

Abstract
Background
Pancreatic cancer is the fourth leading cause of cancer death in the United States. Consequently, identification of clinically relevant biomarkers for the early detection of this cancer type is urgently needed. In recent years, proteomics profiling techniques combined with various data analysis methods have been successfully used to gain critical insights into processes and mechanisms underlying pathologic conditions, particularly as they relate to cancer. However, the high dimensionality of proteomics data combined with their relatively small sample sizes poses a significant challenge to current data mining methodology where many of the standard methods cannot be applied directly. Here, we propose a novel methodological framework using machine learning method, in which decision tree based classifier ensembles coupled with feature selection methods, is applied to proteomics data generated from premalignant pancreatic cancer.
Results
This study explores the utility of three different feature selection schemas (Student t test, Wilcoxon rank sum test and genetic algorithm) to reduce the high dimensionality of a pancreatic cancer proteomic dataset. Using the top features selected from each method, we compared the prediction performances of a single decision tree algorithm C4.5 with six different decision-tree based classifier ensembles (Random forest, Stacked generalization, Bagging, Adaboost, Logitboost and Multiboost). We show that ensemble classifiers always outperform single decision tree classifier in having greater accuracies and smaller prediction errors when applied to a pancreatic cancer proteomics dataset.
Conclusion
In our cross validation framework, classifier ensembles generally have better classification accuracies compared to that of a single decision tree when applied to a pancreatic cancer proteomic dataset, thus suggesting its utility in future proteomics data analysis. Additionally, the use of feature selection method allows us to select biomarkers with potentially important roles in cancer development, therefore highlighting the validity of this method.
Background
Pancreatic cancer is one of the most lethal types of cancer. In United States, there are ~30,000 new cases being diagnosed each year. The mortality rate of pancreatic cancer patients is approaching 100%. Only 4% of the patients survive 5 years or more after being diagnosed. The grim statistics of pancreatic cancer necessitates the urgent development of methods to facilitate their early detection and prevention [1]. Despite the advancement of our knowledge in recent years regarding the pathophysiology of pancreatic cancer [2,3], we still lack an effective method to diagnose this cancer type early enough to impact the treatment outcomes.
Recently, there has been substantial interests in applying proteomics technology to identify clinically useful biomarkers for early-stage pancreatic cancer [4-11]. In a more general sense, many investigators have applied proteomics technology and data mining methods to identify serum proteomic patterns that can distinguish normal from cancer samples. Examples of these include ovarian cancer [12-17], breast cancer [18,19], prostate cancer [20-22], lung cancer [23], brain tumors [24], and head and neck cancer [25].
One of the major challenges for proteomic profiling is the analysis and mining of biologically useful information from the enormous dataset. Due to the high dimensionality of proteomics dataset and their often small sample sizes, non-classical statistical methods for data analysis need to be employed. Therefore, various machine learning classification algorithms have been applied to proteomics data analysis. These include the use of decision tree [26,27], boosted decision tree [28], random forest [29], nearest centroid [30], Bayesian neural network [31], self-organizing map [32], support vector machine [33,34], linear and quadratic discriminant analysis [35] and meta-learners [36,37]. However, there are limitations regarding these studies [38-41]. These include the lack of efficient procedure for biomarker selection and the inability to cope with data noise. More importantly, most of these classification methods were constructed based on a single classifier derived from a single training process. They are not robust enough to handle the great variance inherent in the proteomics data. Thus, a more general machine learning method is needed to overcome these challenges.
Here, we present a computational method to analyze a proteomics dataset obtained from premalignant pancreatic cancer using decision tree based classifier ensembles coupled with three feature selection schemas and show that classifier ensembles always have better performances compared to a single decision tree and other models.
Results
The premalignant pancreatic cancer mass spectrometry dataset used in this study include 181 samples. Of the 181 samples, 101 are control serum samples and 80 are PanIN (pancreatic intraepithelial neoplasias) samples. Control samples are referred to as normal cases while PanIN samples as disease cases. The complete computational procedure used in this study is shown in Figure. 1. After preprocessing, we ran our processed data through a 10 fold cross-validation framework. In each round of the cross validation, 90% of the data were selected randomly as training set to build classifier. Three feature selection methods were applied to select top features (mass to charge ratios, m/z) from the training set only. Classifiers were then tested on the rest of the 10% data using those selected features. The performances of various classifiers were also compared.
Figure 1.

Computation procedure used in this study. In each round of 10 fold cross-validation, the whole dataset was randomly separated into training set and test set. Features that significantly differentiate the control class from the disease class are selected using training set only. Then test sets are classified by decision tree and ensembles using these features. Mass spec: mass spectrometry.
Data preprocessing
To compensate for systematic differences due to sample loadings and instrument errors, raw proteomics data have to be preprocessed before any feature selection method and classification algorithm can be applied. Three major preprocessing procedures were applied to our dataset: baseline adjustment, normalization and kernel smoothing. Using one specific spectrogram as an example, the effects of these processing operations on the raw data are shown in Figure. 2. The original spectrograms consist of 6771 different m/z ratios and they range from 800 to 11992.91 in their values (Figure. 2A). The spectrogram baselines were adjusted based on the group median (Figure. 2B). All data points were smoothed by substituting their values with the weighted average of 5 value points on each side using a Gaussian kernel (Figure. 2C). Using the area under each spectrogram curve (AUG), all spectrograms were normalized and rescaled such that their maximum values equal to 100 (Figure. 2D).
Figure 2.
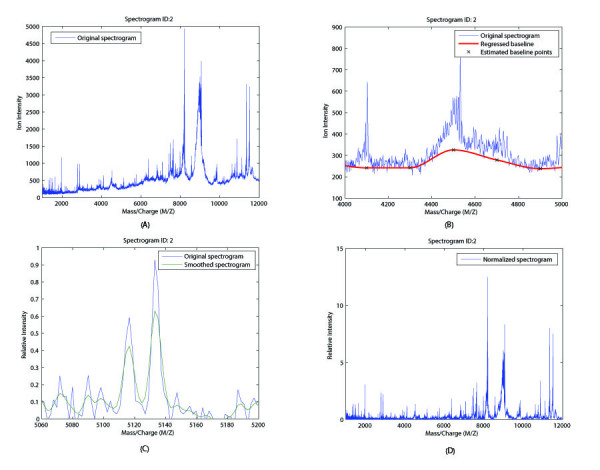
Data preprocessing result. Spectrogram ID 2 is used as an example of data preprocessing procedure. (A) Original spectrogram without any processing. The maximum m/z ratio is 11922.91 and the minimum m/z ratio is 800. (B) Original spectrogram and adjusted baseline. (C) Noise reduction using Gaussian kernel smoothing. (D) Normalization using the area under the curve (AUC).
Biomarker identification
In general, classifiers cannot successfully handle high dimensional dataset generated from proteomics experiments. To overcome this problem, we used three feature selection schemes (Student t test, Wilcoxon rank sum test and genetic algorithm) to reduce the dimensionality of the dataset to a manageable number. Table. 1 lists all the top 10 features generated from each round of two-sample homoscedastic t test. These features are ranked based on their p-values that correspond to the probability of their observed differences in mean intensity between control and disease group being significant. Interestingly, several features (m/z ratios) such as 5798.9 and 5801.2 were repeatedly selected in our 10 rounds of cross validation analyses despite the fact that the training dataset is randomly selected from the whole dataset each time. Regardless of how the data is partitioned, highly significant differences in peptides' m/z intensity between control and disease samples can and will likely be selected each time. Thus, these m/z ratios are considered potentially good biomarkers for disease identification. The use of randomly selected training data provides greater confidence to our results.
Table 1.
Top ten features (m/z ratio) selected by Student t test method in our 10 fold cross validation.
| Rank | Round 1 | Round 2 | Round 3 | Round 4 | Round 5 | Round 6 | Round 7 | Round 8 | Round 9 | Round 10 | Most Frequent |
| 1 | 5798.9 | 5798.9 | 5819.8 | 5798.9 | 5819.8 | 5819.8 | 5798.9 | 5798.9 | 11477 | 5798.9 | 5798.9 |
| 2 | 5801.2 | 5819.8 | 5822.1 | 5801.2 | 5822.1 | 5822.1 | 5801.2 | 11541 | 11774 | 5801.2 | 5801.2 |
| 3 | 5819.8 | 5801.2 | 5798.9 | 5819.8 | 5798.9 | 5798.9 | 5819.8 | 11592 | 11472 | 11592 | 5819.8 |
| 4 | 5796.5 | 5822.1 | 5801.2 | 5822.1 | 5801.2 | 11592 | 5822.1 | 5801.2 | 5798.9 | 11597 | 11541 |
| 5 | 5822.1 | 11541 | 11592 | 5829.1 | 11770 | 11597 | 11541 | 11537 | 11481 | 11587 | 11592 |
| 6 | 11422 | 11592 | 11597 | 5831.4 | 11541 | 11587 | 5831.4 | 11546 | 5819.8 | 11541 | 5822.1 |
| 7 | 5817.4 | 11546 | 11541 | 11592 | 11597 | 5801.2 | 11592 | 11597 | 11770 | 11601 | 11597 |
| 8 | 11774 | 11587 | 11601 | 5803.5 | 11592 | 11541 | 11546 | 11774 | 11514 | 5819.8 | 11546 |
| 9 | 11541 | 11537 | 11546 | 11541 | 11601 | 11643 | 5829.1 | 11587 | 11509 | 11546 | 11601 |
| 10 | 11426 | 11569 | 11639 | 5796.5 | 11606 | 11601 | 11597 | 11601 | 5822.1 | 11606 | 11587 |
Rank is determined by the probability of the two means between disease and control groups in the training set being significantly different. m/z ratios with smaller probability ranks higher. Most frequent features are determined by the frequency of each feature appears in the top 10 list in these ten runs and ranked by their frequency.
While t test assumes that the feature values from two different classes follow normal distributions. In reality, this is often not the case. Therefore, we explored the possibility of using a nonparametric (distribution-free) test to select our top features. The top 10 features selected from Wilcoxon nonparametric rank test are presented in Table 2. Similar to t test, some of the m/z ratios such as 5798.9 and 5801.2 were also selected frequently. Features that are repeatedly selected from independent runs suggest that those features play important roles in discriminating between normal and disease classes.
Table 2.
Top ten features (m/z ratio) selected by Wilcoxon rank test method in our 10 fold cross validation.
| Rank | Round 1 | Round 2 | Round 3 | Round 4 | Round 5 | Round 6 | Round 7 | Round 8 | Round 9 | Round 10 | Most Frequent |
| 1 | 5798.9 | 5798.9 | 4941.6 | 5801.2 | 5822.1 | 5819.8 | 5801.2 | 5798.9 | 4941.6 | 5801.2 | 5798.9 |
| 2 | 5801.2 | 5801.2 | 5819.8 | 5798.9 | 5819.8 | 5822.1 | 5798.9 | 5801.2 | 11774 | 5798.9 | 5801.2 |
| 3 | 4941.6 | 11472 | 5822.1 | 4941.6 | 5798.9 | 5798.9 | 4941.6 | 11472 | 5798.9 | 5796.5 | 11472 |
| 4 | 5796.5 | 5819.8 | 5801.2 | 5819.8 | 5801.2 | 5801.2 | 5803.5 | 11477 | 11770 | 11472 | 5819.8 |
| 5 | 5819.8 | 11477 | 5798.9 | 9706.1 | 11472 | 11592 | 5822.1 | 11774 | 11477 | 5803.5 | 5822.1 |
| 6 | 5822.1 | 5822.1 | 4943.6 | 5822.1 | 11477 | 11587 | 11472 | 11770 | 11472 | 11592 | 11477 |
| 7 | 4943.6 | 11468 | 11592 | 5803.5 | 11468 | 11472 | 5819.8 | 11541 | 5819.8 | 11541 | 11541 |
| 8 | 11472 | 5796.5 | 11541 | 5796.5 | 11770 | 11541 | 11477 | 11537 | 5822.1 | 5819.8 | 4941.6 |
| 9 | 11774 | 11541 | 11472 | 11472 | 4941.6 | 11774 | 5829.1 | 11468 | 5801.2 | 11477 | 11774 |
| 10 | 11477 | 11481 | 11597 | 9710 | 11774 | 5796.5 | 11541 | 11481 | 11481 | 11468 | 5796.5 |
Rank is determined by the probability of the two means between disease and control groups in the training set being significantly different.
To compare with filter feature selection method such as t test and Wilcoxon rank test, we also explored the possible utility of a wrapper method, specifically the use of genetic algorithm coupled with linear discriminant analysis. In contrast to t test and Wilcoxon rank test in which several features were repeatedly selected, genetic algorithm provided a very different result. As shown in Table 3, features selected from each round are quite different, with no feature being selected more than twice in 10 rounds of cross-validation. One possible reason for this result is that the initial population size used by the genetic algorithm was small and that it was randomly selected from the training set. Due to its small population size (10 features in a population), any particular feature is less likely to be selected repeatedly by random sampling.
Table 3.
Ten features (m/z ratio) selected by Genetic algorithm coupled with LDA in our 10 fold cross validation.
| Round 1 | Round 2 | Round 3 | Round 4 | Round 5 | Round 6 | Round 7 | Round 8 | Round 9 | Round 10 |
| 3385 | 4943.6 | 11555 | 1489.9 | 5939.7 | 1859.5 | 5098.2 | 5916 | 9835.1 | 5775.7 |
| 3304.8 | 5775.7 | 1125.2 | 11541 | 5209.5 | 2009.5 | 9578.9 | 2016.7 | 1857.2 | 3833.4 |
| 3186.7 | 4013.8 | 4943.6 | 1644 | 5822.1 | 2951.2 | 3760.5 | 3787.6 | 11940 | 3510.5 |
| 1858.7 | 3915.5 | 3383.7 | 4941.6 | 1063.5 | 11662 | 7553.5 | 5857.1 | 2756.1 | 1857.2 |
| 4256.8 | 1858 | 1528.6 | 5409.1 | 1644.6 | 11546 | 3540.1 | 3727.5 | 1808.1 | 11031 |
| 3790.7 | 1063.1 | 3959.6 | 1936.1 | 1859.5 | 9415.6 | 1860.2 | 1064 | 4532.7 | 5801.2 |
| 4941.6 | 1476.3 | 3726 | 2368.5 | 11463 | 7406.9 | 7966.1 | 5819.8 | 7931.2 | 4318.6 |
| 11027 | 3727.5 | 5829.1 | 3188 | 7592.8 | 6569.5 | 11394 | 9640.4 | 11477 | 11821 |
| 11426 | 11560 | 3188 | 1859.5 | 11472 | 1645.9 | 10183 | 1702.2 | 6511.9 | 5794.2 |
| 7085.3 | 2579.1 | 5949.2 | 3836.4 | 6509.3 | 1411.1 | 9575 | 6506.7 | 4941.6 | 9640.4 |
Classification results and comparisons
After data dimension reduction using methods mentioned above, we tested and compared the performances of a single decision tree algorithm C4.5, six different decision tree-based classifier ensembles, and six different benchmark classification algorithms in a 10 fold cross validation framework. Default parameters were used in all algorithms without any fine-tuning of individual classifier, thus, allowing us to compare the performance of each algorithm. Because no particular classifier is preferred, potential misleading conclusion can be avoided.
Table 4 lists the average performances of all algorithms in a 10-fold cross validation using selected features from t test. In terms of accuracy, all classifier ensembles such as Bagging and Multiboost outperformed single decision tree (64% accurate) or at least had similar results. Moreover, the fact that single decision tree C4.5 has the best prediction sensitivity (0.99) and lowest specificity (0.21) indicates that this model was well adapted to only one class, thus failed to discriminate between both classes. The trade-off between prediction's sensitivity and specificity has been observed in many cases before [42] and was thought to result from the choice of threshold value chosen for making binary predictions. Similar trade-off effect in prediction is also associated with TP (true positive) and FP (false positive) rate.
Table 4.
Classification results using features selected by Student t test.
| Algorithm | Accuracy(%) | TP rate | FP rate | TN rate | FN rate | Sensitivity | Specificity | Precision | Fmeasure | RMSE |
| C4.5 | 0.6444 | 0.99 | 0.79 | 0.21 | 0.01 | 0.99 | 0.21 | 0.61 | 0.76 | 0.4687 |
| Random Forest | 0.6500 | 0.79 | 0.53 | 0.48 | 0.21 | 0.79 | 0.48 | 0.65 | 0.71 | 0.4569 |
| Bagging | 0.6833 | 0.78 | 0.44 | 0.56 | 0.22 | 0.78 | 0.56 | 0.69 | 0.73 | 0.4285 |
| Logitboost | 0.6889 | 0.83 | 0.49 | 0.51 | 0.17 | 0.83 | 0.51 | 0.69 | 0.75 | 0.4402 |
| Stacking | 0.6444 | 0.99 | 0.79 | 0.21 | 0.01 | 0.99 | 0.21 | 0.61 | 0.76 | 0.4761 |
| Adaboost | 0.6444 | 0.77 | 0.51 | 0.49 | 0.23 | 0.77 | 0.49 | 0.69 | 0.69 | 0.4412 |
| Multiboost | 0.6889 | 0.81 | 0.46 | 0.54 | 0.19 | 0.81 | 0.54 | 0.70 | 0.74 | 0.5175 |
| Logistic | 0.7500 | 0.79 | 0.30 | 0.70 | 0.21 | 0.79 | 0.70 | 0.78 | 0.78 | 0.4224 |
| Naivebayes | 0.6833 | 0.64 | 0.26 | 0.74 | 0.36 | 0.64 | 0.74 | 0.76 | 0.68 | 0.5289 |
| Bayesnet | 0.6722 | 0.63 | 0.28 | 0.73 | 0.37 | 0.63 | 0.73 | 0.74 | 0.67 | 0.5308 |
| Neural Network | 0.7000 | 0.70 | 0.30 | 0.70 | 0.30 | 0.70 | 0.70 | 0.75 | 0.72 | 0.4517 |
| RBFnet | 0.6722 | 0.76 | 0.44 | 0.56 | 0.24 | 0.76 | 0.56 | 0.69 | 0.71 | 0.4632 |
| SVM | 0.6944 | 0.71 | 0.33 | 0.68 | 0.29 | 0.71 | 0.68 | 0.74 | 0.71 | 0.5489 |
TP rate: True positive rate, FP rate: False positive rate, TN rate: True negative rate, FN rate: False negative rate, RMSE: Root Mean Squared Error. RBFnet: Radio Basis Function network, SVM: Support Vector Machine.
Besides accuracy, mean squared error of prediction (MSE) is another important measure of performance. MSE is the expected value of the square of "error" and consists of two components – prediction variance and the square of the prediction bias. In many contexts, variance and bias of a single classifier can be effectively reduced by constructing classifier ensemble such as Bagging and Adaboost [43,44]. Our results support this observation. For example, RMSE (Root Mean Squared Error) of single decision tree C4.5 is 0.4687, which is higher than those of Random Forest (0.4569), Bagging (0.4285), Logitboost (0.4402) and Adaboost (0.4412), but interestingly smaller compared to those of Stacked generalization (0.4761) and Multiboost (0.5175) (Table 4). Most of the benchmark algorithms have higher RMSE compared to either single decision tree or classifier ensembles.
Similarly, using our top 10 features selected from the Wilcoxon rank test (Table 5), the prediction accuracy (66.67%) of a single decision tree is lower than those of Random forest, Logitboost and Multiboost, but similar to those from Stacked generalization and Bagging. The trade-off between prediction's sensitivity and specificity still exist for C4.5 and other classifiers. This effect is even more obvious for Stacked generalization. In general, the classification results from t test and Wicoxon rank test have no significant difference, indicating that both feature selection methods work equally well in this context.
Table 5.
Classification results using features selected by Wilcoxon rank test.
| Algorithm | Accuracy(%) | TP rate | FP rate | TN rate | FN rate | Sensitivity | Specificity | Precision | Fmeasure | RMSE |
| C4.5 | 0.6667 | 0.90 | 0.63 | 0.38 | 0.10 | 0.90 | 0.38 | 0.65 | 0.75 | 0.4683 |
| Random Forest | 0.7000 | 0.79 | 0.41 | 0.59 | 0.21 | 0.79 | 0.59 | 0.71 | 0.74 | 0.4401 |
| Bagging | 0.6667 | 0.68 | 0.35 | 0.65 | 0.32 | 0.68 | 0.65 | 0.72 | 0.69 | 0.4484 |
| Logitboost | 0.6833 | 0.76 | 0.41 | 0.59 | 0.24 | 0.76 | 0.59 | 0.70 | 0.73 | 0.4499 |
| Stacking | 0.6667 | 0.93 | 0.66 | 0.34 | 0.07 | 0.93 | 0.34 | 0.64 | 0.76 | 0.4639 |
| Adaboost | 0.6611 | 0.76 | 0.46 | 0.54 | 0.24 | 0.76 | 0.54 | 0.68 | 0.71 | 0.4805 |
| Multiboost | 0.7000 | 0.73 | 0.34 | 0.66 | 0.27 | 0.73 | 0.66 | 0.74 | 0.73 | 0.5187 |
| Logistic | 0.6556 | 0.77 | 0.49 | 0.51 | 0.23 | 0.77 | 0.51 | 0.67 | 0.71 | 0.4362 |
| Naivebayes | 0.6944 | 0.70 | 0.31 | 0.69 | 0.30 | 0.70 | 0.69 | 0.77 | 0.72 | 0.4969 |
| Bayesnet | 0.6778 | 0.73 | 0.39 | 0.61 | 0.27 | 0.73 | 0.61 | 0.71 | 0.71 | 0.5232 |
| Neural Network | 0.6778 | 0.66 | 0.30 | 0.70 | 0.34 | 0.66 | 0.70 | 0.73 | 0.68 | 0.4606 |
| RBFnet | 0.5944 | 0.74 | 0.59 | 0.41 | 0.26 | 0.74 | 0.41 | 0.62 | 0.67 | 0.4556 |
| SVM | 0.6611 | 0.71 | 0.40 | 0.60 | 0.29 | 0.71 | 0.60 | 0.71 | 0.70 | 0.5760 |
In contrast, features selected from the genetic algorithm show large variations compared to those features selected from t test and Wilcoxon rank test. However, it is unclear whether the classification results using genetic algorithm also vary significantly. In our study, we observed a similar pattern in prediction accuracy and RMSE value for genetic algorithm (Table 6). Classifier ensembles usually outperform a single decision tree. For example, a single decision tree has the lowest prediction accuracy (59%) compared to other classifier ensembles. Interestingly, the general performances of classifiers based on the feature selection method of genetic algorithm are considerably lower than those from t test and Wilcoxon rank test, possibly because the heuristic nature of wrapper method can not guarantee that the best features will be selected.
Table 6.
Classification results using features selected by genetic algorithm.
| Algorithm | Accuracy(%) | TP rate | FP rate | TN rate | FN rate | Sensitivity | Specificity | Precision | Fmeasure | RMSE |
| C4.5 | 0.5944 | 0.61 | 0.43 | 0.58 | 0.39 | 0.61 | 0.58 | 0.64 | 0.62 | 0.5718 |
| Random Forest | 0.6000 | 0.71 | 0.54 | 0.46 | 0.29 | 0.71 | 0.46 | 0.63 | 0.66 | 0.5047 |
| Bagging | 0.6111 | 0.64 | 0.43 | 0.58 | 0.36 | 0.64 | 0.58 | 0.66 | 0.65 | 0.4965 |
| Logitboost | 0.6167 | 0.68 | 0.46 | 0.54 | 0.32 | 0.68 | 0.54 | 0.65 | 0.66 | 0.5153 |
| Stacking | 0.6056 | 0.66 | 0.46 | 0.54 | 0.34 | 0.66 | 0.54 | 0.65 | 0.65 | 0.4892 |
| Adaboost | 0.6167 | 0.67 | 0.45 | 0.55 | 0.33 | 0.67 | 0.55 | 0.65 | 0.65 | 0.5960 |
| Multiboost | 0.6111 | 0.68 | 0.48 | 0.53 | 0.32 | 0.68 | 0.53 | 0.65 | 0.66 | 0.6147 |
| Logistic | 0.6056 | 0.67 | 0.48 | 0.53 | 0.33 | 0.67 | 0.53 | 0.63 | 0.65 | 0.5122 |
| Naivebayes | 0.6000 | 0.76 | 0.60 | 0.40 | 0.24 | 0.76 | 0.40 | 0.62 | 0.67 | 0.5251 |
| Bayesnet | 0.5611 | 0.73 | 0.65 | 0.35 | 0.27 | 0.73 | 0.35 | 0.59 | 0.65 | 0.5110 |
| Neural Network | 0.5944 | 0.61 | 0.43 | 0.58 | 0.39 | 0.61 | 0.58 | 0.65 | 0.62 | 0.5814 |
| RBFnet | 0.6000 | 0.69 | 0.51 | 0.49 | 0.31 | 0.69 | 0.49 | 0.63 | 0.65 | 0.5038 |
| SVM | 0.6333 | 0.72 | 0.48 | 0.53 | 0.28 | 0.72 | 0.53 | 0.66 | 0.68 | 0.5985 |
Recently, the area under ROC (Receiver Operating Characteristic) curve (AUG) has been widely used as a measure to compare the performance of different classifiers. Theoretically, AUG value equals the probability of correctly classified one pair of samples (each from one class). Therefore, one classifier is considered better if it has a larger area under the ROC curve compared to a different classifier. Thus, the AUG value under the ROC curve provides another measure of classifier performance. For example, the AUGs of classifiers using t test selected features are summarized in Table 7. Single decision tree C4.5 has the lowest AUG value (0.5625) while Random Forest has the largest AUG value (0.9375) among all classifier tested. These results strongly suggest the need to construct classifier ensembles to analyze proteomics data.
Table 7.
AUG results of classifiers
| Algorithm | AUG | Algorithm | AUG | Algorithm | AUG | Algorithm | AUG |
| C4.5 | 0.5625 | Logitboost | 0.8438 | Bayes Net | 0.8563 | RBFnet | 0.9 |
| Random Forest | 0.9375 | Stacking | 0.5625 | Logistic | 0.925 | SVM | 0.7 |
| Random Tree | 0.825 | Adaboost | 0.85 | Neural Network | 0.85 | ||
| Bagging | 0.85 | Multiboost | 0.875 | Naïve Bayes | 0.8875 |
Discussion
Sensitive detection of clinically useful biomarkers and the building of a reliable predictor specific to pre-malignant pancreatic cancer will certainly aid the early detection of this deadly disease. Here, we propose the use of a more accurate decision tree-based classifier ensembles combined with feature selection methods to address some of the challenges facing current cancer proteomics data analysis. We are able to build a low bias and a low variance predictor using model-averaging method: classifier ensembles. This method greatly improves the accuracy of classification. Furthermore, the use of three feature selection methods have allowed us to select biomarkers that achieve the best classification performance and at the same time give us potential new insights into disease mechanism involved in cancer development.
Biological data sets generated from proteomics studies typically have a very high number of features compared to their small sample sizes. Many feature selection methods have been used in proteomic data analyses to reduce the high dimensionality of the dataset. These include methods such as information gain [37], Kolmogorov-Smirnov test [34] and random forest [35]. In our study, we used three different feature selection methods: t test, Wilcoxon rank test and genetic algorithm. These methods are derived from the two major schemas in feature selection, namely the filter and wrapper method [45]. Filter method is more efficient, reliable, and not subjected to any learning algorithm. However, this method considers each feature independently without regard to its relevance or the possibility that combination of features can improve classifier performance. In contrast, the Wrapper method chooses a particular learning algorithm as its performance guide to consider how useful some feature combinations are to the predictor. In genetic algorithm, the initial size of the population sampled from the whole dataset significantly affects the output result. Because of this, our repeated runs using genetic algorithm failed to yield similar results. The unreliability of genetic algorithm may limit its future utility in proteomics data analysis. Using the three methods mentioned earlier, we observed a generally consistent performance of all classifiers. Their accuracies range from 50% to 70%. Thus, feature selection methods used here are sufficiently robust for classification purpose.
Over the last two decades, intensive explorations of model-averaging methods for classification purposes produce a group of efficient decision tree-based classifier ensembles. In many different contexts, classifier ensembles outperform decision tree model and other single algorithms because of their superior ability to handle data variance. This is also demonstrated in our result. In all three feature selection method cases, classifier ensembles have better prediction accuracies. Meanwhile, many attempts were made to compare classifier ensemble techniques, but most of them only focused on the two most popular methods: Bagging and Adaboost. Although Stacked generalization, Multiboost and Logitboost have been proposed earlier, only recently these methods gained greater popularity in machine learning and bioinformatics community [46-48]. Until now, no direct comparisons of their performances were made. Our study represents the first attempt in this direction by considering them in the context of pancreatic cancer proteomics analysis.
In general, the performances of classifiers tested on the premalignant pancreatic cancer dataset are lower than we had expected, with the best prediction accuracy of 70% in a single run. There are two possible reasons for this. First, this proteomics dataset comes from mice with histologically confirmed premalignant PanIN but no evidence of invasive or metastatic disease [49]. Therefore, in the early developmental stage of pancreatic cancer, the levels of biomarkers may not exhibit significant differences between the normal and disease group. Secondly, we used the default parameters for all our classifiers without performing any fine-tuning. The advantage of doing this is that it can prevent the problem of "over-fitting" because the parameters we used are not adapted to a specific dataset, thus our method can be generalized to more datasets. The disadvantage of using the default parameters is that our result may not represent the best possible results.
Conclusion
We presented a systematic machine learning method to analyze cancer proteomics data that utilized decision tree based classifier ensembles and three popular feature selection schemas in a cross validation framework. Our method includes three steps: preprocessing, feature selection and classification. The proposed method is general enough that it can be adapted to other proteomics data analysis problems. Our results show that classifier ensembles perform significantly better than single decision tree algorithm, highlighting the utility of classifier ensembles in future proteomics research. Additionally, biomarkers selected in this process may shed new lights on processes and mechanisms underpinning cancer development. Our study represents one of the first attempts to apply and compare decision tree based classifier ensembles in the context of cancer proteomics data analysis. Results presented here will open up other possibilities for further research.
Methods
Premalignant pancreatic cancer mass-spectrometry data
Pancreatic cancer peptide mass-spectrometry data was downloaded from the FDA-NCI Clinical Proteomics Program [50]. This dataset was generated from serums of 33 mice (5.5 ± 0.25 months) that carried low-level burdens of human pancreatic intraepithelial neoplasias (PanINs) cells that were induced by endogenous expression of KRASG12D, and 39 age-matched control mice [49]. There are a total of 80 PanINs serum samples that are referred to as disease group and 101 control serum samples that are referred to as control group. For each serum sample, the data stream was binned using a fraction of 400 parts per million (ppm), thus condensed the data from 350,000 to 6771 data points. The m/z ratios range from 800 to 11992.91.
Mass-spectrometry data preprocessing
In general, a typical mass-spectrometry data set contains several thousands of intensity measurements. Many factors such as system artifacts make mass-spectrometry data extremely noisy. Therefore, low-level preprocessing is critical to the success of data analysis [51]. Theoretically, observed mass spectra can be decomposed into three components [52]:
| f(i, j) = b(i, j) + s(i, j) + ε (i, j) | (1) |
where f(i, j) is the observed value, b(i, j) is the baseline value, s(i, j) is the true signal and ε (i, j) is the noise for ith sample at jth m/z ratio. Baseline is considered to be the low frequency component of the observed signal and its variability arises from different sources such as sample ion dispensing, matrix chemical contamination and data collection. This problem is especially significant at low peak intensity because the noise to signal ratio is larger. Some of the baseline correction algorithms are summarized in [53]. To adjust for our baseline problem, we first estimated our baseline by segmenting the whole spectra into windows with a size of 200 m/z ratio intensities. We then used the mean value of these windows as the estimate of baseline value at that intensity [54]. Then a piecewise cubic interpolation method was used to perform regression, thereby avoiding the problem of sharp boundary. This procedure was applied to all spectrograms.
In mass spectrometry data, systematic differences between replicate experiments are often significant enough to prevent the drawing of any meaningful conclusion. To compensate for these systematic differences, we normalized the intensities of all spectrograms from the downloaded dataset. Many normalization methods developed for mass-spectrometry data are available and some of them have been successfully used in previous analysis [55-57]. Area under curves (AUC) which is defined as , where yi is the signal at ith m/z ratio, is used to measure the protein concentration in mass-spectrometry data. In this study, we standardized each spectrum based on the ratio of its area under curves (AUC) over the median calculated from all spectra [57]. Also, the maximum intensities from each spectrogram are rescaled to 100.
Raw mass spectrometry data typically contains signal and random noise introduced by factors such as instrument measurement error. Thus, it is important to reduce the noise in the data to improve the quality of the spectrograms. This enables feature selection schema to select significant features. We adopted a Gaussian kernel smoothing method to reduce the noise in our data. Assuming the signals are generated from a Gaussian distribution, we substitute each original data point value with a weighted average of all samples close to it. Each nearby data point × contributes according to its distance in a Gaussian form. Weights are determined by where dist is the distance between this point and the center point and dmax is the maximum distance of all points and center point. In this work, we used bandwidth 10 to allow a reasonably large distribution such that there will be a five points on each side of the original data point.
Feature selection
Two-sample student t test considers each feature independently. It assumes both groups of data values are distributed normally and have similar variances. Test statistics is calculated as follows:
| (2) |
Where xd and xc are the mean values of intensities from disease group and control group respectively. vard and varc are variances of two distributions. nd and nc are the numbers of instance in each distribution. This t value follows student t distribution with degree of freedom nd + nc-2. The significance p value is calculated based on test statistics and t distribution.
Wilcoxon rank test is a nonparametric test which has no distribution assumption. All the data are ranked together based on their values. Then the ranks from one class are compared with those from the other class. The U statistics is calculated as:
| (3) |
where nd and Rd are the size and sum of ranks in disease samples. An equally valid formula for U is to replace all values from control sample.
Although Wilcoxon rank test is robust against parameter variation and makes no distribution assumption, in situation such as proteomics data where the sample size is small, the P values calculated by Wilcoxon rank test tend to be higher. Therefore, it is not easy to detect statistically real difference. On the other hand, student t test can differentiate between these cases (if the distribution approximates Normal), thus is more powerful than nonparametric test in this context.
The wrapper method used in this study incorporate genetic algorithm as feature space search procedure. Genetic algorithm is a heuristic method. It adopts ideas from the field of evolutionary genetics, such as population, inheritance, cross-over, mutation and selection. Evolution starts from a group of randomly generated feature sets: the initial population. Individual's fitness (performance) is evaluated by a learning algorithm. The mutation process usually selects the individual with good fitness score from the parent population to form the next generation. Normally, the fitness of descendent population is better than their "parent" population. This process continues until the termination condition where fitness reaches maximum. Features in the final population will be reported. Factors such as the initial population, mutation rate, and local maximum can affect the performance of genetic algorithm. In general, genetic algorithm gives reasonably satisfactory result quickly.
Decision tree ensemble algorithms
Decision tree is one of the most popular predictor used in machine learning community and is commonly used as a base learner in constructing classifier ensemble [58]. In decision tree algorithm, the approximated target function is represented as a tree-like structure. In general, it works by sorting down the tree branch from the root to some leaf nodes. Each internal node represents a specific test of instance attribute, and each branch represents one of the possible test results. The classical decision algorithm C4.5 [59] implements a top-down greedy search schema to search through all possible tree spaces. At each split, they try every possible feature to achieve maximum reduction of impurity. Decision tree is efficient, easy to interpret and robust but may suffer from low accuracy and high variance. Thus, many attempts were made to improve it using model averaging method.
Bagging (Bootstrap Aggregating) represents one of the first successful attempts to use model averaging method. It was originally proposed by Leo Breiman [60]. To build ensembles, Bagging repeatedly samples the training set data to form subset with replacement following a uniform probability distribution. Thus in each subset, one instance can appears more than once. One classifier is trained for each newly formed subset. The final classification result is determined by the unweighted votes of each classifier in the committee, thus aggregating all classifiers:
| (4) |
Where H(x) is the final committee vote result, hi is the result from individual classifier.
AdaBoost [61] is similar to Bagging in that both of them resample from the base of the training set. However, one of the major differences between them is that AdaBoost associates a different weight to each instance based on previous classification result. All instances are assigned equal weights at the beginning. After first round of classification, instances that are classified correctly will receive smaller weights in the next round. Instances that are incorrectly classified will have larger weights. By normalizing these weights to form another distribution, AdaBoost will sample from new distribution to train another classifier. The final result is based on weighted sum of all classifiers' results:
| (5) |
Where H(x) is the final prediction result, αi and hi are weight and result from individual predictors.
In the context of binary classification, the above Adaboost algorithm can be called discrete Adaboost. Friedman et al. [62] proved that Boosting algorithm is a stage-wise estimation procedure for fitting additive logistic regression model by minimizing an exponential criterion. This criterion is equivalent to a second order binomial log-likelihood criterion in the Taylor's series. Based on this discovery, they explored the possibility of using Bernoulli log-likelihood criterion, which in turn, called Logitboost. Friedman et al. showed that Logitboost could achieve equivalent level of performance compared to Adaboost [62] using a collection of datasets from the data repository located at University of California (Irvine).
Since the successful introduction of Bagging and Boosting algorithms, many investigators have tried to combine the power of variance reduction from Bagging and bias reduction from Adaboost. One attempt is Multiboost [63]. It tries to combine the benefits offered from both Bagging and Adaboost by exploring Wagging (Weight Aggregating, a variant of bagging), a set of committee formed by Adaboost. It also has an advantage in computation because these committees can learn in parallel. Wagging, which also repeatedly perturb the training data, does not sample the data to form smaller subsets. Instead, it adds noises to all weights such that it no longer assumes a uniform distribution. Thus, wagging take full advantage of the dataset without leaving any of the data unused. Using University of California (Irvine) repository datasets, Multiboost algorithm was frequently shown to achieve lower error than either Bagging or Boosting [63].
Random forest is another type of tree ensemble [64]. It can be considered as Bagging with random feature selection. In the forest, each tree is built using a bootstrap sample of the data. Candidate feature set is selected randomly at all tree splits. Randomness in the algorithm guarantees that low level of correlation between trees. In addition, each tree grows fully without any pruning. Thus, both variance and bias reduction can be achieved at the same time.
All the above algorithms have only one level in that all parallel trees take the original input data and provide one output prediction result. In contrast, Stacking [65] is a method that combines multiple level models for classification. Usually, there are two levels of classifiers. The first level classifiers are trained on the original input data, and their outputs are collected into a new dataset. This new dataset in turn serves as an input data for a second level learning algorithm that produce the final result. Many different combinations of level one and level two classifiers have been tested. Ting and Witten [66] showed that they can achieve the least error rate compared to other classifiers using output class probability together with least squares linear regression as their second level generalizer.
Also, bench mark algorithms such as Logistic Regression, Naïve Bayes, Bayes Net, Neural Network, Radial Basis Function Network and Support Vector Machine are also used in this study for the purpose of comparison.
Classifier performance measurement
Ten fold cross validation is known to be a good estimator of classifier's performance. Ten percent of the data from the whole dataset are chosen randomly as test set while the remaining 90 percent are used as training set. This process is repeated 10 times and the average result is reported. In each run, prediction results can be classified into four groups: TP (true positive), FP (false positive), TN (true negative) and FN (false negative). Thus
And MSE (Mean Squared Error) can be decomposed as
| (6) |
Where is the estimator of parameter θ.
There is always a trade-off between sensitivity and specificity because of the different threshold values used in binary prediction. Thus, ROC (Receiver Operating Characteristic) curve is used to plot true/false positive rates or sensitivity/1-specificity for different thresholds. The area under the ROC curve (AUG) equals the probability of correctly classified one pair of samples, each one from a separate class. It has been used as an important measurement of classifier performance. A classifier is considered a preferred classifier compared to the other classifier if it has a larger AUG value. A random classifier has an area of approximately 0.5 under the ROC graph, whereas a perfect classifier has an area of 1.
Weka machine learning package was used in the simulation of classifier ensembles [67]. We also used Matlab statistics and bioinformatics toolbox in the data preprocessing and feature selections.
Authors' contributions
GG conceived of the study and carried out the computational experiment, GG and GWW drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We thank our anonymous reviewers for their valuable suggestions and corrections.
Contributor Information
Guangtao Ge, Email: guge@eecs.tufts.edu.
G William Wong, Email: gwwong@jhmi.edu.
References
- Lowenfels AB, Maisonneuve P. Epidemiology and prevention of pancreatic cancer. Jpn J Clin Oncol. 2004;34:238–244. doi: 10.1093/jjco/hyh045. [DOI] [PubMed] [Google Scholar]
- Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- Jafari M, Abbruzzese JL. Pancreatic cancer: future outlook, promising trials, newer systemic agents, and strategies from the Gastrointestinal Intergroup Pancreatic Cancer Task Force. Surg Oncol Clin N Am. 2004;13:751–60, xi. doi: 10.1016/j.soc.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Gronborg M, Bunkenborg J, Kristiansen TZ, Jensen ON, Yeo CJ, Hruban RH, Maitra A, Goggins MG, Pandey A. Comprehensive proteomic analysis of human pancreatic juice. J Proteome Res. 2004;3:1042–1055. doi: 10.1021/pr0499085. [DOI] [PubMed] [Google Scholar]
- Crnogorac-Jurcevic T, Gangeswaran R, Bhakta V, Capurso G, Lattimore S, Akada M, Sunamura M, Prime W, Campbell F, Brentnall TA, Costello E, Neoptolemos J, Lemoine NR. Proteomic analysis of chronic pancreatitis and pancreatic adenocarcinoma. Gastroenterology. 2005;129:1454–1463. doi: 10.1053/j.gastro.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Rosty C, Goggins M. Identification of differentially expressed proteins in pancreatic cancer using a global proteomic approach. Methods Mol Med. 2005;103:189–197. doi: 10.1385/1-59259-780-7:189. [DOI] [PubMed] [Google Scholar]
- Chen R, Pan S, Yi EC, Donohoe S, Bronner MP, Potter JD, Goodlett DR, Aebersold R, Brentnall TA. Quantitative proteomic profiling of pancreatic cancer juice. Proteomics. 2006;6:3871–3879. doi: 10.1002/pmic.200500702. [DOI] [PubMed] [Google Scholar]
- Scarlett CJ, Smith RC, Saxby A, Nielsen A, Samra JS, Wilson SR, Baxter RC. Proteomic classification of pancreatic adenocarcinoma tissue using protein chip technology. Gastroenterology. 2006;130:1670–1678. doi: 10.1053/j.gastro.2006.02.036. [DOI] [PubMed] [Google Scholar]
- Cecconi D, Donadelli M, Rinalducci S, Zolla L, Scupoli MT, Scarpa A, Palmieri M, Righetti PG. Proteomic analysis of pancreatic endocrine tumor cell lines treated with the histone deacetylase inhibitor trichostatin A. Proteomics. 2007;7:1644–1653. doi: 10.1002/pmic.200600811. [DOI] [PubMed] [Google Scholar]
- Mikuriya K, Kuramitsu Y, Ryozawa S, Fujimoto M, Mori S, Oka M, Hamano K, Okita K, Sakaida I, Nakamura K. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int J Oncol. 2007;30:849–855. [PubMed] [Google Scholar]
- Zhou L, Lu Z, Yang A, Deng R, Mai C, Sang X, Faber KN, Lu X. Comparative proteomic analysis of human pancreatic juice: methodological study. Proteomics. 2007;7:1345–1355. doi: 10.1002/pmic.200600086. [DOI] [PubMed] [Google Scholar]
- Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, Liotta LA. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–577. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- Alexe G, Alexe S, Liotta LA, Petricoin E, Reiss M, Hammer PL. Ovarian cancer detection by logical analysis of proteomic data. Proteomics. 2004;4:766–783. doi: 10.1002/pmic.200300574. [DOI] [PubMed] [Google Scholar]
- Conrads TP, Fusaro VA, Ross S, Johann D, Rajapakse V, Hitt BA, Steinberg SM, Kohn EC, Fishman DA, Whitely G, Barrett JC, Liotta LA, Petricoin EF, 3rd, Veenstra TD. High-resolution serum proteomic features for ovarian cancer detection. Endocr Relat Cancer. 2004;11:163–178. doi: 10.1677/erc.0.0110163. [DOI] [PubMed] [Google Scholar]
- Posadas EM, Davidson B, Kohn EC. Proteomics and ovarian cancer: implications for diagnosis and treatment: a critical review of the recent literature. Curr Opin Oncol. 2004;16:478–484. doi: 10.1097/00001622-200409000-00012. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yip C, Ying Y, Wang J, Meng XY, Lomas L, Yip TT, Fung ET. Mass spectrometric analysis of protein markers for ovarian cancer. Clin Chem. 2004;50:1939–1942. doi: 10.1373/clinchem.2004.036871. [DOI] [PubMed] [Google Scholar]
- Wang H, Kachman MT, Schwartz DR, Cho KR, Lubman DM. Comprehensive proteome analysis of ovarian cancers using liquid phase separation, mass mapping and tandem mass spectrometry: a strategy for identification of candidate cancer biomarkers. Proteomics. 2004;4:2476–2495. doi: 10.1002/pmic.200300763. [DOI] [PubMed] [Google Scholar]
- Neubauer H, Fehm T, Schutz C, Speer R, Solomayer E, Schrattenholz A, Cahill MA, Kurek R. Proteomic expression profiling of breast cancer. Recent Results Cancer Res. 2007;176:89–120. doi: 10.1007/978-3-540-46091-6_9. [DOI] [PubMed] [Google Scholar]
- Belluco C, Petricoin EF, Mammano E, Facchiano F, Ross-Rucker S, Nitti D, Maggio CD, Liu C, Lise M, Liotta LA, Whiteley G. Serum Proteomic Analysis Identifies a Highly Sensitive and Specific Discriminatory Pattern in Stage 1 Breast Cancer. Ann Surg Oncol. 2007. [DOI] [PubMed]
- Ornstein DK, Rayford W, Fusaro VA, Conrads TP, Ross SJ, Hitt BA, Wiggins WW, Veenstra TD, Liotta LA, Petricoin EF., 3rd Serum proteomic profiling can discriminate prostate cancer from benign prostates in men with total prostate specific antigen levels between 2.5 and 15.0 ng/ml. J Urol. 2004;172:1302–1305. doi: 10.1097/01.ju.0000139572.88463.39. [DOI] [PubMed] [Google Scholar]
- Petricoin EF, Ornstein DK, Liotta LA. Clinical proteomics: Applications for prostate cancer biomarker discovery and detection. Urol Oncol. 2004;22:322–328. doi: 10.1016/j.urolonc.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Wagner M, Naik DN, Pothen A, Kasukurti S, Devineni RR, Adam BL, Semmes OJ, Wright GL., Jr. Computational protein biomarker prediction: a case study for prostate cancer. BMC Bioinformatics. 2004;5:26. doi: 10.1186/1471-2105-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso P, Catala M, Rico-Morales ML, Durante-Rodriguez G, Moro-Rodriguez E, Fernandez-Garcia H, Escribano JM, Alvarez-Fernandez E, Garcia-Poblete E. Proteomic analysis of lung biopsies: Differential protein expression profile between peritumoral and tumoral tissue. Proteomics. 2004;4:442–447. doi: 10.1002/pmic.200300647. [DOI] [PubMed] [Google Scholar]
- Schwartz SA, Weil RJ, Johnson MD, Toms SA, Caprioli RM. Protein profiling in brain tumors using mass spectrometry: feasibility of a new technique for the analysis of protein expression. Clin Cancer Res. 2004;10:981–987. doi: 10.1158/1078-0432.CCR-0927-3. [DOI] [PubMed] [Google Scholar]
- Roesch-Ely M, Nees M, Karsai S, Ruess A, Bogumil R, Warnken U, Schnolzer M, Dietz A, Plinkert PK, Hofele C, Bosch FX. Proteomic analysis reveals successive aberrations in protein expression from healthy mucosa to invasive head and neck cancer. Oncogene. 2007;26:54–64. doi: 10.1038/sj.onc.1209770. [DOI] [PubMed] [Google Scholar]
- Adam BL, Qu Y, Davis JW, Ward MD, Clements MA, Cazares LH, Semmes OJ, Schellhammer PF, Yasui Y, Feng Z, Wright GL., Jr. Serum protein fingerprinting coupled with a pattern-matching algorithm distinguishes prostate cancer from benign prostate hyperplasia and healthy men. Cancer Res. 2002;62:3609–3614. [PubMed] [Google Scholar]
- Vlahou A, Schorge JO, Gregory BW, Coleman RL. Diagnosis of Ovarian Cancer Using Decision Tree Classification of Mass Spectral Data. J Biomed Biotechnol. 2003;2003:308–314. doi: 10.1155/S1110724303210032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Adam BL, Yasui Y, Ward MD, Cazares LH, Schellhammer PF, Feng Z, Semmes OJ, Wright GL., Jr. Boosted decision tree analysis of surface-enhanced laser desorption/ionization mass spectral serum profiles discriminates prostate cancer from noncancer patients. Clin Chem. 2002;48:1835–1843. [PubMed] [Google Scholar]
- Izmirlian G. Application of the random forest classification algorithm to a SELDI-TOF proteomics study in the setting of a cancer prevention trial. Ann N Y Acad Sci. 2004;1020:154–174. doi: 10.1196/annals.1310.015. [DOI] [PubMed] [Google Scholar]
- Levner I. Feature selection and nearest centroid classification for protein mass spectrometry. BMC Bioinformatics. 2005;6:68. doi: 10.1186/1471-2105-6-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Chen XW. Bayesian neural network approaches to ovarian cancer identification from high-resolution mass spectrometry data. Bioinformatics. 2005;21 Suppl 1:i487–i494. doi: 10.1093/bioinformatics/bti1030. [DOI] [PubMed] [Google Scholar]
- Ning K, Ng HK, Leong HW. PepSOM: an algorithm for peptide identification by tandem mass spectrometry based on SOM. Genome Inform. 2006;17:194–205. [PubMed] [Google Scholar]
- Li L, Tang H, Wu Z, Gong J, Gruidl M, Zou J, Tockman M, Clark RA. Data mining techniques for cancer detection using serum proteomic profiling. Artif Intell Med. 2004;32:71–83. doi: 10.1016/j.artmed.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Yu JS, Ongarello S, Fiedler R, Chen XW, Toffolo G, Cobelli C, Trajanoski Z. Ovarian cancer identification based on dimensionality reduction for high-throughput mass spectrometry data. Bioinformatics. 2005;21:2200–2209. doi: 10.1093/bioinformatics/bti370. [DOI] [PubMed] [Google Scholar]
- Wu B, Abbott T, Fishman D, McMurray W, Mor G, Stone K, Ward D, Williams K, Zhao H. Comparison of statistical methods for classification of ovarian cancer using mass spectrometry data. Bioinformatics. 2003;19:1636–1643. doi: 10.1093/bioinformatics/btg210. [DOI] [PubMed] [Google Scholar]
- Bhanot G, Alexe G, Venkataraghavan B, Levine AJ. A robust meta-classification strategy for cancer detection from MS data. Proteomics. 2006;6:592–604. doi: 10.1002/pmic.200500192. [DOI] [PubMed] [Google Scholar]
- Geurts P, Fillet M, de Seny D, Meuwis MA, Malaise M, Merville MP, Wehenkel L. Proteomic mass spectra classification using decision tree based ensemble methods. Bioinformatics. 2005;21:3138–3145. doi: 10.1093/bioinformatics/bti494. [DOI] [PubMed] [Google Scholar]
- Wulfkuhle JD, Liotta LA, Petricoin EF. Proteomic applications for the early detection of cancer. Nat Rev Cancer. 2003;3:267–275. doi: 10.1038/nrc1043. [DOI] [PubMed] [Google Scholar]
- Diamandis EP. Mass spectrometry as a diagnostic and a cancer biomarker discovery tool: opportunities and potential limitations. Mol Cell Proteomics. 2004;3:367–378. doi: 10.1074/mcp.R400007-MCP200. [DOI] [PubMed] [Google Scholar]
- White CN, Chan DW, Zhang Z. Bioinformatics strategies for proteomic profiling. Clin Biochem. 2004;37:636–641. doi: 10.1016/j.clinbiochem.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Baggerly KA, Morris JS, Edmonson SR, Coombes KR. Signal in noise: evaluating reported reproducibility of serum proteomic tests for ovarian cancer. J Natl Cancer Inst. 2005;97:307–309. doi: 10.1093/jnci/dji008. [DOI] [PubMed] [Google Scholar]
- Lashner BA. Sensitivity-specificity trade-off for capsule endoscopy in IBD: is it worth it? Am J Gastroenterol. 2006;101:965–966. doi: 10.1111/j.1572-0241.2006.00513.x. [DOI] [PubMed] [Google Scholar]
- Quinlan JR. Bagging, Boosting and C4.5. AAAI/IAAI. 1996;1 [Google Scholar]
- Todorovski L, Dzeroski S. Combining Classifiers with Meta Decision Trees. Machine Learning. 2003;50:223–249. doi: 10.1023/A:1021709817809. [DOI] [Google Scholar]
- Guyon I, Elisseeff A. An Introduction to Variable and Feature Selection. Journal of Machine Learning Research. 2003;3:1157–1182. doi: 10.1162/153244303322753616. [DOI] [Google Scholar]
- Cai YD, Feng KY, Lu WC, Chou KC. Using LogitBoost classifier to predict protein structural classes. J Theor Biol. 2006;238:172–176. doi: 10.1016/j.jtbi.2005.05.034. [DOI] [PubMed] [Google Scholar]
- Wang SQ, Yang J, Chou KC. Using stacked generalization to predict membrane protein types based on pseudo-amino acid composition. J Theor Biol. 2006;242:941–946. doi: 10.1016/j.jtbi.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Zhang G, Fang B. LogitBoost classifier for discriminating thermophilic and mesophilic proteins. J Biotechnol. 2007;127:417–424. doi: 10.1016/j.jbiotec.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- Website http://home.ccr.cancer.gov/ncifdaproteomics/ppatterns.asp
- Baggerly KA, Morris JS, Wang J, Gold D, Xiao LC, Coombes KR. A comprehensive approach to the analysis of matrix-assisted laser desorption/ionization-time of flight proteomics spectra from serum samples. Proteomics. 2003;3:1667–1672. doi: 10.1002/pmic.200300522. [DOI] [PubMed] [Google Scholar]
- Coombes KR, Baggerly KA, Morris JS. Pre-Processing Mass Spectrometry Data. In: Dubitzky W, Granzow M and Berrar DP, editor. Fundamentals of Data Mining in Genomics and Proteomics. 1. Springer; 2006. p. 282. [Google Scholar]
- Andrade L, Manolakos E. Signal background estimation and baseline correction algorithms for accurate DNA sequencing. Journal of VLSI Signal Processing. 2003. pp. 229–243.
- Brady D, Kocic M, Miller AW, Karger BL. A maximum-likelihood base caller for DNA sequencing. IEEE Trans Biomed Eng. 2000;47:1271–1280. doi: 10.1109/10.867962. [DOI] [PubMed] [Google Scholar]
- Wang W, Zhou H, Lin H, Roy S, Shaler TA, Hill LR, Norton S, Kumar P, Anderle M, Becker CH. Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal Chem. 2003;75:4818–4826. doi: 10.1021/ac026468x. [DOI] [PubMed] [Google Scholar]
- Marcuson R, Burbeck SL, Emond RL, Latter GI, Aberth W. Normalization and reproducibility of mass profiles in the detection of individual differences from urine. Clin Chem. 1982;28:1346–1348. [PubMed] [Google Scholar]
- Fung ET, Enderwick C. ProteinChip clinical proteomics: computational challenges and solutions. Biotechniques. 2002;Suppl:34–8, 40-1. [PubMed] [Google Scholar]
- Mitchell TM. Machine Learning. McGraw-Hill; 1997. [Google Scholar]
- Quinlan JR. C4.5: Programs for Machine Learning. Morgan Kaufmann; 1993. [Google Scholar]
- Breiman L. Bagging Predictors. Machine Learning. 1996;24:123–140. [Google Scholar]
- Schapire RE. A Brief Introduction to Boosting. 1999. pp. 1401–1406.
- Friedman J, Hastie T, Tibshirani R. Additive Logistic Regression: a Statistical View of Boosting. Technical Report, Department of Statistics, Standford University. 1998. pp. 1–45.
- Webb GI. MultiBoosting: A Technique for Combining Boosting and Wagging. Machine Learning. 2000;40:159–196. doi: 10.1023/A:1007659514849. [DOI] [Google Scholar]
- Breiman L. Random Forests. Machine Learning. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Wolpert DH. Stacked Generalization. Neural networks. 1992;5:241–259. doi: 10.1016/S0893-6080(05)80023-1. [DOI] [Google Scholar]
- Ting KM, Witten IH. Stacked Generalization: when does it work? International Joint Conference on Artificial Intelligence. 1997. pp. 866–873.
- Witten IH, Frank E. Data Mining: Practical Machine Learning Tools and Techniques with Java Implementations. Morgan Kaufmann; 1999. [Google Scholar]