

Abstract
Amyloid β-peptides (Aβ) applied directly from solution to model lipid membranes produced dramatic changes in the material properties of the bilayer when certain oxysterols were present in the bilayer. These effects were dependent on both lipid and peptide composition, and occurred at peptide concentrations as low as 100 nM. Using micropipette manipulation of giant unilamellar vesicles, we directly measured the lysis tension of lipid bilayers of various compositions. The glycerophospholipid 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) constituted the main lipid component at 70 mol %. The remaining 30 mol % was composed of the following pure or mixed sterols: cholesterol (CHOL), 7-ketocholesterol (KETO), or 7β-hydroxycholesterol (OHCHOL). SOPC/CHOL bilayers did not exhibit significant changes in mechanical properties after exposure to either Aβ(1–42) or Aβ(1–40). Partial substitution of CHOL with KETO (5 mol %), however, caused a drastic reduction of the lysis tension after exposure to Aβ(1–42) but not to Aβ(1–40). Partial substitution of CHOL with OHCHOL (5 mol %) caused a drastic reduction of the lysis tension after exposure to Aβ(1–40) and to Aβ(1–42). We attribute these effects to the reduction in intermolecular cohesive interactions caused by the presence of the second dipole of oxysterols, which reduces the energetic barrier for Aβ insertion into the bilayer.
INTRODUCTION
The view of the role of amyloid β-peptide (Aβ) in the etiology of Alzheimer's disease (AD) has undergone significant revision within the last two decades. Focus has shifted from the neurotoxicity associated with the senile plaques composed of aggregated Aβ (the amyloid cascade hypothesis (1)) to the activity of the peptide itself well before the onset of aggregation. Acting monomerically or in small oligomers (2), Aβ has been demonstrated to produce a wide range of effects on model membranes and cells in culture (neuronal, PC12, neuroblastoma, and endothelial); however, no single pathological mechanism has emerged. Indeed, it has become increasingly clear that the neurotoxicity of Aβ may derive from any of a number of divergent yet concurrent pathways including the following: 1), increased oxidative stress via the Aβ-promoted production of reactive oxygen species (3–7); 2), Aβ-induced stimulation of the inflammatory response (8,9); 3), membrane depolarization resulting from the formation of structured cation-selective channels by membrane-spanning aggregates of the peptide (10–14); 4), induction of necrosis (15) or apoptosis (16,17); and 5), nonspecific membrane destabilization (18,19).
The insertion of a peptide into a lipid bilayer is coupled to the work of creating a vacancy in the membrane; this work is dependent on characteristics of the peptide and on the membrane material properties (such as bilayer elasticity) that, in turn, are dependent on the bilayer lipid composition (20). Experimental evidence largely confirms a lipid composition dependence of interactions between Aβ and the lipid bilayer matrix of the cell membrane, although the data are sometimes contradictory. For example, electrostatic interactions between either Aβ(1–40) or Aβ(1–42) and anionic lipid headgroups at acidic pH have been reported to be the primary mediators of the initial interaction between peptide and bilayer, with phosphatidylglycerol, cholesterol (CHOL), and ganglioside all reported to mediate surface binding of Aβ (21–25). Kremer et al. (26,27), however, have reported that specific electrostatic interactions are not required for Aβ-bilayer interactions, citing the indifference of steady-state fluorescence anisotropy measurements on anionic, cationic, and zwitterionic headgroups. Their findings indicated that, regardless of the surface charge of the target bilayer, the transmembrane region of Aβ(1–40) localized in the hydrophobic core and the peptide reduced membrane fluidity in a manner that correlated with the degree of peptide aggregation (26,27). These results suggest the importance of intrabilayer interactions and membrane fluidity in influencing the activity of Aβ. In addition, Curtain et al. (28) reported that 20 mol % of CHOL in mixed phosphatidylcholine and phosphatidylserine bilayers appeared to inhibit peptide insertion.
As we will show in this article, we have demonstrated that Aβ applied directly to model lipid membranes produces dramatic changes in the material properties of the bilayer when certain oxysterols are present. These effects are dependent on both lipid and peptide composition, and occur at lower peptide concentrations (100 nM or less) than has previously been reported in the literature, to our knowledge.
MATERIALS AND METHODS
Lipids and peptides
The glycerophospholipid 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) and CHOL were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. We obtained 3β-hydroxy-5-cholesten-7-one (7-ketocholesterol, KETO) and 5-cholestene-3β,7β-diol (7β-hydroxycholesterol, OHCHOL) from Sigma-Aldrich (St. Louis, MO). Phosphate buffered saline (PBS), its component salts (NaCl, KCl, KH2PO4, and Na2HPO4), sucrose, and bovine serum albumin (BSA, essentially fatty acid-free) also were from Sigma-Aldrich. The PBS was prepared according to a standard preparation protocol (Current Protocols in Cell Biology (1998) A.2A.5). Buffer pH and osmolarity were measured and adjusted to maintain pH 7.4 and isoosmolarity with the vesicle suspensions (see below). All solutions were filtered through 0.2-μm filters (Whatman, Florham Park, NJ) to eliminate large particulate impurities. Two variants of Aβ were obtained from Calbiochem (San Diego, CA): Aβ(1–42) (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA) and Aβ(1–40) (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV). Peptides were obtained as lyophilized powders and used without purification. Stock aliquots of peptide were prepared at 10 μM in ultrapure water (Milli-Q system; Millipore, Billerica, MA) with a resistivity of 18 MΩ cm, frozen at −80°C, and thawed only once, immediately before each experiment.
The activity of Aβ at the level of the cell membrane (both in vivo and in vitro) has been widely reported to depend on the aggregation state of the peptide (26,29,30). We therefore developed a formalism for the consistent preparation and handling of stock solutions of Aβ. Due to the potential membrane perturbative effects of commonly used peptide solvents such as dimethylsulfoxide, acetonitrile, hexafluoroisopropanol, and trifluoroacetic acid, we chose to freeze Aβ as stock aliquots in ultrapure water (18.2 MΩ cm resistivity; Millipore) that were thawed, diluted in aqueous buffer, and used immediately before each experiment. Peptide aggregation in stock preparations was assessed with a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) Western blot assay using 4G8, which is a mouse monoclonal antibody to Aβ residues 17–24 (β amyloid (17–24) mAb Hu; BioSource, Camarillo, CA) (31). Separation was conducted on Bis-Tris gel (NuPAGE Novex Bis-Tris Gel; Invitrogen, Carlsbad, CA) according to the manufacturer's protocols. In this manner, we could monitor the degree of aggregation of peptide samples and ensure that predominantly monomeric forms were used in micropipette manipulation experiments. An example of the results of this assay is shown in Fig. 1. Lane 1 consists of fresh Aβ(1–42) peptide that had been thawed immediately before SDS-PAGE; the sample appears predominantly as a monomer (4.4 kDa), with some peptide appearing as dimer. In contrast to the fresh sample, lane 2 contains an aliquot of Aβ(1–42) that had been intentionally aggregated by incubation at room temperature for 24 h before SDS-PAGE. As a result, multiple bands appear, indicating the onset of peptide aggregation in various oligomeric states (dimers, trimers, and tetramers). It should be noted that the larger plaque-like aggregates have yet to develop, as indicated by the absence of high molecular weight bands at >30. All Aβ samples used in the micromanipulation experiments and subjected to this assay produced results similar to those shown in lane 1. The SDS-PAGE Western blot test thus allowed us to confirm that the aggregation state of Aβ could be replicated for each experiment. To minimize the extent of amyloid peptide aggregation, all solutions were prepared immediately before each micromanipulation experiment, which lasted no longer than 2 h.
FIGURE 1.

Results of SDS-PAGE Western blot assay of Aβ(1–42) aggregation. Lane 1 is freshly thawed Aβ aliquot (10 μM in Milli-Q water; Millipore); Lane 2 is Aβ aliquot incubated at room temperature for 24 h.
Lipid and sterol compositions were confirmed by high-performance liquid chromatography (HPLC), using an assay devised and described by Lang (32). Lipid samples in methanol were eluted through a reversed-phase octadecylsilane column with 100% HPLC-grade methanol at 1.0 mL/min. Injection volumes were from 20 to 80 μL, and UV detection was at 207 nm.
Vesicle preparation
Giant unilamellar vesicles (GUVs) with an average diameter of 10–20 μm were prepared by the electroformation method (33,34). Lipid mixtures in organic solvent (chloroform or chloroform/methanol) were spread onto the surface of platinum electrodes, and the solvent was removed in vacuo overnight. The electrodes were then immersed in a cuvette containing 1 mL aqueous sucrose solution (∼200 mOsm). A 10-Hz alternating current electric field was applied to the electrodes. The field amplitude was increased from 1.0 V to 2.0 V in 0.5 V increments every half hour. After 30 min at 2.0 V, the frequency was lowered to 4 Hz for 30 min. After electroformation, giant vesicles were harvested from the electroformation chamber by gentle aspiration of the suspension with a pasteur pipette. GUVs were diluted and resuspended in equiosmotic PBS. The use of different solutions for the hydrating and suspending media (aqueous sucrose and PBS, respectively) provided two advantages. First, density differences drove vesicles to sink to the bottom of the micromanipulation chamber, where they were then easily located using contrast optics. Secondly, differences in the refractive indices of the two solutions produced an enhanced optical contrast of the lipid vesicle relative to its surroundings.
Micropipette manipulation
The micropipette manipulation technique has been described in detail elsewhere (35,36). In brief, pipettes were fashioned from standard glass capillary tubing, pulled to create a suitable taper using a commercially available pipette puller (Sutter Instrument, Novato, CA), and then further trimmed with a microforge (Narishige, Tokyo, Japan) to produce an open circular tip. Pressure application and monitoring over five orders of magnitude (10−1–104 Pa) were achieved by means of a custom-made, water-filled manometer device with an inline pressure transducer (Validyne Engineering, Los Angeles, CA) connected to the micropipettes by a flexible tubing system. The micromanipulation stage was partitioned into two distinct slot chambers (∼400–500 mL total volume each) mounted side by side and parallel to each other. The chambers were separated by a small air gap that allowed two different chemical environments to be maintained simultaneously. One chamber contained GUVs dispersed in PBS and served as a vesicle storage reservoir. The other chamber constituted the test chamber and contained equiosmotic PBS and Aβ peptide at a known concentration. Both chamber partitions were thermoregulated by a circulating water channel in the jacket housing the chambers. Two micropipettes, mounted coaxially and opposite to each other, were used in each experiment. A primary working pipette (4–6 μm internal diameter) was used to capture and mechanically manipulate individual vesicles, and a transfer pipette (50–60 μm internal diameter) was used as a protective sheath for the working pipette during the transfer of a vesicle between the two chambers. The micromanipulation stage was mounted onto the stage of an inverted microscope (Olympus IX51; Olympus, Melville, NY) equipped with relief contrast (Hoffman modulation contrast) optics (40× objective, 0.6 NA, 40 mm working distance condenser).
Before the experiment, the chamber and the inner and outer surfaces of the pipettes were incubated in 0.2 g % aqueous solution of BSA for 15–30 min to eliminate the sticking of vesicles to the glass (37). The BSA solution was then flushed out of the chamber several times with BSA-free PBS. Aβ solutions were prepared in PBS at appropriate concentrations (10 nM and 100 nM) and used to fill the second (test) chamber. All solution osmolarities were measured in a freezing point osmometer (Advanced Instruments, Norwood, MA) to monitor solute concentrations and ensure isoosmolarity.
In a typical experiment, an individual GUV resting at the floor of the peptide-free storage chamber was selected and transferred into the test chamber using the following protocol. The vesicle was aspirated at a sublytic level of tension (typically 4–5 mN/m) by the working pipette, which—with the captured vesicle—was then inserted inside the bore of the much larger transfer pipette. Both pipettes were kept stationary while the micromanipulation stage assembly was translated laterally on the microscope stage. In this manner, the tips of the pipettes were translocated from one chamber environment into the other. Upon emergence from the transfer pipette, the vesicle was exposed to Aβ peptide under continuous observation with videomicroscopy.
Elastic membrane deformations were studied by the application of mechanical tension to the giant vesicles by changing the suction pressure in the working pipette using the manometer system (∼20 Pa resolution). At any given pressure, the bilayer tension τ(t) followed from the relation in the equation
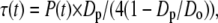 |
(1) |
where P(t) is the instantaneous suction pressure applied to the vesicle at time t by aspiration into the micropipette, Dp is the internal diameter of the pipette, and Do is the diameter of the spherical portion of the vesicle outside the pipette (Fig. 2) (38,39). For small deformations, changes in the parameter Do are beyond the limits of resolution, and so Do is treated as a constant for each vesicle. Under this constraint, a fluid, elastic bilayer membrane that forms a closed vesicle at constant volume experiences relative surface area changes as the bilayer tension is changed (for example, by lifting or lowering the water reservoir in the manometer). Changes in membrane area induced by pipette aspiration are described by the relation in the equation
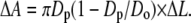 |
(2) |
Under this approximation, changes in membrane area are directly proportional to changes in the length (ΔL) of the portion of the membrane aspirated inside the micropipette. A plot of tension versus relative area change (ΔA divided by initial area Ao) directly provides the elastic area expansion modulus of the lipid bilayer from the slope of the linear fit. The critical lysis tension τc is defined as the maximal tension withstood by the bilayer before failure. Recently, Evans and co-workers (40) demonstrated that the critical lysis tension of lipid bilayers is not solely a function of the bilayer composition. Instead, the critical lysis tension also depends dynamically on the tension loading rate in the bilayer. In this article, our goal was not to map the entire dynamic tension spectrum for each membrane composition but to make a qualitative comparison of the cohesive strengths of bilayers after exposure to Aβ peptide as a function of oxysterol content. We therefore chose to focus on a single loading rate applied to all compositions in the experiment. Using an experimental arrangement identical to that of Evans and co-workers (40), we performed an infusion-withdrawal syringe pump-driven pressure manometer (VWR International, West Chester, PA) to consistently apply tension to the bilayer at a constant ramp on the order of 1 mN m−1 s−1, making adjustments to compensate for slight variations in vesicle and pipette size. A sample tension loading ramp is shown in Fig. 2.
FIGURE 2.

(A) Videomicrograph of a typical GUV, partially aspirated by a micropipette and viewed under Hoffman modulation contrast optics. The scale bar is 5 μm. (B) A sample tension loading ramp. Tension was steadily increased at the rate of ∼1 mN/m/s.
Data were analyzed using image analysis software (Scion Image 4.0; Scion, Frederick, MD) for measurement of vesicle geometric parameters directly from videomicrographic recording of the experiment (obtained using digital video editing software (iMovie; Apple, Cupertino, CA) on a personal computer (PowerPC G4; Apple)). All relevant experimental quantities (including time, pipette pressure, solution osmolarities, and temperature) were recorded. Bilayer material properties were calculated and studied using analysis and graphing software (Origin 7.0; OriginLab, Northampton, MA). Statistical significance of data relative to controls was determined by Student's t-test. Comprehensive treatment of the membrane mechanics used in the micropipette manipulation technique can be found in a monograph by Evans and Skalak (41) and several more recent reviews (35,36,42).
RESULTS
HPLC of pure lipid stocks showed the elution of well-resolved symmetrical peaks (Fig. 3 A). The chromatogram for OHCHOL reveals a secondary peak eluting at ∼1.77 min, at the same point at which KETO elutes, which probably reflects the presence of impurities in the stock. Chromatograms of lipid samples that nominally consisted of SOPC/CHOL (7:3 mol) and were subjected to autooxidation in air revealed the presence of additional components (Fig. 3 B, bottom trace). The autooxidized sample was “spiked” in turn by pure KETO and then pure OHCHOL, resulting in amplification of the corresponding peaks on the chromatogram (Fig. 3 B, middle and top traces). These results indicate the conversion of some CHOL into its major oxidation products, KETO and OHCHOL, consistent with the known oxidative paths of CHOL (43,44). HPLC revealed the presence of these oxysterols as distinct peaks that were well-resolved from SOPC and CHOL; these results are in agreement with those obtained by Lang (32).
FIGURE 3.

HPLC chromatograms of various (A) pure and (B) mixed lipid systems. All samples were in MeOH and eluted at 1.0 mL/min. UV detection was at 207 nm. (B) The chromatogram at the bottom was obtained for a sample of SOPC/CHOL (7:3 mol) autooxidized in air. The middle chromatogram was obtained for the same SOPC/CHOL sample, with the addition of pure KETO. The top chromatogram was obtained for the KETO-spiked sample, with the addition of pure OHCHOL.
GUVs were prepared for the following four compositions: 1), SOPC; 2), SOPC/CHOL (70:30 mol); 3), SOPC/CHOL/KETO (70:25:5 mol); and 4), SOPC/CHOL/OHCHOL (70:25:5 mol). Furthermore, GUVs were prepared from SOPC/CHOL (70:30 mol) stock that was subjected to autooxidation in air for 48 h. GUVs of all lipid compositions tested in this experiment were stable on the timescale of the experiment (typically 2 h). GUV samples that were stored at room temperature overnight and retested the next day showed no significant changes in mechanical properties. As a control, we transferred individual GUVs of each composition from a chamber containing PBS into a second chamber containing PBS. No change in either area expansion modulus or lysis tension was observed after such a transfer. Also, GUVs did not exhibit any gross morphological changes as a consequence of the transfer, such as surface invaginations or vesiculation. We also performed control measurements of GUVs that were not transferred, and we found the mechanical properties to be consistent with values reported in the literature in the case of pure SOPC and SOPC/CHOL.
In the micromanipulation transfer experiment, individual GUVs were transferred into a chamber containing peptide at a concentration of 100 nM. The peptide/lipid ratio (P/L) was roughly estimated based on the bulk peptide concentration, the volume of the micromanipulation chamber, and the geometry of the vesicle. The number of lipids in the GUV was calculated from the diameter and known lipid dimensions, ignoring the presence of “hidden” surface area in the form of surface ruffles below the optically detectible limit. A conservative estimate placed P/L on the order of 104, well above the P/L values reported in other studies (22,24). In the micromanipulation transfer experiment, the GUV bilayer, therefore, is exposed to an excess of peptide relative to lipid.
Pure SOPC bilayers appeared to be unaffected by exposure to either Aβ(1–42) or Aβ(1–40). SOPC GUVs subjected to mechanical tension exhibited elastic area expansion with a corresponding apparent compressibility modulus consistent with values reported in the literature (∼200 mN/m) (45). The same GUV, after transfer to the chamber containing 100 nM Aβ(1–42), showed the same elastic deformation response to aspiration-induced stress. Lysis tension values of pure SOPC bilayers were unchanged by exposure to Aβ (Fig. 4). This finding is consistent with previous studies that indicated a lack of interaction between Aβ and zwitterionic membranes.
FIGURE 4.

Lysis tension values of SOPC GUVs, with and without exposure to either 100 nM Aβ(1–42) or 100 nM Aβ(1–40). Values are mean ± SD (N = 20).
SOPC bilayers containing CHOL and oxysterols (KETO or OHCHOL) were tested in the same manner as pure SOPC bilayers. Lysis tension values for SOPC/CHOL were in agreement with literature values (∼12.6 ± 3.0 mN/m) (Fig. 5), whereas SOPC/CHOL/KETO was indistinguishable from SOPC/CHOL (12.3 ± 1.8 mN/m) (Fig. 6) and SOPC/CHOL/OHCHOL was lower than SOPC/CHOL (8.6 ± 1.9 mN/m) (Fig. 7).
FIGURE 5.

Lysis tension values of SOPC/CHOL (7:3 mol) GUVs, with and without exposure to either 100 nM Aβ(1–42) or 100 nM Aβ(1–40). Values are mean ± SD (N = 13).
FIGURE 6.

Lysis tension values of SOPC/CHOL/KETO (70:25:5 mol) GUVs, with and without exposure to either 100 nM Aβ(1–42) or 100 nM Aβ(1–40). Values are mean ± SD (N = 12). Statistical significance relative to controls was determined by Student's t-test (*p < 0.005).
FIGURE 7.

Lysis tension values of SOPC/CHOL/OHCHOL (70:25:5 mol) GUVs, with and without exposure to either 100 nM Aβ(1–42) or 100 nM Aβ(1–40). Values are mean ± SD (N = 7). Statistical significance relative to controls was determined by Student's t-test (*p < 0.01; **p < 0.005).
Bilayers containing sterols and oxysterols exhibited different responses to Aβ peptides depending on the length of the individual peptide. As with pure SOPC bilayers, the mechanical properties of SOPC/CHOL bilayers did not change after exposure to either Aβ(1–42) or Aβ(1–40). However, partial substitution of CHOL with KETO (5 mol %) caused a drastic reduction of the lysis tension (2.03 ± 0.07 mN/m), under an identical tension loading rate, after exposure to Aβ(1–42) (Fig. 6). In contrast, Aβ(1–40) exposure resulted in only a modest decrease in lysis tension (10.8 ± 0.7 mN/m). This difference, though, was not statistically significant. Partial substitution of CHOL with OHCHOL (5 mol %) caused a drastic reduction of the lysis tension after exposure to Aβ(1–40) (2.05 ± 0.31 mN/m) and to Aβ(1–42) (2.85 ± 0.43 mN/m) (Fig. 7).
Finally, lipid mixtures nominally consisting of SOPC/CHOL (70:30 mol) that were autooxidized in air were hydrated and reconstituted into GUVs. These vesicles demonstrated lower thresholds for lysis (7.45 ± 2.97 mN/m) than SOPC/CHOL GUVs, and a further marked decrease in lysis tension after exposure to Aβ(1–42) (0.69 mN/m) but not to Aβ(1–40) (Fig. 8). These vesicles were also observed to lose optical contrast over time, as shown in Fig. 9. The initial contrast was due to the difference in optical densities of the sucrose solution entrapped inside the vesicle and the PBS outside the vesicle. After exposure to Aβ(1–42), with the vesicles held under very low suction pressure (applied tension on the order of 0.5 mN/m), the contrast weakened rapidly, on a timescale of 1 min, indicating a relatively rapid mixing of internal and external solutes through a permeabilized membrane. Throughout this process, the cohesion of the lipid bilayer itself was maintained, and the bilayer remained faintly visible under Hoffman modulation contrast optics. After the contrast difference was completely lost, the bilayer itself was extremely fragile, failing at low levels of applied tension (0.7–2.8 mN/m).
FIGURE 8.

Lysis tension values of autooxidized SOPC/CHOL (nominal composition 7:3 mol) GUVs, with and without exposure to either 100 nM Aβ(1–42) or 100 nM Aβ(1–40). Values are mean ± SD (N = 4). Statistical significance relative to controls was determined by Student's t-test (*p < 0.005).
FIGURE 9.

Videomicrograph of a GUV composed of autooxidized SOPC/CHOL (7:3 mol), viewed under Hoffman modulation contrast optics. The vesicle was held under very low suction pressure (applied tension on the order of 0.5 mN/m) below the aspiration threshold. 100 nM Aβ(1–42) caused the GUV to become leaky, as indicated by the loss of optical contrast between the sucrose solution inside the vesicle and the PBS outside the vesicle. (Top panel) time = 0; (middle panel) 33 s elapsed; and (bottom panel) 1 min elapsed.
DISCUSSION
Aging is accompanied by compositional changes in brain membranes; for example, transbilayer CHOL distributions tend to equalize with age by mechanisms not fully understood (dysfunction in apolipoprotein E and/or sterol carrier protein-2 have been proposed) (46). Such changes impact a host of interrelated membrane properties including bilayer fluidity, domain structure, and permeability that, in turn, directly affect cell function and viability. We focused on the role of oxysterols on the mechanical stability of lipid bilayers in the model membrane system of the giant bilayer vesicle because of the relative enrichment of CHOL in brain membranes (47,48), and because Aβ has been directly implicated in oxidative stress damage in the brain (4,49). The GUV comprises the simplest model of the cell membrane that retains the basic bilayer structure of the membrane on a size scale comparable to cells (tens to hundreds of microns). SOPC was selected as the major phospholipid component because it is readily reconstituted as GUVs, the mechanical properties of which have been thoroughly characterized by micropipette manipulation techniques (45,50–52). The SOPC bilayer vesicle, therefore, constitutes a useful benchmark for comparisons with GUVs of other lipid compositions. We chose two of the most commonly occurring oxysterol derivatives of CHOL, KETO and OHCHOL (43,44); these compounds have been reported to be cytotoxic (characterized by phosphatidylserine externalization, loss of mitochondrial transmembrane potential, increased membrane permeability to propidium iodide, and membrane fragmentation) (53). The composition of the various lipid bilayers used in the micromanipulation experiments was assayed by HPLC, SOPC, CHOL, KETO, and OHCHOL and exhibited distinctive peaks on chromatograms and so enabled clear identification of each. Furthermore, the chromatographic “fingerprint” of each component was used to identify the components of autooxidized lipid samples.
In the micropipette manipulation experiments, we first established the baseline properties of the reference bilayers. The apparent elastic area expansion modulus and the lysis tension (at 1 mN/m/s loading rate) of pure SOPC and SOPC/CHOL (7:3 mol) bilayers were in agreement with values reported in the literature (42,45). The mechanical properties of these bilayers were unchanged by transfer of the GUVs from one chamber to another if the same buffer was present in both chambers. Furthermore, the mechanical properties were unaffected by exposure of the bilayer to 100 nM Aβ(1–42) or 100 nM Aβ(1–40). These results are consistent with previous findings that suggested that a negative bilayer surface charge was a requisite for peptide interaction with the bilayer and that Aβ peptides did not perturb zwitterionic membranes (22). In addition to surface charge–mediated interactions, CHOL has been shown to condense bilayer molecular area through attractive van der Waals interactions between phospholipid acyl moieties and the sterol ring structure. This cohesivity increases the energetic barrier against insertion of Aβ peptides into the bilayer structure. As shown by Zhelev et al. (20), the work for creating a vacancy for insertion of a peptide into a bilayer is proportional to the bilayer area expansion modulus.
In contrast to the behavior and properties of CHOL-containing bilayers exposed to Aβ peptides, lysis tensions of oxysterol-containing bilayers reflected dramatic destabilization by Aβ peptides in a composition-dependent manner. SOPC/CHOL/KETO (70:25:5 mol) bilayers did not display a significant change in lysis tension relative to SOPC/CHOL; after exposure to 100 nM Aβ(1–42), however, a striking decrease in lysis tension was observed. Under these conditions, the GUVs were extremely fragile, collapsing under very low levels of applied stress. Exposure to Aβ(1–40), however, did not diminish the lysis tension of KETO-containing membranes. In comparison, the lysis tensions of SOPC/CHOL/OHCHOL (70:25:5 mol) bilayers were lower than those of SOPC/CHOL bilayers, showing an ∼32 percent reduction. In addition to this effect, both Aβ(1–42) and Aβ(1–40) further reduced the lysis tension of the OHCHOL-containing bilayers, resulting in their failure under very low applied mechanical stress.
The introduction of an additional dipole to the CHOL ring—e.g., a ketone or hydroxyl group—has important ramifications for bilayer structure that may explain the observed changes in bilayer mechanical properties on addition of Aβ peptides. The single hydroxyl moiety of CHOL anchors the molecule to the bilayer in a configuration in which the plane of the sterol ring is perpendicular to the bilayer surface. This configuration enables the sterol to locally order the acyl chains of neighboring lipid molecules, particularly at the carbons most proximal to the glycerol backbone. This effect strengthens intrabilayer cohesive interactions and increases the energetic barrier to peptide insertion into the bilayer. In comparison, the introduction of a second dipole at the C7 position of the oxysterols used in this study imparts a tilt to the molecule with respect to the bilayer surface, as the dipole seeks exposure to water (54,55). The result is a slight thinning of the bilayer (although this may be partially offset by an increase in the hydration layer of the bilayer), as the free volume is increased within the bilayer, especially at the midplane (56). The increased disorder within the bilayer is reflected in a reduced cohesion in the oxysterol-containing bilayer relative to the CHOL-containing bilayer (57). Simon and co-workers demonstrated that substitution of 6-ketocholestanol (an oxysterol similar to KETO) for CHOL in 1:1 SOPC/CHOL bilayers results in a >50% reduction in the elastic area expansion modulus (54), indicating that such oxysterol-containing bilayers are more easily expanded under isotropic tension than their CHOL-containing counterparts. The reduction in intermolecular cohesive interactions reduces the energetic barrier for Aβ insertion into the bilayer. The hydrophobic effect then drives the insertion of the peptide into the bilayer, even in the absence of surface electrostatic interactions to mediate lipid-peptide attraction. This model is consistent with the results of Kremer et al. (26,27).
The effect of Aβ peptides on the mechanical stability of oxysterol bilayers is consistent with the model of bilayer strength recently described by Evans and co-workers (40). In this model, the rupture of fluid lipid bilayers is described as a two-regime kinetic process that is dependent on the loading rate of tension in the bilayer, with the cutoff between the regimes occurring on the order of 10 mN/m/s. At slow loading rates (including the rate applied in our experiments), the rupture strength is predicted by the classic cavitation model of opening and expanding an unstable hole or pore in the bilayer within the lifetime of the hole. In the case of slow loading rates, membrane edge energy governs the failure tension and can be reduced by the introduction of defects, such as small molecules, to the bilayer. This outcome has been suggested as an explanation, in part, for the membrane effects of small molecules such as salicylate (58) and amphiphilic antimicrobial peptides (59). The propensity of the Aβ peptide to insert itself into the bilayer is driven by the hydrophobic effect, with the transmembrane residues (28–40 or 28–42) adopting an alpha-helical conformation that provides internal hydrogen bonding of the peptide necessary to accommodate the C=O and NH groups within the hydrocarbon core of the bilayer (60). Simulations of Aβ peptide structure based on NMR spectroscopy data indicate that the transmembrane portion of the peptide is approximately the same size as the thickness of the typical lipid monolayer (61) and that the overall structure of the peptide is similar to that of the hemagglutinin peptide fusion domain (62). These results suggest that Aβ localization is keyed to the thickness of the target membrane. Therefore, it is possible that the addition of KETO induces the membrane thinning that accommodates Aβ(1–42) insertion, whereas OHCHOL-induced thinning makes possible the insertion of both Aβ(1–42) and Aβ(1–40). The conversion of CHOL to oxysterols and the accompanying effects on AD membrane stability may explain results reported by Mason et al. (63) that indicate the concurrence of CHOL deficits and membrane thinning in AD cortical membranes.
As demonstrated in this study, the addition of oxysterols to simple PC-CHOL lipid membranes alters the membrane mechanical properties and causes the bilayer to be further destabilized by the exogenous addition of Aβ peptides. In the latter event, the peptides act as point defects from which membrane rupture proceeds at lower levels of applied tension. Furthermore, oxysterols may increase plasma membrane permeability, leading to membrane depolarization. Geddes et al. (64) have demonstrated that mechanical deformation of cultured neurons that simulate physiological stress distributions in the brain results in a transient increase in plasma membrane permeability, which is due to the imposition of membrane tears. Therefore, it is reasonable to expect that structurally destabilizing changes in neuronal membrane composition should increase the cellular damage inflicted by mechanical insults to the brain. It has been reported that Aβ itself promotes the formation of oxysterols and other lipid peroxidation products that are, in effect, catalyzing conditions under which peptide-induced destabilization may occur (65,66). These results are consistent with studies reported by Svennerholm and Gottfries (67) that demonstrate synaptic membrane loss as a hallmark of type 1 AD. Therefore, one of the many pathways by which Aβ leads to neurodegeneration may involve compositional changes in the synaptic plasma membrane, coupled with the local accumulation of membrane-active peptide, that render the bilayer vulnerable to damage at even low levels of mechanical stress to the brain. Investigating how Aβ peptides alter the properties of the bilayer, and how the interactions depend specifically on individual bilayer constituents of biological relevance, will further the general understanding of all diseases involving abnormal protein-cell interactions.
Acknowledgments
The authors thank Dr. Gene Yeh and Ms. Laura Otte for assistance with performing the Western blot assay, and Dr. Mike Nicolaou for assistance with the HPLC assay.
This work was supported by National Institutes of Health (HL40696 and AR40732).
Editor: Anthony Watts.
References
- 1.Hardy, J. A., and G. A. Higgins. 1992. Alzheimer's disease: the amyloid cascade hypothesis. Science. 256:184–185. [DOI] [PubMed] [Google Scholar]
- 2.Lorenzo, A., and B. A. Yankner. 1994. β-Amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc. Natl. Acad. Sci. USA. 91:12243–12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behl, C., J. B. Davis, R. Lesley, and D. Schubert. 1994. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 77:817–827. [DOI] [PubMed] [Google Scholar]
- 4.Huang, X., C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi, and A. I. Bush. 1999. The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 38:7609–7616. [DOI] [PubMed] [Google Scholar]
- 5.Praprotnik, D., M. A. Smith, P. L. Richey, H. V. Vinters, and G. Perry. 1996. Plasma membrane fragility in dystrophic neurites in senile plaques of Alzheimer's disease: an index of oxidative stress. Acta Neuropathol. (Berl.). 91:1–5. [DOI] [PubMed] [Google Scholar]
- 6.Akama, K. T., C. Albanese, R. G. Pestell, and L. J. Van Eldik. 1998. Amyloid β-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proc. Natl. Acad. Sci. USA. 95:5795–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keil, U., A. Bonert, C. A. Marques, I. Scherping, J. Weyermann, J. B. Strosznajder, F. Müller-Spahn, C. Haass, C. Czech, L. Pradier, W. E. Müller, and A. Eckert. 2004. Amyloid β-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J. Biol. Chem. 279:50310–50320. [DOI] [PubMed] [Google Scholar]
- 8.Yan, S. D., X. Chen, J. Fu, M. Chen, H. Zhu, A. Roher, T. Slattery, L. Zhao, M. Nagashima, J. Morser, A. Mighelli, P. Nawroth, D. Stern, and A. M. Schmidt. 1996. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. 382:685–691. [DOI] [PubMed] [Google Scholar]
- 9.Giri, R., Y. Shen, M. Stins, S. D. Yan, A. M. Schmidt, D. Stern, K.-S. Kim, B. Zlokovic, and V. K. Kalra. 2000. β-amyloid–induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am. J. Physiol. Cell Physiol. 279:1772–1781. [DOI] [PubMed] [Google Scholar]
- 10.Arispe, N., H. B. Pollard, and E. Rojas. 1993. Giant multilevel cation channels formed by Alzheimer's disease amyloid β-protein [AßP-(1–40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA. 90:10573–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arispe, N., E. Rojas, and H. B. Pollard. 1993. Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA. 90:567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mirzabekov, T., M. Lin, W. Yuan, P. J. Marshall, M. Carman, K. Tomaselli, I. Lieberburg, and B. L. Kagan. 1994. Channel formation in planar lipid bilayers by a neurotoxic fragment of the beta-amyloid peptide. Biochem. Biophys. Res. Commun. 202:1142–1148. [DOI] [PubMed] [Google Scholar]
- 13.Hirakura, Y., M.-C. Lin, and B. L. Kagan. 1999. Alzheimer amyloid Aβ1–42 channels: effects of solvent, pH, and Congo red. J. Neurosci. Res. 57:458–466. [PubMed] [Google Scholar]
- 14.Arispe, N. 2004. Architecture of the Alzheimer's AßP ion channel pore. J. Membr. Biol. 197:33–48. [DOI] [PubMed] [Google Scholar]
- 15.Behl, C., J. B. Davis, G. F. Klier, and D. Schubert. 1994. Amyloid b peptide induces necrosis rather than apoptosis. Brain Res. 645:253–264. [DOI] [PubMed] [Google Scholar]
- 16.Paradis, E., H. Douillard, M. Koutroumanis, C. Goodyer, and A. LeBlanc. 1996. Amyloid β peptide of Alzheimer's disease downregulates Bcl-2 and upregulates Bax expression in human neurons. J. Neurosci. 16:7533–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kajkowski, E. M., C. F. Lo, X. Ning, S. Walker, H. J. Sofia, W. Wang, W. Edris, P. Chanda, E. Wagner, S. Vile, K. Ryan, B. McHendry-Rinde, S. C. Smith, A. Wood, K. J. Rhodes, J. D. Kennedy, J. Bard, J. S. Jacobsen, and B. A. Ozenberger. 2001. β-amyloid peptide-induced apoptosis regulated by a novel protein containing a G protein activation module. J. Biol. Chem. 276:18748–18756. [DOI] [PubMed] [Google Scholar]
- 18.Kayed, R., Y. Sokolov, B. Edmonds, T. M. McIntire, S. C. Milton, J. E. Hall, and C. G. Glabe. 2004. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279:46363–46366. [DOI] [PubMed] [Google Scholar]
- 19.Ambroggio, E. E., D. H. Kim, F. Separovic, C. J. Barrow, K. J. Barnham, L. A. Bagatolli, and G. D. Fidelio. 2005. Surface behavior and lipid interaction of Alzheimer β-amyloid peptide 1–42: a membrane-disrupting peptide. Biophys. J. 88:2706–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhelev, D. V., N. Stoicheva, P. Scherrer, and D. Needham. 2001. Interaction of synthetic HA2 influenza fusion peptide analog with model membranes. Biophys. J. 81:285–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terzi, E., and G. Hölzemann. 1997. Interaction of Alzheimer β-amyloid peptide(1–40) with lipid membranes. Biochemistry. 36:14845–14852. [DOI] [PubMed] [Google Scholar]
- 22.McLaurin, J., and A. Chakrabartty. 1996. Membrane disruption by Alzheimer β-amyloid peptides mediated through specific binding to either phospholipids or gangliosides. J. Biol. Chem. 271:26482–26489. [DOI] [PubMed] [Google Scholar]
- 23.McLaurin, J., T. Franklin, P. E. Fraser, and A. Chakrabartty. 1998. Structural transitions associated with the interaction of Alzheimer β-amyloid peptides with gangliosides. J. Biol. Chem. 273:4506–4515. [DOI] [PubMed] [Google Scholar]
- 24.Yip, C. M., and J. McLaurin. 2001. Amyloid-β peptide assembly: a critical step in fibrillogenesis and membrane disruption. Biophys. J. 80:1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kakio, A., S. Nishimoto, K. Yanagisawa, Y. Kozutsumi, and K. Matsuzaki. 2001. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 276:24985–24990. [DOI] [PubMed] [Google Scholar]
- 26.Kremer, J. J., M. M. Pallitto, D. J. Sklansky, and R. M. Murphy. 2000. Correlation of β-amyloid aggregate size and hydrophobicity with decreased bilayer fluidity of model membranes. Biochemistry. 39:10309–10318. [DOI] [PubMed] [Google Scholar]
- 27.Kremer, J. J., D. J. Sklansky, and R. M. Murphy. 2001. Profile of changes in lipid bilayer structure caused by β-amyloid peptide. Biochemistry. 40:8563–8571. [DOI] [PubMed] [Google Scholar]
- 28.Curtain, C. C., F. E. Ali, D. G. Smith, A. I. Bush, C. L. Masters, and K. J. Barnham. 2003. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer's disease amyloid-β peptide with membrane lipid. J. Biol. Chem. 278:2977–2982. [DOI] [PubMed] [Google Scholar]
- 29.Pike, C. J., D. Burdick, A. J. Walencewicz, C. G. Glabe, and C. W. Cotman. 1993. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 13:1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dahlgren, K. N., A. M. Manelli, W. B. Stine, Jr., L. K. Baker, G. A. Krafft, and M. J. LaDu. 2002. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 277:32046–32053. [DOI] [PubMed] [Google Scholar]
- 31.Stine, W. B., Jr., K. N. Dahlgren, G. A. Krafft, and M. J. LaDu. 2003. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278:11612–11622. [DOI] [PubMed] [Google Scholar]
- 32.Lang, J. K. 1990. Quantitative determination of cholesterol in liposome drug products and raw materials by high-performance liquid chromatography. J. Chromatogr. 507:157–163. [DOI] [PubMed] [Google Scholar]
- 33.Angelova, M. I., and D. S. Dimitrov. 1986. Liposome electroformation. Faraday Disc. Chem. Soc. 81:303–311. [Google Scholar]
- 34.Angelova, M. I., S. Soleau, P. Meleard, J. F. Faucon, and P. Bothorel. 1992. Preparation of giant vesicles by external AC electric fields. Kinetics and applications. Prog. Colloid Polym. Sci. 89:127–131. [Google Scholar]
- 35.Evans, E., and D. Needham. 1987. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion, and colloidal interactions. J. Phys. Chem. 91:4219–4228. [Google Scholar]
- 36.Needham, D., and D. V. Zhelev. 1996. The mechanochemistry of lipid vesicles examined by micropipet manipulation techniques. In Vesicles. M. Rosoff, editor. Marcel Dekker, New York. 373–444.
- 37.Meltzer, H., and A. Silberberg. 1988. Adsorption of collagen, serum albumin, and fibronectin to glass and to each other. J. Colloid Interface Sci. 126:292–303. [Google Scholar]
- 38.Rand, R. P., and A. C. Burton. 1964. Mechanical properties of the red cell membrane. I. Membrane Stiffness and Intracellular Pressure. Biophys. J. 4:115–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evans, E. A., R. Waugh, and L. Melnik. 1976. Elastic area compressibility modulus of red cell membrane. Biophys. J. 16:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans, E., V. Heinrich, F. Ludwig, and W. Rawicz. 2003. Dynamic tension spectroscopy and strength of biomembranes. Biophys. J. 85:2342–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans, E. A., and R. Skalak. 1980. Mechanics and Thermodynamics of Biomembranes. CRC Press, Boca Raton, FL.
- 42.Kim, D. H., and D. Needham. 2002. Lipid bilayers and monolayers: characterization using micropipette manipulation techniques. In Encyclopedia of Surface and Colloid Science. A. T. Hubbard, editor. Marcel Dekker, New York. 3057–3086.
- 43.Theunissen, J. J. H., R. L. Jackson, H. J. M. Kempen, and R. A. Demel. 1986. Membrane properties of oxysterols. Interfacial orientation, influence on membrane permeability and redistribution between membranes. Biochim. Biophys. Acta. 860:66–74. [DOI] [PubMed] [Google Scholar]
- 44.Smith, L. L. 1987. Cholesterol autoxidation 1981–1986. Chem. Phys. Lipids. 44:87–125. [DOI] [PubMed] [Google Scholar]
- 45.Needham, D., and R. S. Nunn. 1990. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J. 58:997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wood, W. G., F. Schroeder, U. Igbavboa, N. A. Avdulov, and S. V. Chochina. 2002. Brain membrane cholesterol domains, aging and amyloid beta-peptides. Neurobiol. Aging. 23:685–694. [DOI] [PubMed] [Google Scholar]
- 47.Calderon, R. O., B. Attema, and G. H. DeVries. 1995. Lipid composition of neuronal cell bodies and neurites from cultured dorsal root ganglia. J. Neurochem. 64:424–429. [DOI] [PubMed] [Google Scholar]
- 48.Björkhem, I., and S. Meaney. 2004. Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 24:806–815. [DOI] [PubMed] [Google Scholar]
- 49.Hensley, K., J. M. Carney, M. P. Mattson, M. Aksenova, M. Harris, J. F. Wu, R. A. Floyd, and D. A. Butterfield. 1994. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA. 91:3270–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans, E., and W. Rawicz. 1990. Entropy-driven tension and bending elasticity in condensed-fluid membranes. Phys. Rev. Lett. 64:2094–2097. [DOI] [PubMed] [Google Scholar]
- 51.Olbrich, K., W. Rawicz, D. Needham, and E. Evans. 2000. Water permeability and mechanical strength of polyunsaturated lipid bilayers. Biophys. J. 79:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rawicz, W., K. C. Olbrich, T. McIntosh, D. Needham, and E. Evans. 2000. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 79:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lemaire-Ewing, S., C. Prunet, T. Montange, A. Vejux, A. Berthier, G. Bessède, L. Corcos, P. Gambert, D. Néel, and G. Lizard. 2005. Comparison of the cytotoxic, pro-oxidant and pro-inflammatory characteristics of different oxysterols. Cell Biol. Toxicol. 21:97–114. [DOI] [PubMed] [Google Scholar]
- 54.Simon, S. A., T. J. McIntosh, A. D. Magid, and D. Needham. 1992. Modulation of the interbilayer hydration pressure by the addition of dipoles at the hydrocarbon/water interface. Biophys. J. 61:786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smondyrev, A. M., and M. L. Berkowitz. 2001. Effects of oxygenated sterol on phospholipid bilayer properties: a molecular dynamics simulation. Chem. Phys. Lipids. 112:31–39. [DOI] [PubMed] [Google Scholar]
- 56.Phillips, J. E., Y.-J. Geng, and R. P. Mason. 2001. 7-Ketocholesterol forms crystalline domains in model membranes and murine aortic smooth muscle cells. Atherosclerosis. 159:125–135. [DOI] [PubMed] [Google Scholar]
- 57.Rooney, M., W. Tamura-Lis, L. J. Lis, S. Yachnin, O. Kucuk, and J. W. Kauffman. 1986. The influence of oxygenated sterol compounds on dipalmitoylphosphatidylcholine bilayer structure and packing. Chem. Phys. Lipids. 41:81–92. [DOI] [PubMed] [Google Scholar]
- 58.Zhou, Y., and R. M. Raphael. 2005. Effect of salicylate on the elasticity, bending stiffness, and strength of SOPC membranes. Biophys. J. 89:1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shai, Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta. 1462:55–70. [DOI] [PubMed] [Google Scholar]
- 60.White, S. H., and W. C. Wimley. 1994. Peptides in lipid bilayers: structural and thermodynamical basis for partitioning and folding. Curr. Opin. Struct. Biol. 4:79–86. [Google Scholar]
- 61.Coles, M., W. Bicknell, A. A. Watson, D. P. Fairlie, and D. J. Craik. 1998. Solution structure of amyloid β-peptide(1–40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry. 37:11064–11077. [DOI] [PubMed] [Google Scholar]
- 62.Crescenzi, O., S. Tomaselli, R. Guerrini, S. Salvadori, A. M. D'Ursi, P. A. Temussi, and D. Picone. 2002. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 269:5642–5648. [DOI] [PubMed] [Google Scholar]
- 63.Mason, R. P., W. J. Shoemaker, L. Shajenko, and T. E. Chambers. 1992. Evidence for changes in the Alzheimer's disease brain cortical membrane structure mediated by cholesterol. Neurobiol. Aging. 13:413–419. [DOI] [PubMed] [Google Scholar]
- 64.Geddes, D. M., R. S. Cargill II, and M. C. LaPlaca. 2003. Mechanical stretch to neurons results in a strain rate and magnitude-dependent increase in plasma membrane permeability. J. Neurotrauma. 20:1039–1049. [DOI] [PubMed] [Google Scholar]
- 65.Avdulov, N. A., S. V. Chochina, U. Igbavboa, E. O. O'Hare, F. Schroeder, J. P. Cleary, and W. G. Wood. 1997. Amyloid β-peptides increase annular and bulk fluidity and induce lipid peroxidation in brain synaptic plasma membranes. J. Neurochem. 68:2086–2091. [DOI] [PubMed] [Google Scholar]
- 66.Nelson, T. J., and D. L. Alkon. 2005. Oxidation of cholesterol by amyloid precursor protein and β-amyloid peptide. J. Biol. Chem. 280:7377–7387. [DOI] [PubMed] [Google Scholar]
- 67.Svennerholm, L., and C.-G. Gottfries. 1994. Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (type I) and demyelination in late-onset form (type II). J. Neurochem. 62:1039–1047. [DOI] [PubMed] [Google Scholar]