

Abstract
The innate immune systems of humans and other animals are activated by lipopolysaccharides (LPS), which are glucosamine-based phospholipids that form the outer leaflet of the outer membranes of Gram-negative bacteria. Activation involves interactions of LPS with the innate immunity-receptor comprised of toll-like receptor 4 in complex with so-called MD-2 protein and accessory proteins, such as CD14 and LPS binding protein. The Lipid Metabolites and Pathways Strategy (LIPID MAPS) Consortium has isolated in large amounts a nearly homogeneous LPS, Kdo2-Lipid A, and demonstrated that it activates macrophages via toll-like receptor 4. The active form of LPS, monomer or aggregate, is controversial. We have therefore examined the aggregation behavior and other physical properties of Kdo2-Lipid A. Differential scanning calorimetry of Kdo2-Lipid A suspensions revealed a gel-to-liquid crystalline phase transition at 36.4°C (Tm). The nominal critical aggregation concentration, determined by dynamic light scattering, was found to be 41.2 ± 1.6 nM below the Tm (25°C), but only 8.1 ± 0.3 nM above the Tm (37°C). The specific molecular volume of Kdo2-Lipid A, obtained by densitometry measurements was found to be 3159 ± 71 Å3 at 25°C, from which the number of molecules in each aggregate was estimated to be 5.8 × 105. The aggregation behavior of Kdo2-Lipid A in the presence of lipoprotein-deficient serum suggests that Re LPS monomers and multimers are the active units for the immune system in the CD14-dependent and -independent pathways, respectively.
INTRODUCTION
Lipopolysaccharide (LPS) is an amphiphilic glucosamine-based phospholipid found on the outer leaflet of the outer membranes of most Gram-negative bacteria (1,2). LPS activates the immune system in mammals and is considered to play a key role in human septic shock syndrome (1,3), one of the major causes of death in U.S. intensive-care units (4). Recent studies on the mechanism of LPS-induced immunostimulation have significantly advanced our understanding of protein linkage in the signaling pathway (5–7) and focused attention on the roles of monomeric and multimeric forms of LPS in immune activation (8–10). Here we report the results of a study of the aggregation behavior and other properties of an exceptionally pure monomolecular LPS species known as Kdo2-Lipid A, which has recently been shown to activate macrophages via TLR-4 (11).
LPS first associates with LPS binding protein (LBP), an acute-phase protein in serum, and is then transferred to the complex of toll-like receptor 4 (TLR-4) and its co-receptor MD-2 with or without the help of CD14, which is a glycosylphosphatidylinositol-linked protein expressed on the macrophage surface (mCD14) that also exists in a soluble form in serum (sCD14) (12–14). The binding of LPS to TLR-4/MD-2 complex initiates transmembrane signaling via Toll-interleukin 1 receptors, such as myeloid differentiation primary response protein (MyD88), MyD88-like adaptor, TIR-domain-containing adaptor-inducing interferon-β (TRIF), and TRIF-related adaptor molecule. The resulting signals lead to the production of proinflammatory cytokines, including tumor necrosis factor (TNF) and interferon-β (13,15). Depending on the type of recognition, mCD14-dependent or -independent, downstream signaling can be switched between MyD88-dependent and -independent pathways (13,16).
The chemical structure of LPS varies among different species of bacteria, with each species expressing a number of structurally heterogeneous LPSs (17–21). A feature common to the LPS molecules is a hydrophilic polysaccharide attached to a hydrophobic endotoxic principle termed Lipid A. Wild-type bacterial strains express LPS composed of a nonrepeating core oligosaccharide and a distal polysaccharide (O-antigen). LPS of so-called rough mutant strains do not have the O-antigen, but, instead, only 2–15 core oligosaccharides (22), which are characterized by chemotypes with decreasing lengths of the core sugar, known as Ra, Rb, Rc, Rd, and Re (19). The amphiphilic nature of LPS promotes the formation of aggregates above a critical concentration. Because of the high solubility of the oligosaccharide moiety, the critical concentration can be in the nanomolar range (10).
The answer to the fundamental question of whether the active form of LPS is a monomer or a multimer has been controversial. Takayama et al. (10) reported that a disaggregated form of Re LPS from Escherichia coli was more active in stimulating 70Z/3 cells (a LPS-responsive murine pre-B cell line) than the aggregated form. Also, they showed that monomeric LPS induced production of Egr-1 mRNA from thioglycolate-elicited murine peritoneal macrophages 1000-fold more efficiently than aggregated LPS. Furthermore, monomeric Re LPS from E. coli was 179-fold more active than aggregates in the Limulus amebocyte lysate assay (9). On the other hand, Mueller et al. found that Re LPS and Lipid A of E. coli displayed their activities only in multimeric form in the Limulus amebocyte lysate assay and proinflammatory production of TNF-α from human mononuclear cells (8).
The aggregation behaviors of various LPS species have been examined by means of fluorescence spectroscopy (23,24), light scattering (25), and equilibrium dialysis (9,10,23,26) (see review by Brandenburg et al. (27)). The most complete and accurate study is that of Santos et al. (25), who used light scattering to determine the aggregation behavior of a wild-type LPS from E. coli serotype 026:B6. Their work revealed that 026:B6 progresses through three states as LPS concentration is increased: monomers; preaggregate oligomers; and large aggregates (Fig. 1 A). The hydrodynamic radii of preaggregate oligomers and large aggregates were determined to be 60 nm and 95 nm, respectively, but as for all previous studies, the LPS they used was chemically heterogeneous in terms of the length and composition of the terminal glycan chains (1,17,18,22).
FIGURE 1.

The aggregation states of LPS and the structure of Kdo2-Lipid A. (a) Schematic illustration of morphological change of LPS as a function of concentration. The critical concentrations for the transitions between monomer-preaggregate oligomer and between preaggregate oligomer-large aggregate are defined as CMC and CMCa, respectively. (b) Chemical structure of Kdo2-Lipid A, which has the same general structure as Re LPS from E. coli. Native bacteria generally produce structurally heterogeneous LPS, but Kdo2-Lipid A is a homogeneous LPS isolated by the LIPID MAPS Consortium (11).
Recently, a large-scale methodology for preparation of an ultra-pure and chemically homogeneous LPS with two core sugars (Kdo2-Lipid A), one that has the same general structure as the Re LPS from E. coli (Fig. 1 B), has been reported by the LIPID MAPS Consortium (11) (www.lipidmaps.org). We report here the temperature-dependent change in the transition between monomeric and preaggregate oligomer phase of Kdo2-Lipid A, determined using dynamic light scattering. We also determined the partial specific volume of Kdo2-Lipid A from which we estimated the aggregation number of the preaggregate oligomers. Finally, by comparing the multimerization behavior of Kdo2-Lipid A in the presence of lipoprotein-depleted serum with published immunostimulation data, we surmise that Re LPS monomers and multimers are likely the active units for the immune system in the mCD14-dependent and mCD14-independent pathways, respectively.
MATERIALS AND METHODS
Materials
Pure Kdo2-Lipid A was prepared as the tetra-ammonium salt (>99%) by Avanti Polar Lipids (Alabaster, AL) for the LIPID MAPS project. The lipid is commercially available from Avanti. Lipoprotein-deficient fetal bovine serum (FBS), which contains <5% of normal lipoproteins, was obtained from Sigma Chemical (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM) and Dulbecco's PBS (DPBS) buffer without Ca2+ and Mg2+ were purchased from HyClone (Logan, UT). All chemicals were used as purchased without further purification.
Sample preparation
For differential scanning calorimetry (DSC) and dynamic light-scattering experiments, Kdo2-Lipid A was hydrated in DPBS buffer (pH 7.2) to make a 10 mM suspension. The suspension was homogenized by vortexing, followed by 10 freeze (−78°C)/thaw (65°C) cycles. The lipid sample was then extruded with a thermobarrel extruder (Northern Lipids, Vancouver, British Columbia, Canada) 10 times through two stacked 0.2 μm Nucleopore polycarbonate membranes (Whatman, Florham Park, NJ) under N2 pressure of 250 psi at 65°C (28,29). This extruded suspension of Kdo2-Lipid A (10 mM) was proved to be large unilamellar vesicles by using the quenching system with 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) and p-xylene-bis-pyridinium bromide (30–32). The fluorescent ANTS was quenched by p-xylene-bis-pyridinium bromide within Kdo2-Lipid A vesicles, but the leakage of vesicle contents induced by a detergent (Triton X-100) caused an increase in ANTS fluorescence due to dilution into the extravesicular space (data not shown). Previous reports on the aggregation behavior of LPS revealed that the transition from the preaggregate oligomer phase to the large aggregate phase occurs in the micromolar range (23,25). Hence, this fluorophore-leakage result at 10 mM demonstrates that Kdo2-Lipid A exists as vesicles in the large aggregate phase.
Lipid concentrations of the extruded lipid were determined according to the procedure of Bartlett (33). Samples for DSC measurements were prepared by diluting the extruded stock dispersion with DPBS buffer (pH 7.2). For light scattering experiments, we used two procedures for preparation of solutions/dispersions of Kdo2-Lipid A at various concentrations to assure that data were collected for Kdo2-Lipid A in reversible states. In the first (dilution method), several concentrations were made by diluting the extruded stock dispersion with prefiltered (0.2 μm pore) DPBS buffer (pH 7.2) or DMEM with 0.5% (v/v) lipoprotein-deficient FBS. In the second (addition method), appropriate amounts of the stock solution of Kdo2-Lipid A (60 μg/mL) in chloroform-methanol (2:3 v/v) was added to a vial. After evaporation of the solvent under a stream of nitrogen, an appropriate volume of prefiltered DPBS (pH 7.2) was added to the residual pellet and vortexed for 5 min.
For densitometry experiments, Kdo2-Lipid A was carefully weighed on a Mettler H54 balance, and suspended in water. The suspension was homogenized by vortexing, followed by 10 freeze (−78°C)/thaw (65°C) cycles.
DSC measurements
DSC measurements were performed on a MicroCal VP-DSC MicroCalorimeter (Northampton, MA). Three scans were carried out between 5 and 60°C with a scanning rate of 0.2°C/min, and the data of the third scan were collected. DSC data were corrected by subtraction of suitable control data for determination of the gel-to-liquid-crystalline transition temperature.
Dynamic light scattering measurements
Dynamic light scattering measurements at room temperature (∼25°C) and 37°C were performed on a Wyatt Technology (Santa Barbara, CA) DAWN EOS (laser wave length: 690 nm) and a Wyatt Technology DAWN HELEOS (laser wave length: 658 nm), respectively. Samples (1 mL) were injected into an internal flow cell for 15 min and the residual flow allowed to stop. Samples were then equilibrated at each temperature for at least 2 h before data collection. Three measurements were made for each sample at each temperature. The measured intensity of scattered light from each sample was normalized to that of DPBS buffer (pH 7.2) or DMEM. The mean decay rate Γ was determined by fitting the intensity-intensity correlation function g(2)(τ) to the equation
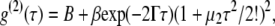 |
where B is the baseline, β is a factor that depends on the experimental geometry, and μ2 is the variance of distribution of Γ (34,35). The averaged hydrodynamic radius (Rh) was determined from Γ by using the Stokes-Einstein equation
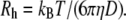 |
The diffusion constant D was calculated using
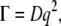 |
in which
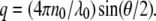 |
In this equation, n0 is the refractive index of the solvent, λ0 is the wavelength of the laser, θ is the scattering angle, kB is the Boltzmann constant, T is the absolute temperature, and η is the viscosity of the solvent. Size distribution analyses were performed with the software ASTRA V (Wyatt Technology).
Densitometry measurements
Density of the lipid solution (dsolution) was determined with a DMA 10 digital precision density meter (Anton Paar, Graz, Austria) by measuring the natural frequency of a hollow oscillator. For calibration of the densitometer, air (dair = 1.184 × 10−3 g/cm3, 25°C, 1 atm) and water (dwater = 0.99707 g/cm3, 25°C, 1 atm) were used. Measurements were performed three times at room temperature. The specific density of the lipid (dL) can be written as
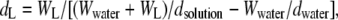 |
where WL and Wwater are the masses of the lipid and water, respectively. Specific molecular volume of the lipid (VL) was determined from dL from
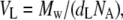 |
where Mw is the molecular weight of the lipid and NA is Avogadro's number.
RESULTS
Fig. 2 shows the DSC heating trace of Kdo2-Lipid A. In addition to a main peak at 36.4°C (Tm), which corresponds to the gel-to-liquid crystalline phase transition, another minor peak was observed as a shoulder at 33.2°C.
FIGURE 2.

DSC heating trace of Kdo2-Lipid A (1 mM) in DPBS (pH 7.2). The relatively sharp peak is consistent with the purity and chemical homogeneity of Kdo2-Lipid A. The principal phase transition temperature (Tm) was determined to be 36.4°C. The minor peak at 33.2°C that appears as a shoulder of the main peak may be due to a pretransition of the alkyl chains.
The aggregation behavior of Kdo2-Lipid A prepared by both the dilution and addition methods (see Materials and Methods) was examined by dynamic light scattering. Dynamic light scattering data from the samples were analyzed by the cumulants method (Fig. 3), which provides the hydrodynamic radius (Rh) and a parameter that correlates with the polydispersity of the solution (μ2) (the higher μ2, the higher the polydispersity). As described in the report by Santos et al. (25), aggregation can be characterized by the increase in hydrodynamic radius and/or the decrease in polydispersity parameter. Fig. 3 shows that μ2 increases over the same range (<200 nM) regardless of the preparation method. Changes in Rh at <200 nM reveal that the transition of Kdo2-Lipid A from the monomeric phase to the preaggregate oligomer phase occurred below 200 nM at room temperature (Fig. 4 a). The aggregation process in this concentration range was found to be highly cooperative by size distribution analyses of the dynamic light scattering data. At concentrations of 20 and 50 nM, monomers (Rh < ∼1 nm) coexisted with intermediate oligomers, whose sizes increased as a function of concentration from Rh∼4 nm to ∼30 nm. (Fig. 4, b and c). At 100 nM, the monomer peak disappeared from distribution histogram (Fig. 4 d). The radius of the steady-state preaggregate oligomers of Kdo2-Lipid A is ∼73 nm (Fig. 4 a), which is only slightly smaller than the nominal radius of the LUV aggregates (∼100 nm). The size of large unilamellar vesicles could not be determined directly by our light scattering instrument, because the concentration at which vesicles were prepared (10 mM; see Materials and Methods) was so high that strongly scattered light from Kdo2-Lipid A was beyond the observable range.
FIGURE 3.

Change in variance of distributions (μ2) at room temperature (∼25°C) of Kdo2-Lipid A prepared by the dilution and addition methods. The data for the dilution and addition methods are depicted by solid and open circles, respectively. Aggregation is indicated by the decrease in the polydispersity parameter (25). These data show that aggregation of Kdo2-Lipid A is independent of method of preparation.
FIGURE 4.

Aggregation behavior of Kdo2-Lipid A. (a) Hydrodynamic radii (Rh; solid circles, left axis) and aggregation numbers (N; open circles, right axis) of Kdo2-Lipid A in DPBS (pH 7.2) at different concentrations at room temperature (∼25°C). The letters refer to neighboring panels. Below 200 nM Kdo2-Lipid A, Rh increased accompanying the transformation of monomers into preaggregate oligomers. The steady-state radius of preaggregate oligomers was 73 nm. Compared with Fig. 3, the multimerizing range (<200 nM) revealed by the change of Rh was consistent with that shown by μ2. Aggregation numbers were calculated from the hydrodynamic radii and the specific molecular volume of Kdo2-Lipid A (3159 ± 71 Å3) obtained by densitometry measurements. The aggregation number of the preaggregate oligomers in steady state was determined to be 5.8 × 105. (b–d) Size-distribution analysis histograms that demonstrate particle-size distributions during aggregation at room temperature. The histograms at concentrations 20 nM, 50 nM, and 100 nM are depicted in panels b, c, and d, respectively. These data reveal that the aggregation process of Kdo2-Lipid A is highly cooperative: monomers coexist with intermediate oligomers whose sizes increase as a function of concentration.
To determine the approximate number of Kdo2-Lipid A molecules in the preaggregates, we determined the specific molecular volume (VL) of Kdo2-Lipid A in water. A general method for determining absolute specific volumes of lipids is the neutral density procedure, carried out using H2O/D2O mixtures (36–40). But this method was not appropriate for Kdo2-Lipid A, because the lipid was found to be denser than D2O (dD2O[25°C] = 1.10445 g/cm3). We therefore used a digital density meter to obtain the specific density (dL) of the lipid. The numerical values of dL and VL were determined to be 1.21 ± 0.03 g/cm3 and 3159 ± 71 Å3, respectively, at room temperature. Assuming that Kdo2-Lipid A is approximately spherical, we estimated the apparent radius Rmonomer 0.91 ± 0.01 nm. Assuming that the preaggregate oligomers of Kdo2-Lipid A are micelles or multilamellar vesicles, we calculated from Rmonomer the aggregation number (N) of steady-state preaggregate oligomers at room temperature to be ∼5.8 × 105 (Fig. 4 a). Rmonomer was also used to obtain simulated sigmoidal curves combined with the dynamic light scattering data, shown in Fig. 5.
FIGURE 5.

Sigmoidal light scattering curves of Kdo2-Lipid A in DPBS (pH 7.2) at 25°C and 37°C. These curves were obtained from the dynamic light scattering data and densitometry measurements. The transition between monomers and preaggregate oligomers at 37°C began at a 10-fold lower concentration than at 25°C: ∼0.3 nM at 37°C and ∼3 nM at 25°C. The concentrations at which half-multimerization occurs (C50s) were determined to be 41.2 ± 1.6 nM (25°C) and 8.1 ± 0.3 nM (37°C). The radius of the preaggregate oligomers in steady state (37°C) was 95 nm.
Because biological experiments (e.g., cell stimulation) are usually carried out at the physiological temperature of 37°C, we compared the light scattering curves of Kdo2-Lipid A at 25 and 37°C (Fig. 5). Although Rmonomer of Kdo2-Lipid A at 37°C is probably larger than at 25°C because of greater disorder of the hydrocarbon chains, the 25°C value of Rmonomer was used for sigmoidal fitting, because the difference in the monomer size at different temperatures should be negligible compared to the size change accompanying multimerization. At 37°C, the concentration at which half-multimerization occurs (C50) is 8.1 ± 0.3 nM, which is lower than at 25°C (41.2 ± 1.6 nM). Furthermore, the radii of preaggregate oligomers were 23% larger (Rh = 95 nm) than those at 25°C.
The measurements at 37°C provide a starting point for examining the question of whether the active form of LPS is a monomer or a multimer in cell stimulation experiments. This is a complicated issue, because such experiments are carried out in media containing blood serum, which is rich in lipoproteins. These lipoproteins create two problems. First, they strongly scatter light and, second, they strongly adsorb LPS (41). (Only LPSs not bound to lipoproteins can stimulate the immune system (42).) As a first step, we carried out dynamic light scattering measurements of Kdo2-Lipid A in the presence of lipoprotein-deficient serum. Fig. 6 shows the multimerization behavior of Kdo2-Lipid A in Dulbecco's modified Eagle's medium (DMEM) in the presence of 0.5% (v/v) fetal bovine serum (FBS) that lacks lipoproteins at 37°C. This FBS concentration minimizes the intense light scattering from numerous FBS proteins, such as albumin and globulin, which readily mask light scattering due to Kdo2-Lipid A. The apparent Rh of the lipoprotein-deficient FBS without Kdo2-Lipid A was found to be ∼5.4 nm. Surprisingly, multimerization in the presence of lipoprotein-deficient FBS occurred at higher concentration than in DPBS buffer: Kdo2-Lipid A began to form multimers at ∼6 nM and C50 was 292 ± 29 nM (Fig. 6).
FIGURE 6.

Hydrodynamic radii (Rh) of Kdo2-Lipid A in DMEM containing 0.5% (v/v) fetal bovine serum (FBS) deficient in lipoproteins at different concentrations at 37°C. These data show that multimerization begins at ∼6 nM and that the C50 was 292 ± 29 nM in the presence of FBS. The interaction of Kdo2-Lipid A with serum components such as LPS binding protein (LBP) and sCD14 probably shifted the critical multimerization concentration (CMC) higher compared to that in DPBS shown in Fig. 5.
DISCUSSION
The natural structural heterogeneity of LPS generally produced by bacteria has hampered accurate physical characterization of the lipids. The availability of Kdo2-Lipid A has made it possible for us to examine, for the first time to our knowledge, the physical properties of a chemically homogeneous LPS. DSC thermograms obtained from Re LPSs from Salmonella minnesota serotype Re595 and E. coli showed very broad peaks (43,44) because of the chemical heterogeneity. The DSC thermogram of Kdo2-Lipid A heating scan, on the other hand, was composed of sharp peaks due to the purity and structural homogeneity of the lipid (Fig. 2). The main transition temperature (Tm) is consistent with the reported values for β↔α transitions of various LPSs, which lie in the range 30–40°C (27). We did not perform any structural analyses to characterize the minor transition at 33.2°C, but considering that the fatty acyl chains of Kdo2-Lipid A are composed of five 14:0 chains and one 12:0 chain (Fig. 1 B), the minor peak may correspond to the gel (Lβ′)-rippled gel (Pβ′) transition (pretransition) of hydrocarbon chains, because phosphatidylcholines with 13:0 or longer acyl chains are known to have pretransitions below the gel (Pβ′)-to-liquid crystalline phase (Lα) transition temperature (45,46). The close proximity of the Lβ′-Pβ′ and Pβ′-Lα peaks of Kdo2-Lipid A suggest that the free energy differences between the Lβ′, Pβ′, and Lα phases are quite small.
LPS monomers are transformed into preaggregate oligomers above a critical concentration (CMC), and at higher concentrations (CMCa) the aggregates form other supramolecular aggregates, which can be multilamellar or nonlamellar depending on the physicochemical environment (23,25,27) (Fig. 1 A). CMCa values of various types of LPS lie in the μM concentration range: 3.2 μM (8 μg/mL) for Re LPS from S. minnesota serotype Re595 (23) and 5.6 μM (14 μg/mL) for wild-type LPS from E. coli serotype 026:B6 (25). We carried out dynamic light scattering measurements in submicromolar range (10–800 nM). Hence, our data shown here demonstrate the multimerization behavior of Kdo2-Lipid A between monomeric phase and preaggregate oligomer phase around the CMC. The size of vesicles formed above the CMCa of Kdo2-Lipid A could not be determined directly by our light scattering instrument, because the concentration at which vesicles form (10 mM; see Materials and Methods) was so high that light scattering from Kdo2-Lipid A was outside the observable range.
If our light scattering experiments were carried out on Kdo2-Lipid A in reversible equilibrated states, then the aggregation behavior of Kdo2-Lipid A as a function of concentration should be identical independent of sample preparation procedure. This was found to be the case. Fig. 3 shows that preparation of the samples by the dilution and addition methods give identical results; multimerization of Kdo2-Lipid A occurred in the identical range (<200 nM) regardless of the preparation procedure. The radius of preaggregate oligomers of Kdo2-Lipid A obtained at room temperature (Fig. 4) is 22% larger than that of wild-type LPS from E. coli serotype 026:B6 at 37°C reported by Santos et al. (25).
As far as we can establish, Fig. 5 presents the first demonstration that the transition between monomers and preaggregate oligomers depends upon temperature. At 37°C, the transition began at a 10-fold lower concentration than at 25°C: ∼0.3 nM at 37°C and ∼3 nM at 25°C. This result differs from that of equilibrium dialysis data: the solubilities of Re LPS from E. coli at 22 and 37°C were almost constant at ∼30 nM (10). However, determination of solubility by equilibrium dialysis does not directly measure multimerization, but rather the maximum concentration at which LPS can be present as monomers (10).
Direct interpretation of immunostimulation data using biophysical LPS multimerization data has been difficult, because the conditions under which physical and cell stimulation experiments are performed are usually different. For example, separation of monomeric and multimeric LPS by means of dialysis has been performed in water or buffer (8,9), while cell stimulation data were collected in cell-culture medium containing blood serum. This raises the possibility that the critical aggregation concentration in culture medium could be different due to the binding of LPS to serum components such as LBP, sCD14, and lipoproteins (5,41). LBP and sCD14 are known to transport LPS to the TLR-4/MD-2 complex and to lipoproteins (42). The LPS that binds to TLR-4/MD-2 complex generates inflammatory responses, but the lipoprotein-bound LPS loses its biological activity (42). The LPS added to serum, therefore, exists as immunostimulative lipoprotein-unbound form or detoxified lipoprotein-bound form. The ratio of the numbers of these two forms has been reported to change depending on time after incorporation into the immune system: LPS incubated with normal human plasma for <10 min accelerated activation of polymorphonuclear leukocytes, while a longer period of incubation (>40 min) resulted in a decrease in stimulation (47). This observation suggests that unbound LPS is the predominant form until LPS is gradually transferred to lipoproteins. This means that conclusions about the active form of LPS should be based upon the aggregation behavior of LPS in the presence of serum before the binding to lipoproteins. Fig. 6 demonstrates that the multimerization in the presence of lipoprotein-deficient FBS occurred at 36-fold higher concentration than in DPBS buffer, probably due to the interaction of Kdo2-Lipid A with LBP and sCD14. Thus one expects that, for FBS with full lipoprotein content, multimerization will occur at an even higher concentration.
There are two modes of LPS recognition by the TLR-4/MD-2 complex: mCD14-dependent and -independent recognition (13,14). Depending on the type of recognition, downstream signaling pathways can be switched between MyD88-dependent and -independent pathways (13,16). Recently, Gangloff et al. (48) examined the TNF-α response of thioglycolate-elicited peritoneal macrophages from normal mice or mCD14-deficient mice, after stimulation with various types of LPS in the presence of 1% autologous serum. They found that murine macrophages with mCD14 started to produce TNF-α in the presence of ∼0.04 nM of Re LPS from E. coli at 37°C, whereas macrophages without mCD14 did not start TNF-α production until the concentration of the lipid increased as much as ∼4 nM (48). Our results, shown by the curve in Fig. 6, suggest that the multimeric form of Re LPS may exist before transfer to lipoproteins in the presence of 0.5% (v/v) FBS if the LPS concentrations are higher than ∼6 nM. Although the conditions under which our light scattering measurements were performed are not exactly the same as for the cell stimulation experiments of Gangloff et al., Fig. 6 is consistent with the idea that Re LPS monomers stimulate the mCD14-dependent pathway and Re LPS multimers stimulate the mCD14-independent pathway.
Acknowledgments
We thank Michael Myers for his editorial assistance and the TEMPO group for useful discussions.
This work was supported by the LIPID MAPS Large-Scale Collaborative Grant No. GM-069338 from the National Institute of Health.
Editor: Thomas J. McIntosh.
References
- 1.Raetz, C. R. H., and C. Whitfield. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikaido, H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glauser, M. P., G. Zanetti, J.-D. Baumgartner, and J. Cohen. 1991. Septic shock: pathogenesis. Lancet. 338:732–736. [DOI] [PubMed] [Google Scholar]
- 4.Parrillo, J. E., M. M. Parker, C. Natanson, A. F. Suffredini, R. L. Danner, R. E. Cunnion, and F. P. Ognibene. 1990. Septic shock in humans: advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann. Intern. Med. 113:227–242. [DOI] [PubMed] [Google Scholar]
- 5.Ulevitch, R. J., and P. S. Tobias. 1995. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu. Rev. Immunol. 13:437–457. [DOI] [PubMed] [Google Scholar]
- 6.Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, and S. Akira. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide. Evidence for TLR4 as the LPS gene product. J. Immunol. 162:3749–3752. [PubMed] [Google Scholar]
- 7.Poltorak, A., X. He, I. Smirnova, M.-Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton, and B. Beutler. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in TLR4 gene. Science. 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 8.Mueller, M., B. Lindner, S. Kusumoto, K. Fukase, A. B. Schromm, and U. Seydel. 2004. Aggregates are the biologically active units of endotoxin. J. Biol. Chem. 279:26307–26313. [DOI] [PubMed] [Google Scholar]
- 9.Takayama, K., D. H. Mitchell, Z. Z. Din, P. Mukerjee, C. Li, and D. L. Coleman. 1994. Monomeric Re lipopolysaccharide from Escherichia coli is more active than the aggregated form in the Limulus amebocyte lysate assay and in inducing Egr-1 m-RNA in murine peritoneal macrophages. J. Biol. Chem. 269:2241–2244. [PubMed] [Google Scholar]
- 10.Takayama, K., Z. Z. Din, P. Mukerjee, P. H. Cooke, and T. N. Kirkland. 1990. Physicochemical properties of the lipopolysaccharide unit that activates B lymphocytes. J. Biol. Chem. 265:14023–14029. [PubMed] [Google Scholar]
- 11.Raetz, C. R. H., T. A. Garrett, C. M. Reynolds, W. A. Shaw, J. D. Moore, D. C. Smith, Jr., A. A. Ribeiro, R. C. Murphy, R. J. Ulevitch, C. Fearns, D. Reichart, C. K. Glass, C. Benner, S. Subramaniam, R. Harkewicz, R. C. Bowers-Gentry, M. W. Bucyzynski, J. A. Cooper, R. A. Deems, and E. A. Dennis. 2006. Kdo2-lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J. Lipid Res. 47:1097–1111. [DOI] [PubMed] [Google Scholar]
- 12.Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell. 124:783–801. [DOI] [PubMed] [Google Scholar]
- 13.Godowski, P. J. 2005. A smooth operator for LPS responses. Nat. Immunol. 6:544–546. [DOI] [PubMed] [Google Scholar]
- 14.Huber, M., C. Kalis, S. Keck, Z. Jiang, P. Georgel, X. Du, L. Shamel, S. Sovath, S. Mudd, B. Beutler, C. Galanos, and M. A. Freudenberg. 2006. R-form LPS, the master key to the activation of TLR4/MD-2-positive cells. Eur. J. Immunol. 36:701–711. [DOI] [PubMed] [Google Scholar]
- 15.Pålsson-McDermott, E. M., and L. A. J. O'Neill. 2004. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 113:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang, Z., P. Georgel, X. Du, L. Shamel, S. Sovath, S. Mudd, M. Huber, C. Kalis, S. Keck, C. Galanos, M. Freudenberg, and B. Beutler. 2005. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6:565–570. [DOI] [PubMed] [Google Scholar]
- 17.Peterson, A. A., A. Haug, and E. J. McGroarty. 1986. Physical properties of short- and long-O-antigen-containing fractions of lipopolysaccharide from Escherichia coli 0111:B4. J. Bacteriol. 165:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson, A. A., and E. J. McGroarty. 1985. High-molecular weight components in lipopolysaccharides of Salmonella typhimurium, Salmonella minnesota, and Escherichia coli. J. Bacteriol. 162:738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkinson, S. G. 1996. Bacterial lipopolysaccharides—themes and variations. Prog. Lipid Res. 35:283–343. [DOI] [PubMed] [Google Scholar]
- 20.Miller, S. I., R. K. Ernst, and M. W. Bader. 2005. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 3:36–46. [DOI] [PubMed] [Google Scholar]
- 21.Erridge, C., E. Bennett-Guerrero, and I. R. Poxton. 2002. Structure and function of lipopolysaccharides. Microbes Infect. 4:837–851. [DOI] [PubMed] [Google Scholar]
- 22.Brade, H., S. M. Opal, S. N. Vogel, and D. C. Morrison, editors. 1999. Endotoxin in Health and Disease. Marcel Dekker, New York.
- 23.García-Verdugo, I., F. Sánchez-Barbero, K. Soldau, P. S. Tobias, and C. Casals. 2005. Interaction of SP-A (surfactant protein A) with bacterial rough lipopolysaccharide (Re-LPS), and effects of SP-A on the binding of Re-LPS to CD14 and LPS-binding protein. Biochem. J. 391:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aurell, C. A., and A. O. Wistrom. 1998. Critical aggregation concentrations of Gram-negative bacterial lipopolysaccharides (LPS). Biochem. Biophys. Res. Commun. 253:119–123. [DOI] [PubMed] [Google Scholar]
- 25.Santos, N. C., A. C. Silva, M. A. R. B. Castanho, J. Martins-Silva, and C. Saldanha. 2003. Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. ChemBioChem. 4:96–100. [DOI] [PubMed] [Google Scholar]
- 26.Din, Z. Z., P. Mukerjee, M. Kastowsky, and K. Takayama. 1993. Effect of pH on solubility and ionic state of lipopolysaccharide obtained from the deep rough mutant of Escherichia coli. Biochemistry. 32:4579–4586. [DOI] [PubMed] [Google Scholar]
- 27.Brandenburg, K., J. Andrä, M. Müller, M. H. J. Koch, and P. Garidel. 2003. Physicochemical properties of bacterial glycopolymers in relation to bioactivity. Carbohydr. Res. 338:2477–2489. [DOI] [PubMed] [Google Scholar]
- 28.Mayer, L. D., M. J. Hope, and P. R. Cullis. 1986. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta. 858:161–168. [DOI] [PubMed] [Google Scholar]
- 29.Allende, D., and T. J. McIntosh. 2003. Lipopolysaccharides in bacterial membranes act like cholesterol in eukaryotic plasma membranes in providing protection against melittin-induced bilayer lysis. Biochemistry. 42:1101–1108. [DOI] [PubMed] [Google Scholar]
- 30.Ellens, H., J. Bentz, and F. C. Szoka. 1984. pH-induced destabilization of phosphatidylethanolamine- containing liposomes: role of bilayer contact. Biochemistry. 23:1532–1538. [DOI] [PubMed] [Google Scholar]
- 31.Ladokhin, A. S., M. E. Selsted, and S. H. White. 1997. Bilayer interactions of indolicidin, a small antimicrobial peptide rich in tryptophan, proline, and basic amino acids. Biophys. J. 72:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wimley, W. C., M. E. Selsted, and S. H. White. 1994. Interactions between human defensins and lipid bilayers: evidence for the formation of multimeric pores. Protein Sci. 3:1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartlett, G. R. 1959. Phosphorus assay in column chromatography. J. Biol. Chem. 234:466–468. [PubMed] [Google Scholar]
- 34.Patty, P. J., and B. J. Frisken. 2003. The pressure-dependence of the size of extruded vesicles. Biophys. J. 85:996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frisken, B. J. 2001. Revisiting the method of cumulants for the analysis of dynamic light-scattering data. Appl. Opt. 40:4087–4091. [DOI] [PubMed] [Google Scholar]
- 36.Greenwood, A. I., S. Tristram-Nagle, and J. F. Nagle. 2006. Partial molecular volumes of lipids and cholesterol. Chem. Phys. Lipids. 143:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koenig, B. W., and K. Gawrisch. 2005. Specific volumes of unsaturated phosphatidylcholines in the liquid crystalline lamellar phase. Biochim. Biophys. Acta. 1715:65–70. [DOI] [PubMed] [Google Scholar]
- 38.Mannock, D. A., T. J. McIntosh, X. Jiang, D. F. Covey, and R. N. McElhaney. 2003. Effects of natural and enantiomeric cholesterol on the thermotropic phase behavior and structure of egg sphingomyelin bilayer membranes. Biophys. J. 84:1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petrache, H. I., S. Tristram-Nagle, K. Gawrisch, D. Harries, V. A. Parsegian, and J. F. Nagle. 2004. Structure and fluctuations of charged phosphatidylserine bilayers in the absence of salt. Biophys. J. 86:1574–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiener, M. C., S. Tristram-Nagle, D. A. Wilkinson, L. E. Campbell, and J. F. Nagle. 1988. Specific volumes of lipids in fully hydrated bilayer dispersions. Biochim. Biophys. Acta. 938:135–142. [DOI] [PubMed] [Google Scholar]
- 41.Kitchens, R. L., P. A. Thompson, R. S. Munford, and G. E. O'Keefe. 2003. Acute inflammation and infection maintain circulating phospholipid levels and enhance lipopolysaccharide binding to plasma lipoproteins. J. Lipid Res. 44:2339–2348. [DOI] [PubMed] [Google Scholar]
- 42.Miyake, K. 2004. Innate recognition of lipopolysaccharide by Toll-like receptor 4-MD-2. Trends Microbiol. 12:186–192. [DOI] [PubMed] [Google Scholar]
- 43.Koch, P.-J., J. Frank, J. Schüler, C. Kahle, and H. Bradaczek. 1999. Thermodynamics and structural studies of the interaction of Polymyxin B with deep rough mutant lipopolysaccharides. J. Colloid Interface Sci. 213:557–564. [DOI] [PubMed] [Google Scholar]
- 44.Garidel, P., M. Rappolt, A. B. Schromm, J. Howe, K. Lohner, J. Andrä, M. H. J. Koch, and K. Brandenburg. 2005. Divalent cations affect chain mobility and aggregate structure of lipopolysaccharide from Salmonella minnesota reflected in a decrease of its biological activity. Biochim. Biophys. Acta. 1715:122–131. [DOI] [PubMed] [Google Scholar]
- 45.Koynova, R., and M. Caffrey. 1998. Phases and phase transitions of the phosphatidylcholines. Biochim. Biophys. Acta. 1376:91–145. [DOI] [PubMed] [Google Scholar]
- 46.Lewis, R. N. A. H., N. Mak, and R. N. McElhaney. 1987. A differential scanning calorimetric study of the thermotropic phase behavior of model membranes composed of phosphatidylcholines containing linear saturated fatty acyl chains. Biochemistry. 26:6118–6126. [DOI] [PubMed] [Google Scholar]
- 47.Wurfel, M. M., S. T. Kunitake, H. Lichenstein, J. P. Kane, and S. D. Wright. 1994. Lipopolysaccharide (LPS)-binding protein is carried on lipoproteins and acts as a cofactor in the neutralization of LPS. J. Exp. Med. 180:1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gangloff, S. C., U. Zähringer, C. Blondin, M. Guenounou, J. Silver, and S. M. Goyert. 2005. Influence of CD14 on ligand interactions between lipopolysaccharide and its receptor complex. J. Immunol. 175:3940–3945. [DOI] [PubMed] [Google Scholar]