

Abstract
The immunological synapse is a stable intercellular structure that specializes in substance and signal transfer from one immune cell to another. Its formation is regulated in part by the diffusion of adhesion and signaling molecules into, and their binding of countermolecules in the contact area. The stability of immunological synapses allows receptor-ligand interactions to approximate chemical equilibrium despite other dynamic aspects. We have developed a mathematical model that describes the coupled reaction-diffusion process in an established immunological synapse. In this study, we extend a previously described contact area fluorescence recovery after photobleaching (FRAP) experiment to test the validity of the model. The receptor binding activity and lateral mobility of fluorescently labeled, lipid-anchored ligands in the bilayer resulted in their accumulation, as revealed by a much higher fluorescence intensity inside the contact area than outside. After complete photobleaching of the synapse, fluorescence recovery requires ligands to dissociate and rebind, and to diffuse in and out of the contact area. Such a FRAP time course consequently provides information on reaction and diffusion, which can be extracted by fitting the model solution to the data. Surprisingly, reverse rates in the two-dimensional contact area were at least 100-fold slower than in three-dimensional solution. As previously reported in immunological synapses, a significant nonrecoverable fraction of fluorescence was observed with one of two systems studied, suggesting some ligands either dissociated or diffused much more slowly compared with other ligands in the same synapse. The combined theory and experiment thus provides a new method for in situ measurements of kinetic rates, diffusion coefficients, and nonrecoverable fractions of interacting molecules in immunological synapses and other stable cell-bilayer junctions.
INTRODUCTION
Information and substance transfer between cells and their environment often occurs in specialized structures with concentrated and organized adhesion and signaling molecules. Examples include intraepithelial junctions, neural and neuromuscular synapses, and immunological synapses, the last of which are the focus of this study (1,2). The formation, evolution, and maintenance of immunological synapses are thought to be due in part to the work of adhesion molecules, which also may serve as signaling molecules (3). These adhesion receptors diffuse in and out of the cell-cell contact, bind to their ligands across the intercellular gap via the extracellular domain, and link to cytoskeletal and/or adaptor molecules via the cytoplasmic domain (4). Quantitative descriptions of these processes are required for an understanding of various structural and functional aspects of immunological synapses.
We have investigated an experimental model as a first step toward a quantitative description of the physiochemical processes that control the dynamics of adhesion molecules involved in the immunological synapse. Our simplified system consists of a single species of fluorescently labeled, lipid-anchored ligands that are reconstituted into a glass-supported bilayer membrane in contact with a cell that expresses a single receptor species for that ligand species. In a classic study (5), fluorescent IgE was used to form a bridge between a cell expressing laterally mobile high-affinity Fcɛ receptors and an antigen-coated bead and to visualize molecular redistribution of the bridges in the interface. The fluorescent IgE accumulated at the interface, which correlated with increased adhesive strength. Because cell signaling may, and is even expected to, lead to the reorganization of FcɛRI after interaction with ligands, it is difficult to directly correlate the IgE accumulation with bridge formation and diffusion in the contact area. This challenge is overcome by replacing the bead with a supported planar bilayer containing freely mobile fluorescent ligands. When cells bearing receptors interact with the fluorescent ligands on this surface, the accumulation of fluorescence in the interface can be related more directly to bridge formation and diffusion (6,7). The self-assembly of the contact area between the cell and the bilayer is monitored by reflection interference microscopy (RIM), again allowing for quantitative measurement (8–10).
A probable physical mechanism for contact area formation is shown in Fig. 1. Before contacting the lipid bilayer with receptor-expressing cells, all ligands are bright, mobile, and uniformly distributed (Fig. 1 A). After the cell contacts the bilayer, receptors begin to bind ligands. Bond formation converts free ligands into bound ligands, which locally depletes free ligands. The density gradient that is generated can result in a net diffusion of free ligands into the contact area to replenish the free ligands, which can push the forward reaction much further than it would otherwise proceed without diffusion. This leads to the accumulation of a large number of ligands and the formation of a high density of bonds in the contact area (Fig. 1 B). Over time, the fluorescence intensity inside the contact area (proportional to the total density of free and bound ligands combined) may become much higher than that outside (proportional to the density of free ligands), until the process reaches an equilibrium. Like solution binding, the number of bonds at equilibrium should initially increase rapidly with the density of free ligands in the bilayer but would saturate beyond a certain point (7,9). Fitting the equilibrium binding model to such a Langmuir isotherm (cf. Fig. 4 A below) or its linearized transformation, i.e., the Scatchard plot or the Zhu-Golan plot (cf. Fig. 4 B below), allows for evaluation of the two-dimensional (2D) equilibrium dissociation constant Kd (7,9).
FIGURE 1.

Schematic of contact formation and contact area FRAP. (A) Before contact, the cell surface receptors and bright ligands in the bilayer are uniformly distributed. (B) After a period of contact formation lasting 20–40 min, the receptor and bright ligand accumulation in the contact area reaches equilibrium and remains stable for hours. (C) Patterned photobleaching of the entire contact area converts that bound and free bright ligands into dark ligands. (D) Contact recovery involves exchange of dark free ligands for bright free ligands by diffusion and then replacement of bound dark ligands by bound bright ligands by chemical dissociation and binding.
FIGURE 4.

(A) Bond-associated fluorescence intensity versus free ligand-associated fluorescence. (B) Zhu-Golan plot for CD16bNA2-IgG interaction. B is the density of bound ligands within the contact area, F is the density of free ligands in the bilayer, B/F is the ratio of the density of bound ligands density to free ligand density, and P is the ratio of contact area to full cell surface area. The slope of the linear fit and the x-intercept provide respective estimates for the binding affinity Ka and the receptor density before contact area formation, mr0 (indicated). The standard deviations of Ka and mr0 (indicated) are calculated from the 95% confidence interval of the fits.
Contact area formation involves global deformations of the cell from a sphere to an indented sphere and local deformations of the membrane from rough to smooth. Thus, the chemical reaction-diffusion process described above must be coupled to a mechanical process of cell deformation. Signaling-dependent reorganization of cytoskeleton and motor-driven transport of molecules into and away from the contact area also may take place, which can be quite complex. To isolate the chemical process from the mechanical and biological processes, we focused on established contact areas, defined as those contact areas the growth of which has already achieved steady state. RIM observations suggest that the size and shape of an established contact area are often stable and that the cell membrane has already reorganized to align with the lipid bilayer with no further change in time. Fluorescence microscopic observations also indicate that the process of ligand accumulation has reached a steady state, as the fluorescence intensity no longer changes in time; this suggests vanishing net diffusion into the contact area and vanishing net bond formation inside the contact area. However, unlike the mechanical equilibrium, which is static, the chemical equilibrium is dynamic. The dynamic nature of bond formation and dissociation at equilibrium has been demonstrated by a fluorescence recovery after photobleaching (FRAP) experiment in the contact area, hereafter called “contact area FRAP”. This experiment revealed rapid turnover of fluorescent bonds (complete turnover in ∼10 min for the fast dissociating CD2-CD58 interaction, see Figs. 5 B and 7 A below) (9). This difference between the mechanical equilibrium and chemical equilibrium allowed us to separate the two processes, as discussed below.
FIGURE 5.

Comparison of measured (dotted line) and fitted (curves) contact area FRAP time courses of (A) CD16bNA2-expressing CHO cells interacting with bilayers reconstituted with rabbit IgG in four densities (indicated) and (B) CD2-expressing Jurkat cells interacting with bilayers reconstituted with CD58 in three densities (indicated). Representative data are expressed as the spatially averaged, background-subtracted, total ligand-associated fluorescence intensity inside the contact area, FIin(t)–FIbdg, normalized by the background-subtracted, free ligand-associated fluorescence intensity outside the contact area before photobeaching, FIout(0−)–FIbdg. The number of repeats for each ligand density for each system is indicated in Fig. 6 on the top of the parameter value bar for that ligand density for that system.
FIGURE 7.

Representative data (points) and model fits (curves) of contact area FRAP (A) and recovery rate (B) time courses for the respective CD16bNA2-IgG (open circles) and CD2-CD58 (open triangles) systems. The solution of the coupled reaction-diffusion model used in the curve-fitting is described in the companion study (18). The number of repeats for each ligand density for each system is indicated in Fig. 6 on the top of the parameter value bar for that ligand density for that system.
For more than three decades, FRAP (also termed “fluorescence photobleaching recovery” or “FPR”) has been widely used to measure lateral diffusion of membrane proteins on the cell surface (11,12). It has been also appreciated that FRAP can yield information about chemical reactions (13–17). In conventional FRAP, a brief strong laser pulse is used to bleach the fluorescence of a small spot, which creates density gradients, as the bleached (dark) molecules are considered different species from the fluorescent (bright) molecules in terms of diffusion. Such gradients drive diffusion of the bright molecules into the bleached area and dark molecules out of the bleached area. Thus, the fluorescence recovery time course provides information about the molecular diffusivity, which can be extracted by fitting the solution of a diffusion model to the recovery data. In contact area FRAP, the bleached spot coincides with the entire contact area in which cell surface receptors interact with initially bright ligands that are all made dark by the laser pulse (Fig. 1 C). Importantly, contact area FRAP provides a new method for measuring receptor-ligand binding kinetics. The idea is to exploit the dynamic nature of the chemical equilibrium of receptor-ligand binding. A bound ligand can rebind after dissociation, and it can exchange place with another ligand that is free before dissociation because, either way, the respective densities of free ligands and bonds remain unchanged. After photobleaching, diffusion results in increases of bright free ligands and decreases of the dark free ligands inside the contact area over time. Because of the exchange between the free and bound ligands, the dark bound ligands are gradually replaced by the bright bound ligands (Fig. 1 D). In other words, the turnover of fluorescent bonds in the contact area is a result of dissociation of the dark bound ligands from the cell surface receptors and their formation of new bonds with bright free ligands. The contact area FRAP time courses, therefore, provides diffusion and kinetic information, which may be extracted if analyzed by a mathematical model for the reaction-diffusion process involved, which is the goal of this work. The theoretical development, solution methods, and mathematical properties of the model are presented in a companion article (18). This article also presents a refinement of the original contact area FRAP assay to generate data to experimentally test the model validity, which also allows evaluation of 2D kinetic rates and other parameters.
MATERIALS AND METHODS
Antibodies, protein, and cells
Monoclonal antibodies (mAb) CLBFcRgran1 (anti-CD16) (19) and TS2/9 (anti-CD58) (20) were obtained from hybridoma culture supernatants provided by Dr. P. Selvaraj (Emory University School of Medicine, Atlanta, GA). Alexa 488-conjugated rabbit anti-dinitrophenyl (DNP) immunoglobulin G (IgG) was obtained from Molecular Probes (Eugene, OR). Fluorescein isothiocyanate-conjugated anti-mouse IgG was obtained from Sigma Chemical (St. Louis, MO). Glycosylphosphatidylinositol-anchored CD58 was purified from human erythrocytes using TS2/9 affinity chromatography and labeled with Cy5 while still coupled to the beads as described previously (7).
Jurkat cells (clone JE6.1) constitutively expressing CD2 were obtained from the American Type Culture Collection (Rockville, MD). Chinese hamster ovary (CHO) cells transfected to express CD16bNA2 were a kind gift from Dr. P. Selvaraj (21). Jurkat and CHO cells were cultured in Roswell Park Memorial Institute medium with 10% fetal bovine serum according to standard practices. Transfection media were supplemented with 400 μg/ml hygromycin B as the selection agent.
Preparation of lipid bilayers reconstituted with ligands
Liposomes of 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(2,4-dinitrophenyl) (DNP-PE) or L-a-phosphatidylcholine (PTIC) with or without Cy5-CD58 were prepared via detergent dialysis. DNP-PE and PTIC lipids were purchased from Avanti Polar Lipids (Alabaster, AL). They were dissolved in chloroform at a concentration of 10 mg/ml. PTIC, alone or combined with DNP-PE in a 50/50 mixture (by mole fraction), was dried under vacuum for 1 h. PTIC or DNP-PE lipids were dissolved in TSA (25 mM Tris, 150 mM NaCl, 0.02% NaN3) containing 1.5% OG (n-octyl β-D-glucopyranoside) with or without Cy5-CD58 (sufficient to generate 1000–2000 molecules/μm2) to a concentration of 0.4 mM. The lipid mixture solutions were then injected into respective dialysis cassettes. Liposomes were formed during OG removal by dialysis, which was carried out in 1.5 L TSA for 36 h within a sterile dialysis tank that replaced TSA every 12 h.
Lipid bilayers were formed in a flow chamber (Focht Chamber System 2 (FCS2); Bioptechs, Butler, PA) on glass coverslips. The coverslips were rigorously cleaned previously by boiling in piranha solution (70% reagent grade sulfuric acid, 30% of 30% hydrogen peroxide) for 1 h at 90°C, followed by thorough rinses with deionized water, and then air-dried. Stock DNP- or CD58-containing liposomes were diluted by PTIC liposomes to obtain variable surface densities of DNP and CD58. A 2-μl drop of properly diluted liposome suspension was sandwiched between the Microaqueduct slide (Bioptechs) and a 40-mm diameter circular coverslip that were separated by a 0.25-mm spacer. The liposomes fused on the hydrophilic glass surface to form a continuous bilayer (6). The flow chamber was connected with two syringes, one filled with TSA containing 1% bovine serum albumin (BSA) and the other with HEPES buffer saline solution (HBS) containing 1% human serum albumin (HSA). After 15 min of incubation, the excess liposome solution was washed off the glass coverslip by injecting TSA/1% BSA through the chamber. After an additional 15 min of incubation, the TSA/1% BSA solution was washed off by injecting HBS/1% HSA through the chamber. The DNP lipid bilayers and the Cy5-CD58 bilayers were then ready to use. To coat IgG ligands on the DNP bilayers, rabbit anti-DNP IgG (10 μg/ml) was injected into the flow chamber for incubation for 30 min at room temperature. Unbound IgG was washed off with HBS/1% HSA.
Site density determination
Quantitative fluorescence measurements were used to determine the density of Alexa 488-conjugated IgG on the lipid bilayer. A range of known quantities of fluorescent IgG were added to a set of flow chambers. The chambers were preincubated with casein for 2 h to minimize nonspecific sticking of IgG to the coverslips. The fluorescence intensities were integrated over the height (optical slice z direction) to obtain the fluorescence emitted from the Alexa 488-IgG molecules in the volume above a unit area (1 μm2 in x, y plane), which was plotted against the amount of IgG in the same volume above the same unit area (Fig. 2 A). It is evident that the fluorescence intensity increases linearly with the number of IgG per unit area. Next, a set of bilayers were prepared by diluting the DNP-containing liposomes with PTIC liposomes at different ratios, which were incubated with the same fluorescent anti-DNP IgG to obtain a range of IgG site densities. The fluorescence intensities were measured using the same microscope settings and the same units (Fig. 2 B). After combining the data from Fig. 2, A and B, to eliminate the setting- and unit-dependent fluorescence intensity values, we obtained a calibration curve for determining the IgG site densities in different lipid bilayers prepared using known percentages of DNP liposomes.
FIGURE 2.

(A) Representative of three independent experiments. Fluorescence intensity integrated over the optical slice height emitted from the Alexa 488-conjugated rabbit anti-DNP IgG molecules in the volume above a 1-μm2 area is plotted versus the amount of IgG in the volume above a unit area. The linear relationship between the fluorescence intensity and the number of IgG per unit area provides a calibration curve for determination of site density of IgG bound to the lipid bilayer. (B) Representative of three independent experiments. Fluorescence intensity of Alexa 488-conjugated rabbit anti-DNP IgG bound to bilayers prepared with different dilutions of stock DNP-CAP-PE liposomes. IgG binding increases with increasing DNP concentration nearly linearly when the DNP liposomes is <0.5% but tends to saturate at higher DNP concentrations.
The CD58 densities on lipid bilayers prepared from dilutions of the stock CD58 liposomes were determined via an immunoradiometric assay. Glass-supported lipid bilayers were prepared on circular 12-mm diameter coverslips by sandwiching 8 μl of the liposome solution between two coverslips in a 24-well plate. After 5 min of incubation, the coverslips were covered with 1 ml TSA/1% BSA. The top coverslip was removed and discarded. The bottom coverslip was allowed to incubate for an additional 15 min. The coverslips were then incubated with a saturating concentration (>10 μg/ml) of 125I-radiolabeled anti-CD58 mAb TS2/9 for 60 min at room temperature on a shaker. After seven washes with TSA, the bilayers were dissolved by 1 ml/1 M NaOH and transferred to a tube for counting. Radioactivities of the samples were measured using a gamma counter (Cobra II Auto Gamma; PerkinElmer, Wellesley, MA). The specific activity per nanogram of antibody was determined by measuring the radioactivities of 125I anti-CD58 mAb TS2/9 in a range from 1 ng to 1 μg. The data were then used as calibration to calculate the number of ligands per unit area for glass-supported lipid bilayers made from liposome preparations with varying concentrations of Cy5-CD58.
Using the above calibration curves, we determined the total ligand (IgG or CD58) site density for any bilayer prepared for any experiment. To determine the relative bright ligand density in a contact area FRAP experiment, we normalized the fluorescence intensity measured in a particular region at a particular time of interest by the fluorescence intensity of the total ligand measured for that experiment. This method is based on the assumption that the microscope settings and the choice of the arbitrary units would only affect the slope of the line but not the linearity of the relationship seen in Fig. 2 A, which we believe is a reasonable hypothesis.
Contact area FRAP experiment
CD16bNA2-expressing CHO cells or CD2-expressing Jurkat cells, respectively, were injected into FCS2 chambers (Bioptechs), where glass-supported lipid bilayers reconstituted with fluorescently labeled rabbit IgG or CD58 were prepared. The FCS2 chamber (Bioptechs) was placed on the stage of a previously described custom-made inverted microscope (10), which has fluorescence, RIM, and bright-field imaging capacities and is equipped with photobleaching lasers and image acquisition software. Measurements of contact area size and fluorescence intensity were made using spectrum image analysis software (IPLab; BioVision Technologies, Exton, PA). The formation of contact area was monitored by RIM and fluorescence microscopy. After stable contact areas had been established, as judged by stable contact area size and fluorescence intensity inside the contact area, photobleaching was performed on selected contact areas, and the time course of fluorescence recovery was recorded. Control experiments showed that neither CHO nor Jurkat cells formed a contact area with ligand-free PTIC bilayers.
Before injecting the cells, the quality of the bilayer was assessed by fluorescence microscopy and conventional FRAP. Uniform fluorescence intensity, a diffusivity of Dl > 1 μm2 s−1, and fractional recovery of >0.9 were used as the criteria for a good bilayer. The diffusivity was estimated from the radius of the bleached circle, r0, and the time to achieve half-maximum recovery, t1/2, according to 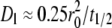 (10,15), which is an approximate formula that has been shown to agree well with the results of curve-fitting the entire FRAP time course with the solution of the diffusion equation (22). All contact area FRAP data presented in this study were obtained using bilayers that met these criteria.
(10,15), which is an approximate formula that has been shown to agree well with the results of curve-fitting the entire FRAP time course with the solution of the diffusion equation (22). All contact area FRAP data presented in this study were obtained using bilayers that met these criteria.
Data analysis and curve-fitting
Data were analyzed using the coupled diffusion-kinetics model developed in our companion study (18). We measured the background fluorescence intensity (FIbdg) and the fluorescence intensity far away from the contact area before photobleaching, FIout(0−) (averaged over several spots), as well as the fluorescence intensity inside the contact area before photobleaching and at various time points after photobleaching, FIin(0−) and FI(tn) (n = 1, 2, …, …, N) (averaged over the contact area), respectively. The background-subtracted normalized fluorescent intensity, FI* = (FIin − FIbgd)/[FIout(0−) − FIbgd], corresponds to the dimensionless total bright ligand density, 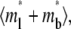 at these time points, tn, solved from the model described in our companion study (18). The nonrecoverable fraction was calculated from fn = 1 − [FIin(∞) − FIbdg]/[FIin(0−) − Fbdg] where FIin(∞) is the steady-state value obtained by extrapolating the FI(tn) data to infinity. The dimensionless recovery rate data R*(tn) were obtained by numerical differentiation of FI*(tn), which corresponded to the dimensionless time rate of replacing the dark ligands by the bright ligands Q*(tn) solved from the previously described model (18). The model solution was fit to the data using the least-squares method by minimizing χ2 as follows:
at these time points, tn, solved from the model described in our companion study (18). The nonrecoverable fraction was calculated from fn = 1 − [FIin(∞) − FIbdg]/[FIin(0−) − Fbdg] where FIin(∞) is the steady-state value obtained by extrapolating the FI(tn) data to infinity. The dimensionless recovery rate data R*(tn) were obtained by numerical differentiation of FI*(tn), which corresponded to the dimensionless time rate of replacing the dark ligands by the bright ligands Q*(tn) solved from the previously described model (18). The model solution was fit to the data using the least-squares method by minimizing χ2 as follows:
 |
(1) |
where wn are weight coefficients. Based on the numerical analysis described in the companion study (18), we chose wn > 0.5 for the early time points to increase the weight of the contact area FRAP recovery data FI*; we used wn < 0.5 for the later time points, however, to increase the weight of the recovery rate data R*. Note that time t has been scaled using the diffusivity Dl of the free ligand in the bilayer and the radius r0 of the contact area to become dimensionless time t* 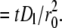 Curve-fitting returns best-fit parameter values for the product mrkf of receptor site density mr and forward rate constant kf, the reverse rate constant kr, and the retarded diffusion fraction ξ.
Curve-fitting returns best-fit parameter values for the product mrkf of receptor site density mr and forward rate constant kf, the reverse rate constant kr, and the retarded diffusion fraction ξ.
Zhu-Golan plot analysis that provides a separate measurement of 2D binding affinity Ka and the initial receptor site density mr0 is described in the Results section.
RESULTS
Equilibrium analysis
Upon establishment of the stable contact area, the receptor-ligand binding kinetics has reached equilibrium. The density of bound ligands inside the contact area is related to the free-ligand density in the bilayer via the 2D binding affinity. A 2D Scatchard plot analysis was used to make the first 2D Kd (≈21 μm−2) measurement for the CD2-CD58 interaction (7). This method was later modified to a Zhu-Golan plot analysis taking into account that only the receptors inside (not those outside) the contact area are capable of interacting with the ligands and only the mobile (not the immobile) fraction of receptors can diffuse from outside into the contact area, which corrected the Kd (≈1–7 μm−2) for the CD2-CD58 interaction (9,23,24). The present study obtained a value of 5 μm−2, which is similar to the value obtained by Dustin and colleagues (9).
The 2D equilibrium dissociation constant was also determined for the interaction between human CD16bNA2 and rabbit IgG Fc. A saturating concentration of Alexa 488-conjugated anti-DNP IgG was allowed to incubate with a series of bilayers prepared by diluting the DNP-containing liposomes with PTIC liposomes at different ratios to obtain a range of total ligand densities. The area of stable contact between the bilayer and a CD16bNA2-expressing CHO cell was found to increase with the bilayer DNP percentage, as expected (Fig. 3). The free ligand density on the bilayer was determined by converting the fluorescence intensity outside the contact area using the calibration curves in Fig. 2. The total number of bonds in the contact area was determined by multiplying the contact area (Ac) by the bond density that was determined from the difference between the fluorescence intensities outside and inside the contact area. The measured relationship between the two is plotted in Fig. 4 A, which resembles a typical saturation binding curve. B is the density of bound ligands within the contact area, F is the density of free ligands in the bilayer, B/F is the ratio of the density of bound ligands density to free ligand density, and P is the ratio of contact area to full cell surface area. To linearize the curve, the bond density was divided by the free-ligand density to obtain the B/F data. The total number of bonds is divided by the total surface area of the cell (AT) to yield the B × P data (P = Ac/AT). To measure AT, the cells were swollen by hypotonic media to smooth their surface roughness, because it is assumed that Ac is a smooth surface in established contact areas (see Discussion). Here, the B/F versus B × P plot is the so-called “Zhu-Golan plot” (Fig. 4 B), where the negative slope of the line is the 2D binding affinity and the x-intercept is the initial receptor density mr0 before the contact area formation (assuming that the receptors were uniformly distributed initially and the mobile fraction fm = 1) (9). The reciprocal of the binding affinity yields a Kd value of 0.16 μm−2.
FIGURE 3.

Contact area Ac measured by RIM is plotted versus the DNP concentration in the bilayer. Ac increases with increasing DNP concentration nearly linearly when DNP < 0.1% but tends to saturate at higher DNP concentrations.
Contact area FRAP time course depends on kinetic rates and ligand density
Contact area FRAP experiments were performed on two systems: 1), CD16bNA2-expressing CHO cells forming contact areas on a DNP bilayer bound with Alexa 488-conjugated rabbit anti-DNP IgG; and 2), CD2-expressing Jurkat cells forming contact areas on a bilayer reconstituted with Cy5-conjugated glycosylphosphatidylinositol-anchored CD58. The basis for the contact area FRAP method is the dynamic nature of the binding reaction as revealed by the recovery of fluorescence after photobleaching (10), which suggests that binding continues to proceed in both directions at equal rates that cancel each other after equilibrium is achieved. After the free and bound ligands were bleached, fluorescence could recover no more than the level outside the contact area should the receptor-ligand bonds be stable. This is because the bound dark ligands would have been constrained within the contact area. Therefore, only free dark ligands could diffuse out of the contact area and be replaced by free bright ligands that diffused into the contact area. In contrast, if the bonds were dynamic (i.e., underwent continuous dissociation and rebinding), fluorescence recovery could reach as high as the prebleached level, which is much higher than the level outside the contact area. These hypotheses were confirmed by the later obtained two receptor-ligand systems studied in this article as shown in Fig. 5, which depicts representative contact area FRAP time courses of the two systems.
It is apparent that the CD16-IgG system (Fig. 5 A) recovered much less rapidly and completely than the CD2-CD58 system (Fig. 5 B). The former required >1 h to regain equilibrium, and the fluorescence recovery was still largely incomplete (<50%); the latter required <10 min, and the fluorescence recovery was nearly complete (>90%). Thus, the measured contact area FRAP time courses were specific to the molecular and cellular systems in question. The contact area FRAP time course was also sensitive to the ligand density ml on the lipid bilayer for both systems, indicating that ml is a controlling parameter of the observed kinetic process. The differences in the contact area FRAP time courses for different ligand densities were not due to contact area changes. Consistent with the data in Fig. 3, the contact area was insensitive to the ligand density in the range of ml tested, with Ac = 45.5 ± 6.6, 41.8 ± 5.8, 36.0 ± 6.0, and 53.5 ± 16.5 μm2 for ml = 490, 630, 650, and 680 μm−2, respectively, for the CD16bNA2-IgG system and Ac = 99 ± 36, 116 ± 28, and 93 ± 12 μm2 for ml = 100, 200, and 400 μm−2, respectively, for the CD2-CD58 system. The coupled reaction-diffusion model described in the companion study (18) predicts that the contact area FRAP time course depends on the kinetic rates kf and kr (binding affinity Ka = kf/kr) of the interacting receptors and ligands, their densities mr and ml, the size of the contact area Ac, and the diffusion coefficients of ligands outside and inside the contact area Dl and ξDl, respectively (18). The observations that the data respond to the changes in the receptor-ligand kinetic rates and the ligand density (above) have provided preliminary qualitative test of the model described in the companion study (18).
Significant fraction of fluorescence is nonrecoverable in the CD16bNA2-IgG system
The contact area FRAP of the CD2-CD58 system reached a plateau that was only slightly lower than the fluorescence level before photobleaching (Fig. 5 B), confirming that these bonds were highly dynamic, as observed previously (10). By comparison, the plateau fluorescence level for the CD16bNA2-IgG system was significantly lower than its prebleached level (Fig. 5 A), suggesting a fraction of the bonds was stable or resides in regions that were inaccessible to diffusion (see Discussion). These nonrecoverable fractions of fluorescence resemble the immobile fractions of conventional FRAP in studies of the lateral mobility of cell surface molecules. On the glass-supported lipid bilayer, however, the immobile fraction was quite low, as nearly complete recovery in the conventional FRAP was used as a criterion for selecting good bilayers for experiments (see Materials and Methods). The nonrecoverable fraction fn is plotted versus the ligand density (ml) for the CD16bNA2-IgG system (Fig. 6 A) and the CD2-CD58 system (Fig. 6 B). As observed in the representative data (Fig. 5), fn is much higher for the former system than the latter system. Importantly, fn is insensitive to the ml changes in both systems.
FIGURE 6.

Fraction of nonrecoverable fluorescence fn for the (A) CD16bNA2-IgG and (B) CD2-CD58 systems. Results are expressed as mean ± SE of data evaluated from 3–6 (indicated) contact area FRAP time courses measured at the indicated ligand densities.
Recovery in different systems is limited by different processes
The contact area FRAP of the two receptor-ligand systems exhibited very different recovery times, which can be more clearly seen when the normalized fluorescence intensity is plotted versus the dimensionless time to account for the effect of different contact area sizes, as shown in Fig. 7 A by representative data. The transient phase of the recovery time course of the CD16bNA2-IgG system (open circles) is much longer than that of the CD2-CD58 system (open triangles), which is comparable to that of the conventional FRAP of the lipid bilayer alone (18). In the companion study (18), we used numerical simulations to study the coupled reaction-diffusion model proposed to govern the contact area FRAP process. Two types of behaviors were identified: reaction-limited and diffusion-limited processes, which respectively correspond to the conditions kr*/ξ ≪ 1 and kr*/ξ ≫ 1, where 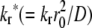 is the dimensionless off-rate and ξ (0 ≤ ξ ≤ 1) is the ratio of diffusivity of free ligands inside the contact area to that of outside the contact area. Reaction-limited recoveries exhibit two timescales: one associated with the fast process of replacing the dark free ligands by diffusion and the other associated with the slow process of replacing the dark bound ligand by reaction. By comparison, diffusion-limited recoveries display a single timescale because reaction has reached equilibrium as soon as diffusion brings free bright ligands into the contact area (18). Both types of behavior can be seen in Fig. 7 A—the CD16bNA2-IgG data show a two-phase recovery, whereas the CD2-CD58 data show a single-phase recovery.
is the dimensionless off-rate and ξ (0 ≤ ξ ≤ 1) is the ratio of diffusivity of free ligands inside the contact area to that of outside the contact area. Reaction-limited recoveries exhibit two timescales: one associated with the fast process of replacing the dark free ligands by diffusion and the other associated with the slow process of replacing the dark bound ligand by reaction. By comparison, diffusion-limited recoveries display a single timescale because reaction has reached equilibrium as soon as diffusion brings free bright ligands into the contact area (18). Both types of behavior can be seen in Fig. 7 A—the CD16bNA2-IgG data show a two-phase recovery, whereas the CD2-CD58 data show a single-phase recovery.
Reaction-diffusion model fits contact area FRAP data
Numerical simulations in the companion study also suggest a strategy to determine the parameters of the coupled kinetic and diffusive process (18). The contract area FRAP data (Fig. 7 A) were numerically differentiated to obtain recovery rate data (Fig. 7 B). The solution of the coupled reaction-diffusion model (curves) was simultaneously fit to the recovery (Fig. 7 A) and rate of recovery (Fig. 7 B) data (points) by minimizing the χ2 defined by Eq. 1. Curves that fit the representative data of different ligand densities in Fig. 5 are plotted with the data points. All the data show excellent agreement for both CD16bNA2-IgG and CD2-CD58 systems, thus supporting the validity of the model.
Determining retarded diffusion and binding kinetics
To further test the validity of the model, we examined dependence (or the lack thereof) of the model parameters that best fit the data on the changes in the ligand density for each system. The self-assembly of a stable contact area might retard diffusion inside the contact area, which could also contribute to the much slower contact area FRAP time courses than the conventional FRAP time course. Whether diffusion is retarded can be determined from the time required for the fluorescence inside the contact area to recover half the fluorescence level outside the contact area, because this initial recovery can be accomplished by diffusion without dissociation of the dark bound ligands. A much longer initial recovery time than the half-time of free diffusion, 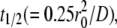 is indicative of retarded diffusion. Examination of the contact area FRAP time courses suggests that diffusion of unbound ligands was indeed retarded inside the stable contact areas formed between the CHO cells expressing CD16bNA2 and the lipid bilayers reconstituted with IgG ligands (Figs. 5 A and 7 A). This observation can be quantified by the fractional diffusivity ξ—the larger the ξ-value, the less retarded the diffusion. The best-fit values that ξ returned from curve-fitting confirm that diffusion is retarded substantially for the CD16bNA2-IgG system (Fig. 8 A) but much less for the CD2-CD58 system (Fig. 8 B). Interestingly, the extent of this retardation increases with increasing ligand density on the bilayer (Fig. 8), presumably because of the increasing density of bonds in the contact area (see Discussion).
is indicative of retarded diffusion. Examination of the contact area FRAP time courses suggests that diffusion of unbound ligands was indeed retarded inside the stable contact areas formed between the CHO cells expressing CD16bNA2 and the lipid bilayers reconstituted with IgG ligands (Figs. 5 A and 7 A). This observation can be quantified by the fractional diffusivity ξ—the larger the ξ-value, the less retarded the diffusion. The best-fit values that ξ returned from curve-fitting confirm that diffusion is retarded substantially for the CD16bNA2-IgG system (Fig. 8 A) but much less for the CD2-CD58 system (Fig. 8 B). Interestingly, the extent of this retardation increases with increasing ligand density on the bilayer (Fig. 8), presumably because of the increasing density of bonds in the contact area (see Discussion).
FIGURE 8.

Fractional diffusivity inside the contact area, or the ratio of diffusion coefficient inside the contact area to that outside, ξ, for the (A) CD16bNA2-IgG and (B) CD2-CD58 systems. Results are expressed as mean ± SE. of data evaluated from 3–6 (indicated) contact area FRAP time courses measured at the indicated ligand densities.
By comparison, the reverse-rate kr is insensitive to the changes in the ligand density ml, as expected (Fig. 9). This result supports the validity of the model described in the companion study (18), which assumes kr and ml to be independent model parameters. Significantly, our data suggest that CD2-CD58 bonds dissociate >100-fold more rapidly than CD16bNA2-IgG bonds.
FIGURE 9.

Off-rate of receptor-ligand dissociation kr for the (A) CD16bNA2-IgG and (B) CD2-CD58 systems. Results are expressed as mean ± SE of data evaluated from 3–6 (indicated) contact area FRAP time courses measured at the indicated ligand densities.
Fitting the contact area FRAP data also returns the dimensionless binding affinity kf*/kr*, which equals the product of the dimensional affinity Ka and the free receptor density mr. Dividing the best-fit mrKa value by the Ka value estimated from the Zhu-Golan plot analysis (Fig. 4 B) provides a value for mr, which decreases with increasing ligand density ml (Fig. 10). This is expected because the higher the ligand density originally reconstituted in the bilayer, the higher the bond density (Fig. 4 A), which lowers the free receptor density in the contact area. A comparison of the mr values (Fig. 10 A) to the initial receptor density mr0 estimated from the Zhu-Golan plot analysis (Fig. 4 B) demonstrates that >90% of the CD16bNA2 in the contact area are engaged by IgG.
FIGURE 10.

Free receptor density inside the contact area mr for the (A) CD16bNA2-IgG and (B) CD2-CD58 systems calculated using the model (18). Results are shown as mean ± SE of data evaluated from 3–6 (indicated) contact area FRAP time courses measured at the indicated ligand densities.
DISCUSSION
This article has a tripartite objective: 1), validate the model developed in the companion study (18); 2), apply the model to make in situ measurement of parameters important to the formation of an immunological synapse; and 3), explore the unexpected biology of contact areas as revealed by contact area FRAP.
Validity of the model
To test the validity of the model, we varied the experimental parameters and examined whether the recovery time course responded properly and whether the mathematical model fit the data appropriately. The varied parameters were kinetic rates of the molecular system and ligand density on the bilayer. To vary the kinetic rates, two receptor-ligand systems—CD16bNA2-IgG and CD2-CD58 interactions—were used because previous solution measurements suggest that these interactions have different 3D kinetics (25–27). Different recovery timescales were correspondingly observed: the CD16bNA2-IgG system had slow recovery time on the order of tens of minutes whereas the CD2-CD58 system had a fast recovery time on the order of minutes (Figs. 5 and 7). Such a correlation qualitatively supports the conceptualization of the model as depicted in Fig. 1. Furthermore, the model was able to fit well these recovery data of different timescales and returned from curve-fitting fast and slow kinetic rates (Fig. 9), thereby quantitatively supporting the validity of the model.
To further test the model validity and to ensure confidence in the parameters estimated from the model fits, contact area FRAP experiments were performed using lipid bilayers reconstituted with different densities of ligands before contact area formation. Higher ligand density resulted in a more rapid initial time rate of increase and a higher equilibrium level of the contact area FRAP time course (Fig. 5), as predicted by the law of mass action. However, these data should be described by the same model with the same or related parameters. Indeed, with independently measured ligand densities and contact area sizes as input, the model fit all data equally well (Fig. 5 and data not shown). The estimated model parameters make good sense—the fractional diffusion retardation ξ decreases with increasing ligand density ml (Fig. 8). The reverse-rate kr does not depend on ml (Fig. 9). The free receptor density mr inside the contact area decreases with increasing ml (Fig. 10). Together, these data provide strong support for the validity of the coupled reaction-diffusion model developed in the companion study (18).
Comparison with published kinetic rates
Because one objective of this work is to measure the kinetic rates and binding affinities, we must compare the reported values to those measured previously by different 3D and 2D techniques. The principal method for determining 3D kinetic rates (in which one or both ligands diffuses in 3D) is surface plasmon resonance (SPR). By SPR, the 3D CD2-CD58 binding affinity is 5 × 105 M−1 and a reverse-rate of ≥ 6 s−1 (9,25). A flow chamber method reported an extrapolated zero-force reverse-rate of 7.8 s−1 (28). In this work, we found a reverse-rate of 0.074 ± 0.005 s−1 for this interaction and a calculated (by multiplying by the binding affinity of 0.2 μm2) forward-rate of 0.015 ± 0.001 μm2 · s−1. The kinetic rates and binding affinities of soluble CD16bNA2 interacting with human IgG were measured by two SPR studies (26,27). One study (26) reported respective binding affinities for human IgG1 and IgG3 of 13 and 2.6 × 105 M−1, respectively, for glycosylated CD16bNA2 and of 11 and 4.2 × 105 M−1, respectively, for aglycosylated CD16bNA2. The reverse-rates were similarly slow, ranging from 0.001–0.002 s−1 (26). The other study (27), which was performed by the same group reporting CD2-CD58 data, demonstrated comparable binding affinities of 5–7 × 105 M−1, but >2 orders of magnitude faster reverse-rates of 0.7 s−1 for aglycosylated CD16bNA2. These SPR data suggest that there may be two binding modes for CD16bNA2 to IgG Fc—a fast dissociating mode and a slow dissociating mode. We also found two modes of interaction between CD16bNA2 and rabbit IgG, one had a kr = 0.0011 ± 0.0001 s−1 and kf = 0.0070 ± 0.0007 μm2·s−1 and the other was not reversible on the timescale of our contact FRAP measurements (see discussion of nonrecoverable fraction below). The data for CD2-CD58 demonstrate that 2D reverse-rates are ∼100-fold slower than corresponding 3D reverse-rates. Taking the 3D reverse-rate from the same group that determined the 3D reverse-rate for CD2-CD58, the 2D reverse-rate for CD16bNA2 interaction with rabbit IgG Fc is 600-fold slower than the 3D reverse-rate. One possible explanation for this very slow 2D reverse-rate in an equilibrated contact area is that the chemical reverse-rate remains unchanged, but the receptor and ligand tend to remain together and rebind hundreds of times before diffusion away from the encounter complex. Another possibility, which seems less likely, is that the chemistry of the bond changes in the contact area such that it dissociates more slowly.
The 2D kinetics of several glycosylated CD16 membrane anchor isoforms interacting with IgG of different isotypes and from different species have been measured by micropipette (29–34) and centrifugation (35) methods. These studies reported 2D effective binding affinities ranging from 0.1–8 × 10−6 μm4 for CD16 expressed on CHO or K562 cells (29–35), which correlate with the 3D binding affinities for the same IgGs of the same CHO cell CD16 isoforms measured by competitive inhibition (30). We have previously discussed the 3–4 orders of magnitude difference in 2D affinities measured by mechanical methods like micropipette and centrifugation and fluorescence methods like those described in this study (36). The mechanical methods are calibrated for statistical analysis of small numbers of bonds. By comparison, the fluorescence methods are calibrated to work with thousands of interactions in an equilibrated contact area that persist when the relevant contact area is estimated. Contact area FRAP and Golan-Zhu analysis of the CD16bNA2 interaction with rabbit IgG Fc confirms this dramatic difference in equilibrium binding and now, based on this study, in kinetic rates between fluorescence-based methods and mechanical methods. The major difference between the two situations that we previously envisioned was the higher degree of order leading to much faster forward rates in the equilibrated contact area (36). We believe that this is still the most likely explanation for these differences; however, we did not anticipate the 100–600-fold slower reverse-rate in the contact area, which we had argued would remain the same in 3D and different 2D settings. As discussed above, we cannot determine from our data whether the longer interaction in the 2D setting is due to many cycles of dissociation and rebinding before complete separation or due to a chemical change in the contact area.
Possible explanations for nonrecoverable fraction
Nearly full fluorescence recovery is expected for conventional FRAP performed on lipid-anchored molecules reconstituted in glass-supported synthetic bilayer membranes. By comparison, a substantial immobile fraction usually exists when proteins on a live cell surface are examined. Similarly, depending on the molecular systems examined, we and others have found variable fractions of nonrecoverable fluorescence in the contact area FRAP experiment (Fig. 6). We were surprised to observe significant nonrecoverable fluorescence in CD16bNA2-IgG-mediated contact areas. This is mechanistically different from the immobile fraction in the conventional FRAP experiment because, in this study, the ligands remained anchored to the lipid bilayer, which presumably does not change its fluidic properties even in a contact area where some of the ligands bind with the cell surface receptors. The failure to dissociate has been seen previously in the immunological synapse between T cells and supported planar bilayers, in which supramolecular activation clusters are formed (37) that in some way render TCR-pMHC interactions nonrecoverable (38). TCR and pMHC ligands have been shown previously to undergo extensive aggregation in solution (39), which could explain the nonrecoverable fluorescence through structural/chemical changes leading to a slowing of the 3D reverse-rate. Similarly, the measurement of 100-fold different reverse-rates for CD16bNA2 interaction with Fc suggests that two modes of interaction are possible. If the interactions in equilibrated contact areas have reverse-rates that are at least 100-fold slower than in 3D, then the slower measured rate of 0.001–0.002 s−1 would translate to a rate of 0.000015 s−1 in a contact area. This would correspond to a recovery half-time of ∼12 h, which would appear nonrecoverable in our experiments. Another possible explanation for nonrecoverable fluorescence is that certain areas become inaccessible to free ligands, thus precluding exchange. Although we have observed reductions of 20–40% in free ligand density in contact areas, we have never observed complete exclusion in light microscopy experiments; however, we cannot rule out that functionally relevant exclusion zones exist on smaller length scales in the contact area. Biologically, the existence of nonrecoverable modes of interaction may enhance adhesion and some modes of signaling, but poses a challenge for fast-moving immune cells that would need to resolve these apparently less dynamic interactions to move within tissues.
Possible explanation for retarded diffusion
The diffusion within the contact area was reduced 2 orders of magnitude for the CD16 system, which seemed surprising at first glance. However, closer examination of the geometry reveals that diffusion within the contact area requires an unbound IgG ligand of the dimension of ∼15 nm to maneuver around many posts of similar size through a gap of ∼20 nm spanning the cell membrane and the lipid bilayer. Obviously, this is a much more difficult task than diffusion outside the contact area where the low ceiling and the dense posts are absent. Therefore, the retarded diffusion may simply be caused by a “traffic jam” within the contact area. This hypothesis is consistent with the observation that the fractional diffusion retardation ξ decreases with increasing ligand density (Fig. 8 A) and that the much higher affinity CD16bNA2-IgG interaction results in much lower ξ-values than the much lower affinity CD2-CD58 interaction (Fig. 8).
CONCLUSIONS
We have validated the model (18) for contact FRAP using two biological systems over a range of ligand densities. To our surprise, we have learned that the effective reverse-rate is at least 100-fold slower in an equilibrium contact area than in solution. We have confirmed by direct comparison that affinity and kinetic measurements in stable cell contacts mediated by many interactions have a 3–4 orders of magnitude higher affinity than the same system measured by mechanical methods that form one or a few interactions. We also found evidence for two modes of CD16bNA2 interaction with Fc that lead to recoverable and nonrecoverable fractions. The nonrecoverable fraction may be biologically relevant in forming more stable platforms for signaling or adhesion.
Acknowledgments
We thank S. K. Bromley for teaching the contact area FRAP measurement method to T.P.T. and P. Selvaraj for providing CD16bNA2-expressing CHO cells.
This work was supported by the National Institutes of Health (AI38282 to C.Z. and AI44931 to M.L.D.), a grant from the Whitaker Foundation (M.L.D. and C.Z.), as well as a grant from the National Science Foundation of China (10372118) and a grant from the Science Foundation of Guangdong, China (04009767 to J.W.). T.P.T. was also partially supported by the National Institutes of Health (GM08433).
Timothy P. Tolentino and Jianhua Wu contributed equally to this work.
Editor: Elliot L. Elson.
References
- 1.Dustin, M. L., and D. R. Colman. 2002. Neural and immunological synaptic relations. Science. 298:785–789. [DOI] [PubMed] [Google Scholar]
- 2.Vasioukhin, V., C. Bauer, M. Yin, and E. Fuchs. 2000. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 100:209–219. [DOI] [PubMed] [Google Scholar]
- 3.Somersalo, K., N. Anikeeva, T. N. Sims, V. K. Thomas, R. K. Strong, T. Spies, T. Lebedeva, Y. Sykulev, and M. L. Dustin. 2004. Cytotoxic T lymphocytes form an antigen-independent ring junction. J. Clin. Invest. 113:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dustin, M. L., M. W. Olszowy, A. D. Holdorf, J. Li, S. Bromley, N. Desai, P. Widder, F. Rosenberger, P. A. van der Merwe, P. M. Allen, and A. S. Shaw. 1998. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell. 94:667–677. [DOI] [PubMed] [Google Scholar]
- 5.McCloskey, M. A., and M. M. Poo. 1986. Contact-induced redistribution of specific membrane components: local accumulation and development of adhesion. J. Cell Biol. 102:2185–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McConnell, H. M., T. H. Watts, R. M. Weis, and A. A. Brian. 1986. Supported planar membranes in studies of cell-cell recognition in the immune system. Biochim. Biophys. Acta. 864:95–106. [DOI] [PubMed] [Google Scholar]
- 7.Dustin, M. L., L. M. Ferguson, P.-Y. Chan, T. A. Springer, and D. E. Golan. 1996. Visualization of CD2 interaction with LFA-3 and determination of the two-dimensional dissociation constant for adhesion receptors in a contact area. J. Cell Biol. 132:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Izzard, C. S., and L. R. Lochner. 1976. Cell-to-substrate contacts in living fibroblasts: an interference reflexion study with an evaluation of the technique. J. Cell Sci. 21:129–159. [DOI] [PubMed] [Google Scholar]
- 9.Dustin, M. L., D. E. Golan, D. M. Zhu, J. M. Miller, W. Meier, E. A. Davies, and P. A. van der Merwe. 1997. Low affinity interaction of human or rat T cell adhesion molecule CD2 with its ligand aligns adhering membranes to achieve high physiological affinity. J. Biol. Chem. 272:30889–30898. [DOI] [PubMed] [Google Scholar]
- 10.Dustin, M. L. 1997. Adhesive bond dynamics in contacts between T lymphocytes and glass-supported planar bilayers reconstituted with the immunoglobulin-related adhesion molecule CD58. J. Biol. Chem. 272:15782–15788. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson, K., Z. Derzko, E. S. Wu, Y. Hou, and G. Poste. 1976. Measurement of the lateral mobility of cell surface components in single, living cells by fluorescence recovery after photobleaching. J. Supramol. Struct. 5:565–576. [DOI] [PubMed] [Google Scholar]
- 12.Elson, E. L., J. Schlessinger, D. E. Koppel, D. Axelrod, and W. W. Webb. 1976. Measurement of lateral transport on cell surfaces. Prog. Clin. Biol. Res. 9:137–147. [PubMed] [Google Scholar]
- 13.Axelrod, D., D. E. Koppel, J. Schlessinger, E. Elson, and W. W. Webb. 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 16:1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Icenogle, R. D., and E. L. Elson. 1983. Fluorescence correlation spectroscopy and photobleaching recovery of multiple binding reactions. II. FPR and FCS measurements at low and high DNA concentrations. Biopolymers. 22:1949–1966. [DOI] [PubMed] [Google Scholar]
- 15.Elson, E. L., and H. Qian. 1989. Interpretation of fluorescence correlation spectroscopy and photobleaching recovery in terms of molecular interactions. Methods Cell Biol. 30:307–332. [DOI] [PubMed] [Google Scholar]
- 16.Oancea, E., M. N. Teruel, A. F. Quest, and T. Meyer. 1998. Green fluorescent protein (GFP)-tagged cysteine-rich domains from protein kinase C as fluorescent indicators for diacylglycerol signaling in living cells. J. Cell Biol. 140:485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Presley, J. F., T. H. Ward, A. C. Pfeifer, E. D. Siggia, R. D. Phair, and J. Lippincott-Schwartz. 2002. Dissection of COPI and Arf1 dynamics in vivo and role in Golgi membrane transport. Nature. 417:187–193. [DOI] [PubMed] [Google Scholar]
- 18.Wu, J., Y. Fang, V. I. Zarnitsyna, T. P. Tolentino, M. L. Dustin, and C. Zhu. 2008. A coupled diffusion-kinetics model for analysis of contact area FRAP experiment. Biophys. J. 95:910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selvaraj, P., O. Carpen, M. L. Hibbs, and T. A. Springer. 1989. Natural killer cell and granulocyte FcgRIII (CD16) differ in membrane anchor and signal transduction. J. Immunol. 143:3283–3288. [PubMed] [Google Scholar]
- 20.Tiefenthaler, G., M. L. Dustin, T. A. Springer, and T. Hunig. 1987. Serologic cross-reactivity of T11 target structure and lymphocyte function-associated antigen 3. Evidence for structural homology of the sheep and human ligands of CD2. J. Immunol. 139:2696–2701. [PubMed] [Google Scholar]
- 21.Nagarajan, S., S. Chesla, L. Cobern, P. Anderson, C. Zhu, and P. Selvaraj. 1995. Ligand binding and phagocytosis by CD16 (Fc gamma receptor III) isoforms. Phagocytic signaling by associated zeta and gamma subunits in Chinese hamster ovary cells. J. Biol. Chem. 270:25762–25770. [DOI] [PubMed] [Google Scholar]
- 22.Tolentino, T. P. 2002. Measuring ligand diffusivity and receptor binding kinetics within a cell membrane contact area. M.S. thesis. Georgia Institute of Technology, Atlanta.
- 23.Zhu, D. M., M. L. Dustin, C. W. Cairo, and D. E. Golan. 2007. Analysis of two-dimensional dissociation constant of laterally mobile cell adhesion molecules. Biophys. J. 92:1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu, D. M., M. L. Dustin, C. W. Cairo, H. S. Thatte, and D. E. Golan. 2006. Mechanisms of cellular avidity regulation in CD2–CD58-mediated T cell adhesion. ACS Chem. Biol. 1:649–658. [DOI] [PubMed] [Google Scholar]
- 25.van der Merwe, P. A., A. N. Barclay, D. W. Mason, E. A. Davies, B. P. Morgan, M. Tone, A. K. C. Krishnam, C. Ianelli, and S. J. Davis. 1994. The human cell-adhesion molecule CD2 binds CD58 with a very low affinity and an extremely fast dissociation rate but does not bind CD48 or CD59. Biochemistry. 33:10149–10160. [DOI] [PubMed] [Google Scholar]
- 26.Galon, J., M. W. Robertson, A. Galinha, N. Mazieres, R. Spagnoli, W. H. Fridman, and C. Sautes. 1997. Affinity of the interaction between Fc gamma receptor type III (Fc gammaRIII) and monomeric human IgG subclasses. Role of Fc gammaRIII glycosylation. Eur. J. Immunol. 27:1928–1932. [DOI] [PubMed] [Google Scholar]
- 27.Maenaka, K., P. A. van der Merwe, D. I. Stuart, E. Y. Jones, and P. Sondermann. 2001. The human low affinity Fcgamma receptors IIa, IIb, and III bind IgG with fast kinetics and distinct thermodynamic properties. J. Biol. Chem. 276:44898–44904. [DOI] [PubMed] [Google Scholar]
- 28.Pierres, A., A. M. Benoliel, P. Bongrand, and P. A. van der Merwe. 1996. Determination of the lifetime and force dependence of interactions of single bonds between surface-attached CD2 and CD48 adhesion molecules. Proc. Natl. Acad. Sci. USA. 93:15114–15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chesla, S. E., P. Selvaraj, and C. Zhu. 1998. Measuring two-dimensional receptor-ligand binding kinetics with micropipette. Biophys. J. 75:1553–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chesla, S. E., S. Nagarajan, P. Selvaraj, and C. Zhu. 2000. The membrane anchor influences ligand binding 2D kinetics rates and 3D affinity of FcγRIII (CD16). J. Biol. Chem. 275:10235–10246. [DOI] [PubMed] [Google Scholar]
- 31.Williams, T. E., S. Nagarajan, P. Selvaraj, and C. Zhu. 2000. Concurrent and independent binding of Fcgamma receptors IIa and IIIb to surface-bound IgG. Biophys. J. 79:1867–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams, T. E., P. Selvaraj, and C. Zhu. 2000. Concurrent binding to multiple ligands: kinetic rates of CD16b for membrane-bound IgG1 and IgG2. Biophys. J. 79:1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams, T. E., S. Nagarajan, P. Selvaraj, and C. Zhu. 2001. Quantifying the impact of membrane microtopology on effective two-dimensional affinity. J. Biol. Chem. 276:13283–13288. [DOI] [PubMed] [Google Scholar]
- 34.Huang, J., J. Chen, S. E. Chesla, T. Yago, R. P. McEver, C. Zhu, and M. Long. 2004. Quantifying the effects of molecular orientation and length on two-dimensional receptor-ligand binding kinetics. J. Biol. Chem. 279:44915–44923. [DOI] [PubMed] [Google Scholar]
- 35.Li, P., P. Selvaraj, and C. Zhu. 1999. Analysis of competition binding between soluble and membrane-bound ligands for cell surface receptors. Biophys. J. 77:3394–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dustin, M. L., S. K. Bromley, M. M. Davis, and C. Zhu. 2001. Identification of self through two-dimensional chemistry and synapses. Annu. Rev. Cell Dev. Biol. 17:133–157. [DOI] [PubMed] [Google Scholar]
- 37.Monks, C. R., B. A. Freiberg, H. Kupfer, N. Sciaky, and A. Kupfer. 1998. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 395:82–86. [DOI] [PubMed] [Google Scholar]
- 38.Grakoui, A., S. K. Bromley, C. Sumen, M. M. Davis, A. S. Shaw, P. M. Allen, and M. L. Dustin. 1999. The immunological synapse: a molecular machine controlling T cell activation. Science. 285:221–227. [DOI] [PubMed] [Google Scholar]
- 39.Reich, Z., J. J. Boniface, D. S. Lyons, N. Borochov, E. J. Wachtel, and M. M. Davis. 1997. Ligand-specific oligomerization of T-cell receptor molecules. Nature. 387:617–620. [DOI] [PubMed] [Google Scholar]