EYFP-Stim1 expressing HEK293 cells that were treated with nocodazole for 20 min, followed by fixation and immunostaining with antibodies against GFP (left panel; green) and α-tubulin (middle panel; red). Scalebars = 10 μm.
Colchicine, another drug known to disrupt microtubules (Oka et al. 2005), had similar effects on Stim1 distribution and microtubule structure, although the effects on microtubule structure were somewhat less extensive than for nocodazole (not shown, and Supplemental Figure 1).
Microtubule depolymerization had a significant impact on EYFP-Stim1 localization, so it was therefore of interest to evaluate the effect of microtubule depolymerizing drugs on Stim1-mediated SOCE. SOCE was activated by the SERCA inhibitor, thapsigargin. As shown in Figure 4, pretreatment of HEK293 cells with either 100 μM colchicine or 10 μM nocodazole resulted in significant inhibition of the sustained [Ca2+]i elevation following thapsigargin addition, suggesting inhibition of store-operated entry. Consistent with the morphological observations, the extent of inhibition was greater for nocodazole than for colchicine, and thus nocodazole was used for the majority of subsequent experiments.
Figure 4. Microtubule depolymerization inhibits SOCE.

A) Wildtype HEK293 cells were treated with 100 μM colchicine for 20 min (red trace) or left untreated (control; black trace), and thapsigargin (Tg; 2 μM) was then added in the presence of 1.8 mM extracellular Ca2+. Intracellular Ca2+ concentration was monitored throughout the experiments, and SOCE became activated soon after Tg addition as evidenced by the second peak in Ca2+ concentration, and remained active based on the sustained elevation of intracellular Ca2+ above baseline. Each trace represents the average response of all cells on a single coverslip (20-30 cells). B) The average difference between the 340/380 value 15 minutes following Tg addition (sustained SOCE) and the 340/380 value just prior to Tg addition (baseline) was calculated for untreated control (n = 150 cells, 3 coverslips) and colchicine-treated (n = 145 cells, 3 coverslips) cells for experiments performed as described in (A). C) SOCE was analyzed as described in (A) for cells treated for 20 min with 10 μM nocodazole (NZL; red trace) and for control cells treated with 0.1 % DMSO (black trace). Because NZL treatment alone caused some store depletion and activation of SOCE (see figure 7), NZL and DMSO treatments were performed in nominally Ca2+-free extracellular solution. D) The average difference between the 340/380 value 15 minutes following Tg addition (sustained SOCE) and the 340/380 value just prior to Tg addition (baseline) was calculated for DMSO-treated (n = 122 cells, 3 coverslips) and NZL-treated (n = 125 cells, 3 coverslips) cells for experiments performed as described in (C). Data are reported as mean ± SEM; p-values are based on t-tests.
The inhibition by microtubule depolymerizing drugs of the thapsigargin-induced sustained [Ca2+]i elevation seen in Figure 4 may reflect decreased Ca2+ entry through SOCE channels, but could also conceivably result from increased Ca2+ buffering by intracellular or plasma membrane transporters, or by decreased driving force for Ca2+ influx (i.e., by membrane depolarization). Thus, we also tested the ability of nocodazole to inhibit the store-operated current Icrac in HEK 293 cells. Whole-cell currents were measured using a pipette solution containing IP3 to deplete intracellular Ca2+ stores and BAPTA to prevent Ca2+-dependent inactivation; when HEK 293 cells were patched in the presence of 10 mM extracellular Ca2+, a very small inwardly rectifying current on the order of -0.5 pA/pF developed (Figure 5A). Cells pre-treated with 10 μM nocodazole for 20 min prior to patching exhibited a smaller Ca2+ current, although this decrease was not statistically significant (Figure 5A). Thus, given the very small Ca2+ current in HEK 293 cells, it was difficult to ascertain whether nocodazole had an inhibitory effect. We therefore switched to a protocol using a divalent cation-free extracellular solution, which allows for a more accurate assessment of Icrac currents in these cells (DeHaven et al. 2007). When cells are patched in the presence of extracellular Ca2+ and then switched to a divalent-free solution, a readily discernible Na+ current of ∼-3 pA/pF develops (Figure 5B). When the divalent-free protocol was applied to cells pre-treated with nocodazole for 20 min, the peak Na+ current was approximately 50 % less compared to untreated controls (Figure 5B and C). Thus, as observed for SOCE, Icrac is inhibited in HEK 293 cells by the microtubule-depolarizing drug, nocodazole.
Figure 5. Microtubule depolymerization inhibits Icrac.

A) Whole-cell currents were measured in control (black trace) and nocodazole-treated (10 μM, 20 min; red trace) wildtype HEK 293 cells in the presence of 10 mM extracellular Ca2+; stores were depleted with a pipette solution containing IP3 (25 μM). Shown are the averaged responses (± SEM) of 7 control and 8 nocodazole-treated cells. The means were not significantly different at any point along the traces. B) Currents were measured as in D, except the extracellular solution was switched from 10 mM Ca2+ to divalent-free at the time indicated. Averaged (± SEM) traces from 8 control (black trace) and 6 nocodazole-treated (red trace) cells are shown; the maximal Na+ current was significantly smaller in nocodazole-treated compared to control cells (t-test, p < 0.01). C) Averages of the peak Na+ currents from 8 control and 6 nocodazole-treated cells. Data are reported as mean ± SEM; p-value is based on t-test.
The results of the experiments showing inhibition of SOCE by nocodazole are not directly comparable to the morphological findings, because in the latter case, the cells overexpressed EYFP-Stim1. Thus, we carried out experiments comparing the effects of nocodazole on Ca2+ entry in wild-type HEK293 cells to cells overexpressing EYFP-Stim1. For these experiments, SOCE was monitored by emptying intracellular Ca2+ stores with thapsigargin in the presence of nominal extracellular Ca2+, followed by readdition of 1.8 mM extracellular Ca2+. Consistent with the results in Figure 4, when HEK293 cells were treated with 10 μM nocodazole for 20 min prior to store depletion with thapsigargin, Ca2+ entry was reduced by approximately 70 % when compared to cells treated with DMSO alone (Figure 6A and C; p < 0.01). Surprisingly, in HEK293 cells overexpressing EYFP-Stim1, nocodazole had relatively little effect on SOCE in these cells (Fig. 6B and C). And although the inhibition in EYFP-Stim1 cells was statistically significant compared to DMSO-treated controls at the p < 0.05 level, the degree of inhibition was only 15 %. Thus, overexpression of EYFP-Stim1 appears to significantly rescue the inhibition of SOCE caused by microtubule depolymerization, consistent with the conclusion that the primary cause of inhibition by nocodazole is related to Stim1 dysfunction.
Figure 6. Overexpression of Stim1 rescues inhibition by nocodazole.

To evaluate SOCE, changes in intracellular Ca2+ concentration were monitored in HEK293 cells in which Ca2+ stores were depleted with thapsigargin (Tg; 2 μM) in the presence of nominally Ca2+-free extracellular solution, followed by addition of 1.8 mM Ca2+. In wildtype HEK293 cells overexpressing unconjugated EYFP (A) and cells overexpressing EYFP-Stim1 (B), SOCE was evaluated following a 20 min incubation in 10 μM nocodazole (NZL; red traces) or 0.1 % DMSO (black traces). Each trace represents the average response of all cells on a single coverslip (20-30 cells). C) The average difference between the peak 340/380 value following Ca2+ addition and the 340/380 value just prior to Ca2+ addition was calculated for EYFP-expressing cells treated with DMSO (n = 89 cells, 3 coverslips) or 10 μM NZL (n = 110 cells, 4 coverslips) and in EYFP-Stim1 expressing cells treated with DMSO (n = 77, 3 coverslips) or with 10 μM NZL (n = 84 cells, 3 coverslips) for experiments performed as described in (A) and (B). Data are reported as the mean ± SEM; p-values are based on t-tests.
We also carried out experiments whereby Ca2+ readdition was performed in the absence of store depletion with thapsigargin. Unexpectedly, nocodazole treatment alone for 20 min was sufficient to activate a significant Ca2+ entry upon Ca2+ readdition in EYFP-Stim1 overexpressing cells as compared to cells treated with DMSO alone (Figure 7A and C). As shown in Figure 7E, all the EYFP-Stim1 overexpressing cells treated with nocodazole responded with Ca2+ entry, although the magnitude of this entry varied widely among the cells; in contrast, only very few of the DMSO-treated cells exhibited an observable Ca2+ entry (Figure 7D). Little or no nocodazole-activated entry was seen in cells overexpressing unconjugated EYFP (Figure 7B). The Ca2+ entry induced by nocodazole in EYFP-Stim1 cells was attributable to activation of physiological SOCE channels by two criteria. First, reduction of Orai1 expression by RNAi significantly reduced nocodazole-activated Ca2+ entry (Figure 7A and C). Second, the increased entry in nocodazole-treated EYFP-Stim1 cells is associated with increased Icrac. EYFP-Stim1 cells, control or treated with nocodazole, were patched using a pipette solution in which Ca2+ was strongly buffered to low levels to passively deplete stores, and the extracellular solution was switched to divalent-free solution at 1 minute intervals (Figure 7G and H). Because the magnitude of the observed increase in Ca2+ entry in nocodazole-treated cells was somewhat variable, we selected nocodazole-treated cells that showed visible EYFP-Stim1 punctae (see below). With this passive depletion protocol (i.e., no IP3 in the pipette), there is a delay before the onset of development of Icrac permitting evaluation of the constitutive current following break-in. As shown in Figure 7G and H, in EYFP-Stim1 cells treated with nocodazole (10 μM, 20 min), the Na+ current at the time of the first switch to divalent-free solution was substantially greater than that seen in untreated controls (nocodazole-treated cells, 9/10 had initial Na+ currents >4pA/pF; control cells, 1/6 had similarly large currents). Thus, nocodazole-treated cells exhibited an Icrac current that was active prior to store depletion induced by the patch-clamp protocol.
Figure 7. Nocodazole treatment activates SOCE and Icrac in EYFP-Stim1 overexpressing cells.

A) EYFP-Stim1 overexpressing cells were treated with 10 μM nocodazole (NZL; red trace) or 0.1 % DMSO (black trace) for 20 min in nominally Ca2+-free extracellular solution followed by addition of 1.8 mM extracellular Ca2+ in the absence of store depletion. Also shown is NZL-treated EYFP-Stim1 overexpressing cells in which Orai1 expression was reduced by RNAi (blue trace). Each trace represents the average response of all cells on a single coverslip (20-30 cells). B) EYFP-expressing cells were treated with 10 μM NZL (red trace) or 0.1 % DMSO (black trace) and Ca2+ entry was evaluated in the absence of store depletion as described in (A). C) The average difference between the peak 340/380 value following Ca2+ addition and the 340/380 value just prior to Ca2+ addition was calculated for EYFP-Stim1 expressing cells treated with 0.1 % DMSO (n = 85 cells, 3 coverslips), 10 μM NZL (n = 167 cells, 6 coverslips), and in EYFP-Stim1 expressing, Orai1 knockdown cells treated with NZL (n = 62 cells, 3 coverslips) for experiments performed as described in (A). Data are represented as mean ± SEM; p-values are based on one-way ANOVA. D-F) The responses of all the cells measured on a single coverslip are shown for the averaged traces shown in (A); D: EYFP-Stim1 cells treated with DMSO; E: EYFP-Stim1 cells treated with 10 μM nocodazole; F: EYFP-Stim1 overexpressing, Orai1 knockdown cells treated with nocodazole. G) EYFP-Stim1 overexpressing cells were patched with an intracellular solution clamped at 0 Ca2+. Break-in was performed in 10 mM extracellular Ca2+, and the extracellular solution was switched to divalent-free at 1 minute intervals with 30 second intervals of 10 mM Ca2+ in between. H) The same protocol in C was performed using 0 Ca2+ intracellular solution on a NZL-treated, EYFP-Stim1 overexpressing cell.
Nocodazole Does not Prevent EYFP-Stim1 Reorganization into Punctae
The various effects of nocodazole on SOCE and Icrac led us to investigate the influence of microtubule depolymerization on the formation of EYFP-Stim1 punctae near the plasma membrane. Store depletion with thapsigargin in nocodazole-treated cells caused formation of EYFP-Stim1 punctae, as evaluated by confocal microscopy (Fig. 8A). To provide a more quantitative analysis of EYFP-Stim1 punctae formation, we performed similar experiments using TIRFM. As shown in Figure 8B, the 20 min time course of nocodazole treatment caused a marked increase in near-plasma membrane EYFP-Stim1 punctae; punctae formation was further increased upon store depletion with thapsigargin. DMSO treatment alone had no effect (Fig. 8C). Quantitative analysis of the TIRFM fluorescence over time indicates that near plasma membrane EYFP-Stim1 fluorescence began to increase soon after nocodazole application (Fig. 8D); at the culmination of the 20 min nocodazole treatment (ii), there was a statistically significant increase in fluorescence intensity compared to cells treated with DMSO (Fig. 8E; p < 0.01). Furthermore, the thapsigargin-induced fluorescence intensity increase was significantly greater in the nocodazole-treated versus DMSO-treated cells (Figure 8E; p < 0.05). In fact, the nocodazole-induced and thapsigargin-induced fluorescence intensity increases appeared to be additive, implying that the cause of the nocodazole-induced increase is independent of that of thapsigargin (i.e., store depletion).
Figure 8. Microtubule depolymerization does not inhibit punctate EYFP-Stim1 localization.

A) Confocal images of HEK293 cells in the presence of 1.8 mM extracellular Ca2+ (left panel), following a 20 min incubation in 10 μM nocodazole (NZL; middle panel), and 15 min following store depletion with thapsigargin (Tg; 2 μM) in the continued presence of nocodazole (right panel). B) TIRFM images of EYFP-Stim1 expressing cells in 1.8 mM extracellular Ca2+ (left panel; i) following a 20 min treatment in 10 μM NZL (middle panel; ii), and 15 min following store depletion in the continued presence of NZL (right panel; iii). The same experiment is shown in C, except that cells were treated with 0.1 % DMSO instead of NZL. D) Fluorescence intensity profiles over time for the cells shown in B and C; each trace represents the average fluorescence intensity measured in a region of interest encompassing a single cell. Red traces: NZL-treated cells; black traces: DMSO-treated cells. The labeling of i, ii, and iii in the graph represent the times at which the still images in B and C were taken. E) Average maximal TIRFM fluorescence intensity measured in EYFP-Stim1 cells following the 20 min treatment in NZL (n = 6 cells over 3 coverslips) or DMSO (n = 5 cells over 3 coverslips), and in the same cells following store depletion with thapsigargin (Tg). Data are reported as mean ± SEM; p-values are based on t-test. Scalebars = 10 μm.
These results indicate that disruption of microtubules by nocodazole can induce Stim1 punctae formation, and activate SOCE and Icrac without the use of Ca2+ store-depleting drugs. However, we noticed that the shape of the Ca2+ release response to thapsigargin was altered in nocodazole-treated cells (see for example Figure 6), such that the rate of release appeared faster. This could indicate a subtle effect of nocodazole on endoplasmic reticulum Ca2+ permeability. Thus, we examined the total intracellular ionomycin-releasable Ca2+ pool (as in (Bird et al. 1992; Bird and Putney, Jr. 2005)) in HEK293 cells, and found that the ionomycin-induced 340/380 fluorescence peak was 3.59 ± 0.08 in DMSO-treated EYFP-Stim1 expressing cells (n = 85 cells, 3 coverslips) and 2.77 ± 0.09 in nocodazole-treated cells (n = 84 cells, 3 coverslips). Thus, the total ionomycin-releasable intracellular Ca2+ stores were reduced by 22.8 % by nocodazole (p < 0.01). Because of the additivity of nocodazole and thapsigargin in Figure 8, we thought it unlikely that this small depletion of Ca2+ stores could account for the activation of Stim1 redistribution and SOCE. However, we considered the possibility that this release might be required and that the microtubule disrupting action of nocodazole may in some manner potentiate the response. To address this possibility we took advantage of the fact that colchicine, like nocodazole, disrupts microtubules but does not appear to cause any significant depletion of Ca2+ stores (unpublished). As shown in Figure 9A, treatment of EYFP-Stim1-expressing HEK293 cells with colchicine did not result in constitutive activation of Ca2+ entry. However, when colchicine-treated cells were stimulated with 1 μM cyclopiazonic acid (CPA), a reversible SERCA pump inhibitor that elicits only partial store depletion when used at low micromolar concentrations, an augmentation of Ca2+ entry was observed compared to cells treated with CPA alone (Figure 9A and B). Thus, in cells over-expressing Stim1, disruption of the microtubule cytoskeleton can actually augment Ca2+ entry, but apparently at least some Ca2+ store depletion is necessary.
Figure 9. Colchicine potentiates but does not activate SOCE in cells overexpressing Stim1.

A) EYFP-Stim1-expressing HEK293 cells were treated with 1.0 μM CPA alone (Control; black trace) or with CPA and 100 μM colchicine (red trace) for 20 min in nominally Ca2+-free extracellular solution, followed by addition of 1.8 mM extracellular Ca2+ to reveal SOCE. Also shown are cells treated with colchicine alone (blue trace). Each trace represents the average response of all cells on a single coverslip (20-30 cells). B) The average difference between the peak 340/380 value following Ca2+ addition and the 340/380 value just prior to Ca2+ addition was calculated for CPA-treated control (n = 64 cells, 3 coverslips) and colchicine-treated (n = 97 cells, 3 coverslips) cells for experiments performed as described in (A). Data are reported as mean ± SEM; p-value is based on t-test.
Discussion
We have presented several lines of evidence indicating that Stim1 localization and function in HEK293 cells involves the microtubule cytoskeleton. First and foremost is the observation that overexpressed EYFP-Stim1 colocalizes with α-tubulin in HEK293 cells, as has previously been shown for HeLa cells (Baba et al. 2006). Second, the fibrillar arrangement of EYFP-Stim1 is drastically altered in cells treated with the microtubule depolymerizing agent nocodazole, resulting in a distribution of Stim1 similar to that of a general ER marker. Thus, Stim1 appears to be organized within the ER by the microtubule network. Third, SOCE and Icrac were affected in several ways by microtubule depolymerization. In cells that did not overexpress EYFP-Stim1, nocodazole inhibited SOCE and Icrac. This inhibition of SOCE was almost completely reversed by overexpression of EYFP-Stim1, implying that the inhibitory effect of SOCE by nocodazole is due to an impairment of Stim1 function. We considered that the disruption of microtubules can lead to a retraction of the endoplasmic reticulum away from the plasma membrane (Graier et al. 1998). For this reason, we limited pretreatment with the drugs to times wherein no visible retraction was observed by confocal microscopy. As a more rigorous test of this idea, we also examined the near membrane localization of an endoplasmic reticulum-target green fluorescent protein by TIRF microscopy. Treatment of HEK293 cells expressing this fluorescent protein with nocodazole or colchicine for 20 minutes did not result in significant retraction of the endoplasmic reticulum, but rather the TIRF signal was slightly, although not significantly increased (F/F0 for control, 0.94 ± 0.04, n=4; nocodazole, 1.45 ± 0.28, n=4; colchicine, 1.38 ± 0.12, n=6).
We were surprised to find that nocodazole treatment alone was sufficient to activate SOCE and Icrac in EYFP-Stim1 overexpressing cells. This increase in Ca2+ entry caused by nocodazole is likely initiated by modest Ca2+ store depletion by nocodazole, together with a significant potentiation of Stim1 movements and SOCE activation due to microtubule disaggregation. In support of this interpretation, colchicine, which does not cause any Ca2+ store depletion, did not activate entry on its own but substantially potentiated entry in response to submaximal store depletion. In aggregate, these findings suggest that movement of Stim1 to near membrane punctae in response to store depletion is not a microtubule-dependent process per se. Rather, the organizing effects of the microtubule cytoskeleton on Stim1 localization and movements is required for optimal signaling when Stim1 is limiting or expressed at physiological levels. The microtubule-dependent organization becomes unnecessary at very high levels of expression, and even inhibits excessive access of Stim1 to signaling complexes, presumably with Orai channels.
Several previous studies have reported discordant effects of nocodazole treatment on SOCE and Icrac in a variety of cell types. Bakowski et al (Bakowski et al. 2001) reported a lack of effect of 5 μM nocodazole treatment for 16-26 hours on Icrac in RBL-1 cells, the assumption being that such a treatment should cause retraction of the ER away from the plasma membrane and prevent conformational coupling with the Icrac channel. We also observed little or no effects of nocodazole on Icrac in RBL cells with the protocol used in the current study (data not shown). A lack of effect of nocodazole treatment (10 μM, 30 minutes) on SOCE was also reported in NIH 3T3 cells (Ribeiro et al. 1997). However, a more recent study showed a significant inhibitory effect of nocodazole (0.3-3.0 μM, 15 minutes) and colchicine on SOCE in RBL-2H3 cells and bone marrow-derived mast cells (Oka et al. 2005), although no current measurements were presented. Thus it appears that any number of factors, including cell type and treatment protocol may significantly influence the results of experiments intended to assess the effects of microtubule depolymerization on SOCE and/or Icrac. One intriguing possibility is that differences in Stim1 expression levels in different cell types could have an impact. In HEK293 cells, endogenous Stim1 expression appears to be limiting for activation of SOCE since overexpression of Stim1 results in enhanced entry in this cell type (Roos et al. 2005; DeHaven et al. 2007). Furthermore, overexpression of EYFP-Stim1 significantly rescues nocodazole-inhibited SOCE, implying that microtubule depolymerization is far less deleterious under conditions in which Stim1 expression is not limiting. The most likely explanation for these results is that microtubule depolymerization results in mislocalization of endogenous Stim1, as we have shown by confocal imaging with overexpressed EYFP-Stim1 (unfortunately, a lack of Stim1 antibodies suitable for immunofluorescence has prevented us from monitoring the distribution of endogenous Stim1). Such mislocalization likely prevents Stim1 from efficiently sensing Ca2+ store depletion and/or from efficiently coupling with the SOCE signaling machinery and possibly members of the Orai family of SOCE channels. When Stim1 is overexpressed, the excess protein permits near full activation even when the efficiency of the process is compromised.
Interestingly, we also observed a nocodazole-induced activation of SOCE and Icrac in EYFP-Stim1-expressing cells, as well as an increase in near-plasma membrane-localized EYFP-Stim1. Nocodazole causes minor Ca2+ store depletion, which is likely necessary for this effect. However, in the TIRFM experiments, the increase in near-plasma membrane EYFP-Stim1 induced by nocodazole was additive with that induced by full store depletion with thapsigargin, indicating that nocodazole and store depletion also influence rearrangement of EYFP-Stim1 by independent mechanisms. In other words, if nocodazole caused formation of EYFP-Stim1 punctae only because of store depletion, then it would be expected that the extent of EYFP-Stim1 punctae formation in response to full store depletion with thapsigargin in nocodazole-treated cells should not be any greater than that seen in cells in which stores are depleted with thapsigargin alone. An alternative interpretation is that EYFP-Stim1 localization is less restricted when microtubules are depolymerized, such that when limited discharge of Ca2+ store mobilizes EYFP-Stim1, more EYFP-Stim1 is able to interact with and activate the SOCE machinery; this effect is not seen in wildtype cells to any appreciable degree because endogenous Stim1 expression is much less than in the EYFP-Stim1 overexpressing cells. This is an interesting possibility since, if true, it implies that the quantitative relationship between Stim1 mobilization and activation of SOCE can be regulated by factors that influence its localization and mobility independently of the degree of Ca2+ store depletion.
In a recent report, Baba et al. (Baba et al. 2006) also documented a dependence of Stim1 localization on microtubules, wherein it was shown that overexpressed Stim1 exhibits a fibrillar organization in DT40 B-lymphocytes that is lost upon treatment with nocodazole. In that study nocodazole-treated cells exhibited unimpaired SOCE; however, the experiments of Baba et al. were performed on Stim1 overexpressing cells and the effect of nocodazole on SOCE in wildtype cells was not reported. Interestingly, Baba et al. (Baba et al. 2006) also reported that a mutant Stim1 lacking the C-terminal, cytoplasmic serine/threonine-rich domain exhibited a localization that was not fibrillar but instead was very similar to ER localization; this is reminiscent of effects we have reported of nocodazole on EYFP-Stim1 and ER localization. Stim1 lacking the serine/threonine-rich region did not rearrange into punctae upon Ca2+ store depletion, and was unable to support SOCE. Thus, this serine/threonine-rich region may be involved in microtubule association of Stim1 as well as in downstream signaling from Stim1 to Orai channels.
An effect of microtubule depolymerization on SOCE and Icrac involving mitochondria transport was reported by Quintana et al. (Quintana et al. 2006). In that study it was shown that mitochondria are recruited toward the plasma membrane in response to initiation of Ca2+ entry, and that these translocated mitochondria act as Ca2+ buffers that attenuate Ca2+ dependent inactivation of SOCE. However, this is unlikely to account for the inhibition that we observed with nocodazole for two reasons. First, in our study nocodazole inhibited Icrac in electrophysiological experiments performed under conditions of high intracellular Ca2+ buffering, such that Ca2+-dependent inactivation is prevented and mitochondrial effects are not generally seen. Second, the nocodazole-induced inhibition of entry could be rescued by EYFP-Stim1 overexpression, a finding that is not consistent with a mitochondrial effect.
Exactly how Stim1 localization and function depend on microtubules is not clear at this time. The major conclusion from this study is that the microtubular cytoskeleton facilitates and perhaps organizes Stim1 movements, but the translocation and aggregation of Stim1 does not absolutely depend upon a microtubular mechanism. What drives this important translocation step in the signaling mechanism for store-operated calcium channels, and how Stim1 acts to open the channels will be topics of future investigations.
Supplementary Material
Acknowledgements
We thank Dr. Patricia Wadsworth and Dr Nasser Rusan for helpful discussions and advice throughout the completion of this study. We also thank Jeff Reece and Jeff Tucker for technical assistance with TIRF and confocal microscopy, and Rebecca Boyles for plasmid preparations. Dr. Steven Shears and Dr. David Miller read the manuscript, and provided helpful comments. This research was supported by the Intramural Program, National Institute of Environmental Health Sciences, NIH.
Reference List
- 1.Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M, Kurosaki T. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc.Nat.Acad.Sci.USA. 2006;103:16704–16709. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakowski D, Glitsch MD, Parekh AB. An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current Icrac in RBL-1 cells. J.Physiol.(Lond.) 2001;532:55–71. doi: 10.1111/j.1469-7793.2001.0055g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bird GS, Putney JW., Jr. Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells. J.Physiol. 2005;562:697–706. doi: 10.1113/jphysiol.2004.077289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bird G. St. J., Obie JF, Putney JW., Jr. Functional homogeneity of the non-mitochondrial Ca2+-pool in intact mouse lacrimal acinar cells. J.Biol.Chem. 1992;267:18382–18386. [PubMed] [Google Scholar]
- 5.DeHaven WI, Smyth JT, Boyles RR, Putney JW. Calcium inhibition and calcium potentiation of orai1, orai2 and orai3 calcium-release-activated calcium channels. J Biol Chem. 2007 doi: 10.1074/jbc.M611374200. in press, doi/10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- 6.Dziadek MA, Johnstone LS. Biochemical properties and cellular localisation of STIM proteins. Cell Calcium. 2007 doi: 10.1016/j.ceca.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Graier WF, Paltauf-Doburzynska J, Hill BJF, Fleischhacker E, Hoebel BG, Kostner GM, Sturek M. Submaximal stimulation of porcine endothelial cells causes focal Ca2+ elevation beneath the cell membrane. J.Physiol.(Lond.) 1998;506:109–125. doi: 10.1111/j.1469-7793.1998.109bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG, Rao A. Biochemical and functional characterization of Orai family proteins. J.Biol.Chem. 2007 doi: 10.1074/jbc.M609630200. M609630200. [DOI] [PubMed] [Google Scholar]
- 9.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr., Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr.Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J.Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercer JC, DeHaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr. Large store-operated calcium-selected currents due to co-expression of orai1 or orai2 with the intracellular calcium sensor, stim1. J.Biol.Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oka T, Hori M, Ozaki H. Microtubule disruption suppresses allergic response through the inhibition of calcium influx in the mast cell degranulation pathway. J Immunol. 2005;174:4584–4589. doi: 10.4049/jimmunol.174.8.4584. [DOI] [PubMed] [Google Scholar]
- 13.Patterson RL, van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- 14.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJS, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quintana A, Schwarz EC, Schwindling C, Lipp P, Kaestner L, Hoth M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J Biol Chem. 2006;281:40302–40309. doi: 10.1074/jbc.M607896200. [DOI] [PubMed] [Google Scholar]
- 16.Ribeiro CMP, Reece J, Putney JW., Jr. Role of the cytoskeleton in calcium signaling in NIH 3T3 cells. An intact cytoskeleton is required for agonist-induced [Ca2+]i signaling, but not for capacitative calcium entry. J.Biol.Chem. 1997;272:26555–26561. doi: 10.1074/jbc.272.42.26555. [DOI] [PubMed] [Google Scholar]
- 17.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J.Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smyth JT, Lemonnier L, Vazquez G, Bird GS, Putney JW., Jr. Dissociation of regulated trafficking of TRPC3 channels to the plasma membrane from their activation by phospholipase C. J.Biol.Chem. 2005;281:11712–11720. doi: 10.1074/jbc.M510541200. [DOI] [PubMed] [Google Scholar]
- 19.Terasaki M, Chen LB, Fujiwara K. Microtubules and the endoplasmic reticulum are highly interdependent structures. J.Cell Biol. 1986;103:1557–1568. doi: 10.1083/jcb.103.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr.Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. The Journal of Cell Biology. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu P, Lu J, Li Z, Yu X, Chen L, Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem.Biophys.Res Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 23.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc.Natl.Acad.Sci U.S.A. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.